

Abstract
The mechanism by which the adenoviral (Ad) E1A oncogene induces cellular susceptibility to lysis by killer lymphocytes involves interactions between its first exon and different second-exon accessory regions. Mutational analysis showed that two first-exon regions—one in the N terminus and one in the conserved region 1 (CR1) domain—are necessary for this activity. E1A complex formation with cellular p300 protein through these first-exon-encoded regions correlated with induction of the cytolytic susceptible phenotype but was only effective in the context of E1A second-exon expression. An E1A first-exon deletion that prevented p300 binding eliminated both oncoprotein-induced cytolytic susceptibility and rejection of transfected sarcoma cells by immunocompetent animals. These results suggest that the E1A oncogene induces cytolytic susceptibility and tumor rejection by interactions with cellular proteins of the p300 family that affect transcription of genes involved in the cellular response to injury inflicted by host killer cells.
Oncogenes of the small DNA tumor viruses encode oncoproteins that immortalize mammalian cells from many species (reviewed in ref. 1). However, these oncogene-immortalized cells are usually rejected when inoculated into immunocompetent animals and can only form tumors in animals that lack normal cellular immune responses. These observations led to studies of differences in the susceptibilities of oncogene-immortalized cells to the cytolytic effects of the cellular components of the antineoplastic immune response.
One example of an oncogene-controlled cellular trait that limits tumorigenicity in immunocompetent animals is adenovirus type 2 or type 5 E1A oncogene induction of immortalized cell susceptibility to lysis by killer lymphocytes (2, 3, 4, 5, 6, 7). E1A-induced conversion of cells to a cytolytic susceptible phenotype occurs with hamster, rat, mouse, and human cells and eliminates sarcoma cell tumorigenicity in animals with competent natural killer lymphocyte (NK cell) responses (5). Recent studies showed that E1A-induced cytolytic susceptibility requires expression of different second-exon accessory regions to complement function(s) of the E1A first exon (Fig. 1) (8). Similar collaborations between the two exons are required for E1A activities that control viral and cellular gene transcription (9, 10, 11, 12) through interactions with cellular protein intermediaries (reviewed in ref. 1) and that immortalize primary cells (13) or cause complete neoplastic transformation of cells in collaboration with the Ad type 2 or type 5 E1B oncogene.
Figure 1.
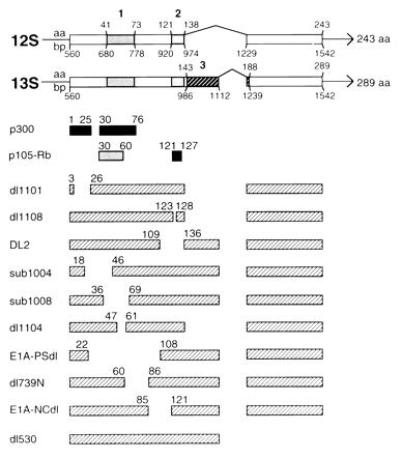
Genetic map locations of mutations spanning the E1A first exon and E1A–cell protein binding sites. The top two bar diagrams represent sequences (exons) from which E1A 12S and 13S mRNAs are transcribed; interruptions (indicated by carets) represent introns. E1A CR1, CR2, and CR3 are indicated by the boldface numbers above shaded areas. Amino acid and base pair positions of conserved regions, and initiation, splice, and termination sites are shown. The next two sets of bars beneath the E1A transcription map indicate consensus sites for E1A binding with cellular p300 and p105-retinoblastoma (Rb) proteins. The lighter of the two Rb-binding sites (amino acids 30–60) is of secondary importance (reviewed in ref. 1). Mutant E1A oncogene names are listed in the left column. Hatched bars next to these names represent coding regions of mutant polypeptides. Numbers above these bars indicate the last amino acid expressed at the ends of deletions.
The E1A first exon contains three regions that are highly conserved (CR1, CR2, and CR3) in the oncoproteins of different Ad (Fig. 1) (14) and a nonconserved, N-terminal region. To evaluate mechanisms of E1A-induced cytolytic susceptibility, we tested the effects of overlapping, in-frame deletions spanning the first exon. Two E1A regions—one in the N terminus and one in CR1—were required to collaborate with the second exon to induce cytolytic susceptibility. Protein binding studies indicated that interactions through these E1A regions with cellular proteins of the p300 family are involved in the mechanism through which the cytolytic phenotype is controlled. The importance of these E1A–p300 interactions for control of tumor development was demonstrated by the finding that sarcoma cells expressing mutant E1A proteins that fail to bind p300 formed progressive tumors in immunocompetent animals, whereas cells expressing p300-binding E1A proteins were rejected.
MATERIALS AND METHODS
Cells and Cell Lines.
Hamster embryo cells (HEC) were prepared and used as described (15). BHK-D5 cells expressing the Ad5 E1 gene region (E1A and E1B oncogenes) and BHK-neo cells expressing only the neor selection marker have been described (5). Cell lines were maintained in DMEM containing antibiotics and 5% calf serum (Sterile Systems, Logan, UT); 10% calf serum was used for HEC. Cells expressing neor were maintained in 200 μg/ml G418 (GIBCO/BRL) and were cultured in G418-free medium for 1 week before cytolysis assays. Cell lines were negative for mycoplasma using the Mycotect assay (GIBCO/BRL).
BHK-21 cells expressing the mutant E1A genes E1A-PSdl (16) and E1A-NCdl (17) were selected in G418 after cotransfection with the plasmids pRSV-PSdl and pRSV-NCdl, respectively, and pSVE1Bneo (18). Mutant E1A gene inserts containing the restriction enzymes sites BclI and HindIII were synthesized by PCR and inserted into the plasmid pRSVneo (19) after digestion with BglII and HindIII. The neor gene in pRSVneo was destroyed as a result of these insertions. G418-resistant clones were screened for E1A expression by Western blotting using the E1A-specific monoclonal antibody M73 (20). Subclones BHK-PSdl8.2T and BHK-NCdl3.3 were selected for study because of the >95% purity for E1A-positive cells and high-level E1A expression.
Viruses.
Viral mutants (Fig. 1) contained in-frame deletions of the E1A first exon of either Ad2 or Ad5 and, except for dl530, expressed a normal second exon. “12S viruses” express only the E1A 12S mRNA. “13S viruses” express only E1A 13S mRNA. Other viruses express the indicated regions of both E1A mRNAs.
The Ad5 12S viruses, dl1101, dl1104, and dl1108, were provided by Stan Bayley (21). The Ad2 13S virus, DL2, was provided by Margaret Fahnestock (22). The Ad5 E1A 13S viruses, sub1004/13S and sub1008/13S, were provided by Ed Ziff (23). The Ad5 mutants, E1A-PSdl (16) and E1A-NCdl (17), were provided by Betty Moran. The Ad5 mutant, dl739N, was provided by Ed Harlow (24). The Ad5 mutant, dl530, was provided by Nic Jones (25). The wild-type Ad5 viruses, dl309 (26) and dl520 (a 12S virus) (27), and the E1A-negative Ad5 virus, dl312, were provided by Tom Shenk. All virus pools were grown and titered on 293 cells. HEC were infected in suspension for 1 hr and incubated overnight as adherent cell cultures before use.
Detection of E1A Oncoprotein Expression and Binding to Cellular p300 Protein.
Quantitative Western blots were used to measure steady-state levels of E1A expression in infected and transfected cells as described (6) except detection was by enhanced chemiluminescence (Amersham). Cellular proteins complexed with E1A were coimmunoprecipitated using M73 (which recognizes an epitope in the E1A second exon) or M37 (for dl530-infected HEC only; which recognizes an E1A epitope in the first exon) antibodies (20) and resolved on SDS/7.5% polyacrylamide gels (28). p300 proteins in coimmunoprecipitates were detected by immunoblotting using p300-specific antibody (RW128; Upstate Biotechnology, Lake Placid, NY). Preliminary studies showed that this anti-human p300 antibody cross-reacts with hamster cell p300 (data not shown).
Cytolysis Assays.
Hamster NK cell killing of target cells was assessed as described (5, 29) using 6-hr 51Cr release assays. The significance of the differences in the cytolytic susceptibilities of control and E1A-expressing target cells was estimated by analysis of variance.
Tumor Induction Assays.
Male golden Syrian hamsters (2–4 months old) were obtained from the National Jewish Biological Resources Center (Denver). Female nude mice (athymic NCr-nu; 6–8 weeks old) were obtained from the Frederick Cancer Research Center. Subcutaneous tumor induction by BHK-21 cell lines was tested as described (5). To ensure that tumors were caused by E1A-positive cells, tumor cells from the lowest tumor-producing dose were tested for E1A expression by indirect immunofluorescence at the first tissue culture passage. The 50% end points for tumor production (TPD50) were calculated by the method of Karber (30).
RESULTS AND DISCUSSION
Sequences in the E1A N Terminus and CR1 Domain Must Be Expressed for Induction of Cytolytic Susceptibility in Infected HEC.
Transient E1A oncoprotein expression in cells to be tested as targets of NK killing was accomplished using short-term, nonlytic viral infection. This technique favors E1A expression in the majority of target cells and has the added advantage of allowing manipulation of oncoprotein expression level by altering the multiplicity of infection (i.e., numbers of viral plaque-forming units per cell) (31). These experiments were designed to achieve expression of each type of mutant E1A oncoprotein at a level that was comparable to or exceeded the levels of expression of native E1A oncoproteins in cells infected with wild-type control viruses (dl520 for E1A 12S viruses and dl309 for viruses expressing E1A 13S mRNA) known to induce susceptibility to NK killing (Fig. 2) (29). This approach ensured that reduced induction of cytolytic susceptibility was caused by the E1A first-exon mutation and not by inadequate oncoprotein expression. The Ad5 virus that deletes most of the E1A gene and is incapable of E1A expression or induction of cytolytic susceptibility, dl312 (Fig. 2 Right) (29), was used as a negative control infection.
Figure 2.
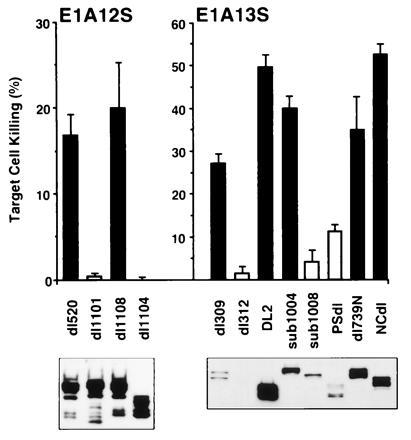
E1A oncoprotein expression and susceptibility to NK killing of infected HEC. (Upper) Bars represent three to eight cytotoxicity assays in which NK-cell-induced lysis was measured at the optimal 100:1 lymphocyte/target cell ratio. Cell killing is expressed as the net lysis (mean ± SEM) over that observed with same-day, mock-infected HEC (15.2 ± 1.5% lysis in 42 assays). Killing shown with black bars was significantly greater than that of uninfected HEC (P < 0.05). (Lower) Quantitative Western blot analysis of E1A oncoprotein expression in infected HEC. Multiplicities of infection (plaque-forming units per cell) for both types of assay were as follows: wild-type viruses, dl520 = 200 and dl309 = 100; mutant viruses, 400.
The E1A 12S virus that deletes E1A N-terminal sequences encoding amino acids 4–25, dl1101 (Fig. 1), failed to induce cytolytic susceptibility in infected HEC, despite mutant E1A oncoprotein expression comparable to that detected in the positive control infection (dl520-infected HEC; Fig. 2 Left). The dl1101 mutation eliminates one E1A region required for binding to the cellular transcriptional coactivator proteins p300 and cAMP response element binding protein (CREB) binding protein (hereafter referred to as p300-family proteins) (28, 32, 33, 34). The other E1A binding sites for p300-family proteins are located in CR1 (Fig. 1). In addition to its effects on p300-family protein binding, the dl1101 mutation also reduces but does not eliminate E1A binding to cellular Rb-family proteins (p105-Rb, p107, and p130) (33). Rb-family proteins bind E1A primarily through the CR2 domain (Fig. 1) but have secondary binding through the E1A N terminus (33, 35). Mutants with deletions only in E1A CR2 (dl1108 and DL2; Fig. 1) were tested for induction of cytolytic susceptibility to evaluate cell protein binding through this region independently of the E1A N terminus (Fig. 2). Dl1108, which deletes CR2 amino acids 124–127, which in turn are required to bind all Rb-family proteins (33), induced high level susceptibility to NK killing. The observation that E1A CR2 expression was not required to induce cytolytic susceptibility was confirmed by the similar result using HEC infected with DL2 (Fig. 2 Right), which deletes almost all of CR2 (Fig. 1). In summary, these data indicated that E1A-induced cytolytic susceptibility requires expression of E1A N-terminal sequences that are required for p300-family protein binding and that expression of E1A CR2 is neither necessary (dl1108 and DL2) nor sufficient (dl1101) for induction of this phenotype.
To further define E1A first-exon expression requirements for induction of cytolytic susceptibility, viruses with overlapping mutations between the E1A N terminus and CR2 were tested. HEC infected with sub1004/13S that deletes E1A amino acids 19–45 were highly susceptible to NK killing (Fig. 2 Right). Comparison of the mutations in sub1004/13S and dl1101 suggested that only E1A amino acids 4–18 in the E1A N terminus are necessary for this E1A activity. In contrast to sub1004/13S, sub1008/13S (amino acids 37–68 deleted; Fig. 1) eliminated E1A-induced susceptibility to NK killing (Fig. 2). The fact that both of these mutants express E1A CR3 domain (23) indicates that this E1A region that is unique to the 13S mRNA (Fig. 1) is both unnecessary as reported (2, 3, 8, 29) and insufficient for this E1A activity. Comparison of the mutations in sub1004/13S and sub1008/13S also suggested that expression of amino acids 46–68 in E1A CR1 (Fig. 1) is required for E1A-induced cytolytic susceptibility. Refinement of this observation was obtained using the mutant, dl1104, that deletes amino acids 48–60 (Fig. 1) and failed to induce cytolytic susceptibility (Fig. 2 Left). The fact that the dl1104 mutation also removes the second E1A binding site for cellular p300-family proteins (33) supported the inference from the dl1101 results that E1A–p300 complex formation is required for this activity.
Two other E1A CR1 mutations were used to confirm the importance of this region for induction of cytolytic susceptibility and to test the effect of deleting CR1 C-terminal sequences. E1A-PSdl, which deletes all of CR1 but does not affect expression of the N-terminal 22 amino acids (16), did not induce significant cytolytic susceptibility in HEC, despite expression of mutant E1A protein at a level comparable to (Fig. 2 Right) or in excess of (data not shown) the dl309 control. This confirmed the conclusion that expression of E1A CR1 is required for induction of cytolytic susceptibility independently of the N-terminal region of E1A. Dl739N-infected HEC were used to test the requirement for sequences at the C-terminal boundary of E1A CR1 for induction of cytolytic susceptibility. The dl739N deletion (amino acids 61–85) begins where that of dl1104 ends (Fig. 1). Dl739N-infected HEC expressed large amounts of E1A and were highly susceptible to lysis by NK cells (Fig. 2). Therefore, only the portion of CR1 defined by the dl1104 mutation was required for E1A-induced cytolytic susceptibility. These results and those with dl1101 defined two first-exon regions required for induction of susceptibility to NK killing—amino acids 4–18 and 48–60.
As noted, the E1A nucleotide sequence between CR1 and CR2 is not conserved among Ad serotypes. However, preservation of this spacer region is common among E1A genes. Wang et al. (36) reported that this E1A spacer sequence is required for E1A to bind simultaneously to p300 and p105-Rb. Both of these cellular proteins have secondary binding sites in CR1. Wang et al. used the E1A mutation that deletes most of the spacer sequences, NCdl, to define these cell protein binding characteristics of E1A. In the present study, HEC infected with E1A-NCdl were highly susceptible to lysis by NK cells (Fig. 2 Right). Therefore, maintenance of this E1A spacer region is not required for E1A-induced cytolytic susceptibility. This conclusion is compatible with the observation that E1A mutations (i.e., dl1108 and DL2) that remove the Rb binding region in CR2 but retain the p300 binding regions in the N terminus and CR1 retain the ability to induce susceptibility to NK killing (Fig. 2).
E1A Oncoprotein Binding to p300 Must Be Complemented by E1A Second-Exon Functions to Induce Cytolytic Susceptibility.
E1A oncoproteins affect cellular gene expression and associated phenotypes indirectly through interactions with transcriptionally active cellular proteins, such as those of the p300/CREB binding protein family and Rb family (reviewed in refs. 1, 28, 33, 37, and 38). The observation that E1A forms complexes with similar cellular proteins in hamster, rat, mouse, and human cells (38) suggests that these same intermediary interactions may be used to regulate cellular gene transcription in all mammalian cells.
The data in Fig. 2 suggested a requirement for E1A oncoproteins to complex with cellular p300 protein to induce cytolytic susceptibility in hamster cells. To test this correlation directly, E1A–p300 coprecipitation studies were done using hamster cells infected with selected E1A mutant viruses (Fig. 3). The E1A mutations that abrogated E1A-induced cytolytic susceptibility, dl1101, dl1104, and PSdl (Fig. 2), also eliminated p300 protein binding. Conversely, E1A mutations that did not prevent E1A-induced cytolytic susceptibility retained the ability to coprecipitate p300 (Figs. 2 and 3). These results supported the hypothesis that E1A–p300 complex formation is required for E1A-induced cytolytic susceptibility (Fig. 3).
Figure 3.
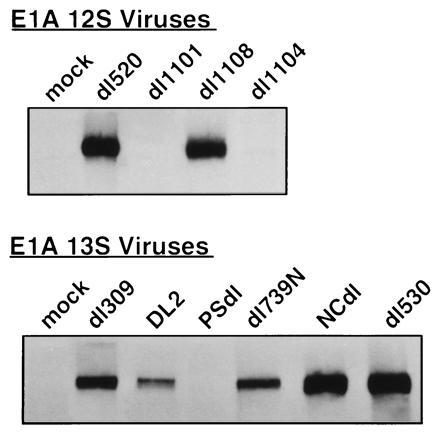
Immunoprecipitations of E1A–p300 protein complexes from infected HEC. Immune complexes formed by E1A-specific antibody (not cross-reactive with p300) were resolved by SDS/PAGE, blotted, probed with antibody specific for cellular p300 proteins, and detected by enhanced chemiluminescence.
There was one apparent discrepancy between these E1A–p300 coprecipitation results and those reported for infected human cells. Whyte and Harlow (24) did not detect p300 protein in dl739N-infected HeLa cells when [35S]methionine-labeled proteins were coprecipitated with E1A and visualized by SDS/PAGE analysis. In contrast, we detected p300 proteins in E1A complexes in dl739N-infected HEC using a Western blotting assay. The deletion in the dl739N virus used to infect HEC was confirmed by PCR analysis (data not shown) to ensure identity with the virus used by Whyte and Harlow. These differences could be species-related. However, it is also possible that Western blotting of p300 protein in E1A–cell protein complexes is a more sensitive method of detection. Our unpublished data showing that p300 proteins are not detected in immunoprecipitated E1A complexes in metabolically labeled, dl739N-infected HEC support the latter explanation.
Our previous observation that second-exon expression is necessary for E1A-induced cytolytic susceptibility (8) suggested the hypothesis that E1A–p300 complex formation is necessary but not sufficient for this E1A activity unless the E1A second exon is also expressed. This possibility was tested using the Ad5 mutant that expresses only the E1A 13S first exon (Fig. 1) and fails to induce cytolytic susceptibility (8), dl530 (25). The observation that p300 protein is present in E1A–cell protein complexes in dl530-infected HEC suggests that the mechanism by which E1A increases the cytolytic susceptibility of expresser cells involves two steps—binding of p300-family proteins through the E1A first exon and modification of the E1A–cell protein complex by accessory activities encoded by the second exon. Formal analysis of this proposal will require further functional studies and perhaps structural analysis of these E1A–cell protein complexes.
Abrogation of E1A Oncoprotein Binding to p300 Prevents E1A-Induced Rejection of Sarcoma Cells by Immunocompetent Animals.
E1A oncogene expression in BHK-21 cells induces susceptibility to NK killing and reduces tumor-forming capacity in animals with competent NK cell responses (5, 18). For example, immunocompetent adult hamsters are highly susceptible to tumor induction by E1A-negative BHK-21 cells (requiring approximately 103 cells for a 50% end point of tumor formation, i.e., TPD50 = 3.0; see Table 1 footnote) but develop no tumors after a challenge with 107 E1A-positive cells (i.e., TPD50 ≥ 7.5) (5, 18). In contrast, nude mice, which lack T lymphocyte responses and whose NK cells are incompetent to lyse E1A-positive hamster cells (39), are highly susceptible to tumor formation by E1A-positive BHK-21 cells (5). Using this model, we tested the correlation among E1A–p300 complex formation, E1A-induced cytolytic susceptibility, and E1A-induced tumor rejection. BHK-PSdl8.2T cells expressing the loss-of-function, p300(−) mutation, PSdl (Figs. 1, 2, 3), were compared with cells expressing either wild-type E1A (BHK-D5) or the E1A-NCdl mutation that retained the ability to bind p300 and induce cytolytic susceptibility. BHK-21 cells expressing these mutant E1A oncoproteins exhibited cytolytic phenotypes and patterns of E1A–p300 binding identical to those observed with infected HEC (Fig. 4, compare with Fig. 3). BHK-NCdl3.3 cells were susceptible to lysis by NK cells and contained E1A–p300 protein complexes, whereas BHK-PSdl8.2T cells were resistant to lysis by NK cells and lacked detectable E1A–p300 complexes. Furthermore, these in vitro data predicted the patterns of tumor development in immunocompetent hamsters. These animals rejected challenges with large numbers of cytolytic susceptible BHK-NCdl3.3 cells (TPD50 ≥ 7.4) but developed tumors when challenged with 100-fold fewer cytolytic resistant BHK-PSdl8.2T cells (TPD50 = 5.2). Whereas BHK-NCdl3.3 cells failed to induce tumors in immunocompetent hamsters, these cells retained high-level tumorigenicity in immunodeficient, nude mice (TPD50 = 4.5 cells). This observation is consistent with the conclusion that the E1A-NCdl mutation does not affect the wild-type E1A oncogene activity that eliminates sarcoma cell tumorigenicity (as evidenced by its ability to increase the TPD50) and that rejection of BHK-NCdl3.3 cells, like rejection of BHK-D5 cells expressing wild-type E1A (5), depends on the competence of the cellular immune response of the host. Cumulatively, these results support the hypothesis that the E1A-induced cytolytic susceptibility and the related rejection of E1A-positive sarcoma cells by immunocompetent animals is mediated by E1A oncoprotein activities that require E1A–p300-family complex formation in neoplastic cells.
Table 1.
Tumorigenicity of BHK-21 transfectants expressing mutant E1A oncoproteins differing in their abilities to form p300 complexes and induce cytolytic susceptibility
| Cell line | Recipient | Tumor
incidence*
|
TPD50† | ||||
|---|---|---|---|---|---|---|---|
| No. of cells
innoculated
| |||||||
| 103 | 104 | 105 | 106 | 107 | |||
| BHK-neo | Hamster | 1/3 | 3/3 | 3/3 | 3/3 | 3/3 | 3.2 |
| BHK-D5 | Hamster | — | — | — | — | 0/3 | ≥7.5 |
| BHK-NCd13.3 | Hamster | — | — | 0/3 | 0/3 | 1/9 | ≥7.4 |
| BHK-NCd13.3 | Nude mouse | 0/3 | 0/3 | 3/3 | 3/3 | 3/3 | 4.5 |
| BHK-PSd18.2T | Hamster | 1/6 | 0/6 | 3/6 | 5/6 | 4/5 | 5.2 |
| BHK-PSd18.2T | Nude mouse | 0/3 | 0/3 | 3/3 | 3/3 | 2/2 | 4.5 |
BHK-D5 cells consistently form tumors in nude mice (5). The tumor that developed in a hamster challenged with BHK-NCd13.3 cells was E1A-negative and likely represents a contaminant of the clone. Differences in tumor producing efficiency of less than one log (e.g., BHK-PSd18.2T—hamster vs. nude mouse) are not significant.
Tumor incidence, number of animals with progressive tumors per number of challenged animals.
TPD50, logarithm of the cell number required to cause tumors in 50% of animals (30).
Figure 4.
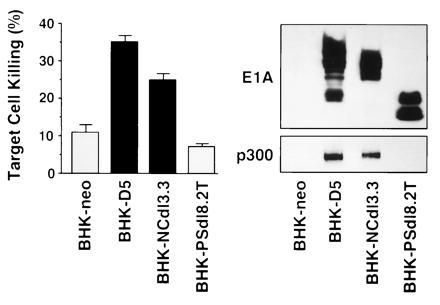
Susceptibility to NK killing (Left), E1A expression (Upper Right), and E1A–p300 complex formation (Lower Right) in transfected BHK-21 cells. Bars represent the results (mean ± SEM) of eight NK cell assays. BHK-D5 and BHK-NCdl3.3 cells were significantly more susceptible to NK killing than the control transfectant, BHK-neo (P < 0.05). E1A oncoprotein expression was detected by quantitative Western blotting. E1A–p300 complexes were detected by the stepwise procedure described in the Fig. 2 legend.
Correlations Between First-Exon Requirements for E1A-Induced Cytolytic Susceptibility and Other E1A Activities.
These and other (8) data indicate that noncontiguous regions of the E1A oncoprotein—amino acids 4–18 in the N terminus, amino acids 48–60 in CR1, and either of two accessory regions in the second exon—are required for the mechanism by which E1A converts cells to the cytolytic susceptible phenotype. Other reports implicate similar regions in different E1A activities, including cell protein interactions (reviewed in refs. 1, 33, 35, 36, and 40, 41, 42) and transcriptional activation (12, 43, 44) and repression (9, 42, 45, 46) of viral and cellular genes.
Detailed comparisons of mapping studies reveal both similarities and differences in E1A expression requirements for induction of cytolytic susceptibility and other E1A effects. One example is the comparison with E1A sequence requirements for sensitization to tumor necrosis factor α (TNFα) cytotoxicity (3, 47, 48). E1A 12S mRNA products induce susceptibility to both NK killing (e.g., see dl520-infected HEC, Fig. 2) (2, 3, 29) and TNF cytotoxicity. Therefore, E1A CR3 function (Fig. 1) is not required for either activity. Both activities require expression of sequences within E1A CR1 (Fig. 1). E1A amino acids 48–60 are required to induce NK susceptibility in hamster cells (Figs. 2, 3, 4), and amino acids 31–60 are required to sensitize mouse cells to TNFα (47). Like E1A-induced susceptibility to lysis by killer lymphocytes, TNFα-induced cytotoxicity has also been related to E1A second-exon function (49). At this point in the comparison between E1A-induced susceptibility to NK killing and TNFα cytotoxicity, the relationship in sequence requirements diverges. E1A N-terminal sequences must be expressed for E1A-induced susceptibility of hamster cells to NK killing (Figs. 1 and 2), whereas deletion of these sequences has no effect on E1A-induced sensitivity to TNFα in mouse cells (47). This may be explained by a difference in the role of E1A complex formation with Rb-family proteins in these two activities. Expression of the E1A CR2 region that binds Rb-family proteins is sufficient, in the absence of the E1A N terminus, to sensitize mouse cells to TNFα (48) but is insufficient to induce susceptibility to NK cells (e.g., see dl1101, Figs. 1 and 2). This could reflect differences in either the species tested or the mechanisms through which E1A controls these cytolytic phenotypes. There are also differences in the cytolytic mechanisms used by TNFα and killer lymphocytes to kill E1A-positive target cells (7) that may contribute to these differences.
Another consideration is whether E1A-induced cytolytic susceptibility is mediated through the same pathways by which E1A regulates the cell cycle and immortalizes cells. The effects of selected E1A mutations on stimulation of cellular DNA synthesis and immortalization suggests that these E1A activities are regulated differently. The mutation in sub1004/13S eliminates E1A stimulation of cellular DNA synthesis (23), as do deletions in E1A CR2 (28); however, none of these mutations prevented E1A-induced cytolytic susceptibility (Figs. 1 and 2). Conversely, expression of the E1A first exon in the absence of the second exon is sufficient to induce cellular DNA synthesis (16) but not cytolytic susceptibility (8). It is therefore not surprising that there are E1A mutations, such as sub1004/13S (23) and E1A-NCdl (36), that block viral immortalizing activity but do not affect induction of cytolytic susceptibility (Fig. 2), because the ability of E1A to immortalize cells is linked to E1A-induced cellular DNA synthesis.
Possible Mechanisms of Action.
Among the mechanisms by which E1A expression could induce cytolytic susceptibility is alteration in target cell surface expression of major histocompatibility complex (MHC) class I molecules. In some studies, target cell surface expression of MHC class I molecules conveys an antilytic “off signal” to NK cells (50). Alterations in this NK-inhibitory signal, by either down-regulation of MHC molecule expression or MHC molecule binding to certain peptides can increase NK cell cytolytic activity (50, 51). The available data do not support either of these mechanisms. E1A of Ad type 2 or type 5 does not predictably alter MHC class I molecule expression (52, 53). Furthermore, some E1A-transfected, NK-susceptible target cells actually express more MHC class I molecules than the NK-resistant cells from which they were derived (6, 54). Regarding the peptide binding hypothesis, our recent E1A mapping data failed to identify any single E1A peptide coding region linked to induction of cytolytic susceptibility (8). Therefore, it does not appear that the pathway through which E1A induces conversion of cells to the cytolytic susceptible phenotype involves quantitative or qualitative regulation of MHC class I molecules.
Our recently published data suggest that the major mechanism through which E1A induces cytolytic susceptibility involves a postrecognition stage in the interaction with killer lymphocytes rather than an E1A effect on target cell recognition (7). Killer cells use two major types of mechanisms to lyse their targets (reviewed in refs. 55 and 56). One involves killer cell degranulation and is mediated by the joint activities of perforin and granzymes. The other involves killer cell expression of Fas-ligand which signals injury through interactions with its receptor, Fas-antigen, on the target cell surface. E1A sensitizes cells to both of these cytolytic mechanisms (7). Since both degranulation-dependent and Fas-dependent injury can cause apoptotic cell death (56, 57) and since E1A can sensitize cells to drug-induced apoptosis (58), it is possible that E1A-induced cytolytic susceptibility involves an E1A effect on cellular apoptotic pathways. Our unpublished data support this association. The molecular mechanism(s) through which this postrecognition effect is mediated by E1A remain to be defined. However, based on the data presented in this report, these mechanisms appear to involve E1A interactions with p300-family proteins.
p300 is a nuclear phosphoprotein that is homologous to CREB binding protein (13), which binds to the CREB transcription factor. This “p300-family” of proteins interacts with a variety of enhancer-binding transcription factors and appears to function by linking these factors to components of the basal transcription machinery. For this reason, p300 and CREB binding protein have been described as transcriptional adaptor or coactivator proteins (34, 59, 60, 61). E1A interactions with p300-family transcriptional coactivators may either induce (62, 63, 64) or repress (34, 46, 60, 61, 65, 66, 67, 68) expression of cellular genes, depending on the transcription factors involved. In this context, the data presented here suggest that the mechanism of E1A-induced cytolytic susceptibility and associated tumor rejection involves transcriptional modulation of key cellular genes whose function affects the response to killer cell injury. Definition of the cellular pathways through which E1A oncoproteins control cytolytic and tumorigenic phenotypes will require identification of these postulated cellular genes and studies of their regulation by E1A. These future studies may provide an explanation for the lack of oncogenicity of cells transformed by DNA tumor viruses in immunocompetent animals and may also contribute to a basic understanding of the molecular mechanisms that control the outcome of tumor cell interactions with killer cells.
Acknowledgments
We thank Drs. S. Bayley, M. Fahnestock, N. Jones, E. Moran, T. Shenk, and E. Ziff for providing the indicated viral mutants; Dr. Harlow for E1A-specific antibodies; and L. Landskröner and B. Silverstein for artwork and photography. This work was supported by Public Health Service Grant CA43187 from the National Cancer Institute and American Cancer Society Grant IM-730. J.L.C. is the C. L. C. Kramer Foundation Scientist in Medicine at National Jewish Center for Immunology and Respiratory Medicine.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: Ad, adenovirus; E1A, early region 1A gene of the adenovirus genome; HEC, hamster embryo cells; NK cell, natural killer cell; CR1, CR2, and CR3, conserved regions 1, 2, and 3 in the first exon of the Ad E1A gene; Rb, retinoblastoma; TNFα, tumor necrosis factor α; MHC, major histocompatibility complex; TPD50, 50% end point of tumor production; CREB, cAMP response element binding protein.
References
- 1.Bayley S T, Mymryk J S. Int J Oncol. 1994;5:425–444. doi: 10.3892/ijo.5.3.425. [DOI] [PubMed] [Google Scholar]
- 2.Cook J L, Walker T A, Lewis A M, Jr, Ruley H E, Graham F L, Pilder S H. Proc Natl Acad Sci USA. 1986;83:6965–6969. doi: 10.1073/pnas.83.18.6965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cook J L, May D L, Wilson B A, Holskin B, Chen M-J, Shalloway D, Walker T A. J Immunol. 1989;142:4527–4534. [PubMed] [Google Scholar]
- 4.Kenyon D J, Dougherty J, Raska J K. Virology. 1991;180:818–821. doi: 10.1016/0042-6822(91)90099-w. [DOI] [PubMed] [Google Scholar]
- 5.Walker T A, Wilson B A, Lewis A M, Jr, Cook J L. Proc Natl Acad Sci USA. 1991;88:6491–6495. doi: 10.1073/pnas.88.15.6491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Routes J M, Cook J L. Virology. 1995;210:421–428. doi: 10.1006/viro.1995.1358. [DOI] [PubMed] [Google Scholar]
- 7.Cook J L, Potter T A, Bellgrau D, Routes B A. Oncogene. 1996;13:833–842. [PubMed] [Google Scholar]
- 8.Krantz C K, Routes B A, Quinlan M P, Cook J L. Virology. 1996;217:23–32. doi: 10.1006/viro.1996.0089. [DOI] [PubMed] [Google Scholar]
- 9.Velcich A, Ziff E. Mol Cell Biol. 1988;8:2177–2183. doi: 10.1128/mcb.8.5.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bautista D S, Hitt M, McGrory J, Graham F L. Virology. 1991;182:578–596. doi: 10.1016/0042-6822(91)90599-7. [DOI] [PubMed] [Google Scholar]
- 11.Linder S, Przemyslaw P, Svensson C, Marshall H, Bondesson M, Akusjarvi G. Oncogene. 1992;7:439–443. [PubMed] [Google Scholar]
- 12.Bondesson M, Svensson C, Linder S, Akusjarvi G. EMBO J. 1992;11:3347–3354. doi: 10.1002/j.1460-2075.1992.tb05413.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quinlan M P, Douglas J L. J Virol. 1992;66:2020–2030. doi: 10.1128/jvi.66.4.2020-2030.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Ormondt H, Maat J, Dijkema R. Gene. 1980;12:63–76. doi: 10.1016/0378-1119(80)90016-5. [DOI] [PubMed] [Google Scholar]
- 15.Cook J L, Lewis A M., Jr Cancer Res. 1979;39:1455–1461. [PubMed] [Google Scholar]
- 16.Moran B, Zerler B. Mol Cell Biol. 1988;8:1756–1764. doi: 10.1128/mcb.8.4.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moran E, Zerler B, Harrison T M, Mathews M B. Mol Cell Biol. 1986;6:3470–3480. doi: 10.1128/mcb.6.10.3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cook J L, Wilson B A, Wolf L A, Walker T A. Oncogene. 1993;8:625–635. [PubMed] [Google Scholar]
- 19.Gorman C, Padmanabhan R, Howard B H. Science. 1983;221:551–553. doi: 10.1126/science.6306768. [DOI] [PubMed] [Google Scholar]
- 20.Harlow E, Franza R, Jr, Schley C. J Virol. 1985;55:533–546. doi: 10.1128/jvi.55.3.533-546.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jelsma T N, Howe J A, Evelegh C M, Cunniff N F, Skiadopoulos M H, Floroff M R, Denman J E, Bayley S T. Virology. 1988;163:494–502. doi: 10.1016/0042-6822(88)90290-5. [DOI] [PubMed] [Google Scholar]
- 22.Fahnestock M L, Lewis J B. J Virol. 1989;63:1495–1504. doi: 10.1128/jvi.63.4.1495-1504.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith D H, Ziff E B. Mol Cell Biol. 1988;8:3882–3890. doi: 10.1128/mcb.8.9.3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whyte P, Williamson N M, Harlow E. Cell. 1989;56:67–75. doi: 10.1016/0092-8674(89)90984-7. [DOI] [PubMed] [Google Scholar]
- 25.Schneider J F, Fisher F, Goding C R, Jones N C. EMBO J. 1987;6:2053–2060. doi: 10.1002/j.1460-2075.1987.tb02470.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones N, Shenk T. Cell. 1979;17:683–689. doi: 10.1016/0092-8674(79)90275-7. [DOI] [PubMed] [Google Scholar]
- 27.Haley K P, Overhauser J, Babiss L E, Ginsberg H S, Jones N C. Proc Natl Acad Sci USA. 1984;81:5734–5738. doi: 10.1073/pnas.81.18.5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Howe J A, Bayley S T. Virology. 1992;186:15–24. doi: 10.1016/0042-6822(92)90057-v. [DOI] [PubMed] [Google Scholar]
- 29.Cook J L, May D L, Lewis A M, Jr, Walker T A. J Virol. 1987;61:3510–3520. doi: 10.1128/jvi.61.11.3510-3520.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karber G. Arch Exp Pathol Pharmakol. 1931;162:480–483. [PubMed] [Google Scholar]
- 31.Cook J L, Lewis A M., Jr Science. 1984;224:612–615. doi: 10.1126/science.6710160. [DOI] [PubMed] [Google Scholar]
- 32.Egan C, Jelsma T N, Howe J A, Bayley S T, Ferguson B, Branton P E. Mol Cell Biol. 1988;8:3955–3959. doi: 10.1128/mcb.8.9.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barbeau D, Charbonneau R, Whalen S G, Bayley S T, Branton P. Oncogene. 1994;9:359–373. [PubMed] [Google Scholar]
- 34.Arany Z, Newsome D, Oldread E, Livingston D M, Eckner R. Nature (London) 1995;374:81–84. doi: 10.1038/374081a0. [DOI] [PubMed] [Google Scholar]
- 35.Fattaey A R, Harlow E, Helin K. Mol Cell Biol. 1993;13:7267–7277. doi: 10.1128/mcb.13.12.7267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang H-G H, Moran E, Yaciuk P. J Virol. 1995;69:7917–7924. doi: 10.1128/jvi.69.12.7917-7924.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yee S-P, Branton P E. Virology. 1985;147:142–153. doi: 10.1016/0042-6822(85)90234-x. [DOI] [PubMed] [Google Scholar]
- 38.Harlow E, Whyte P, Franza R, Jr, Schley C. Mol Cell Biol. 1986;6:1579–1589. doi: 10.1128/mcb.6.5.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cook J L, Lewis A M., Jr J Virol. 1987;61:2155–2161. doi: 10.1128/jvi.61.7.2155-2161.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ikeda M-A, Nevins J R. Mol Cell Biol. 1993;13:7029–7035. doi: 10.1128/mcb.13.11.7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang H-G H, Rikitake Y, Carter M C, Yaciuk P, Abraham S E, Zerler B, Moran E. J Virol. 1993;67:476–488. doi: 10.1128/jvi.67.1.476-488.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lewis B A, Tullis G, Seto E, Horikoshi N, Weinmann R, Shenk T. J Virol. 1995;69:1628–1636. doi: 10.1128/jvi.69.3.1628-1636.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kannabiran C, Morris G F, Labrie C, Mathews M B. J Virol. 1993;67:507–515. doi: 10.1128/jvi.67.1.507-515.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wong H, Ziff E B. J Virol. 1994;68:4910–4920. doi: 10.1128/jvi.68.8.4910-4920.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Datta P K, Bagchi S. J Biol Chem. 1994;269:25392–25399. [PubMed] [Google Scholar]
- 46.Kirshenbaum L A, Schneider M D. J Biol Chem. 1995;270:7791–7794. doi: 10.1074/jbc.270.14.7791. [DOI] [PubMed] [Google Scholar]
- 47.Duerksen-Hughes P, Hermiston T W, Wold W S M, Gooding L R. J Virol. 1991;65:1236–1244. doi: 10.1128/jvi.65.3.1236-1244.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shisler J, Duerksen-Hughes P, Hermiston T M, Wold W S, Gooding L R. J Virol. 1996;70:68–77. doi: 10.1128/jvi.70.1.68-77.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsuji Y, Ninomiya-Tsuji J, Torti S V, Torti F M. J Immunol. 1993;150:1897–1907. [PubMed] [Google Scholar]
- 50.Karrë K. Semin Immunol. 1993;5:127–145. doi: 10.1006/smim.1993.1016. [DOI] [PubMed] [Google Scholar]
- 51.Chadwick B S, Sambhara S R, Sasakura Y, Miller R G. J Immunol. 1992;149:3150–3156. [PubMed] [Google Scholar]
- 52.Haddada H, Lewis A M, Jr, Sogn J A, Coligan J E, Cook J L, Walker T A, Levine A S. Proc Natl Acad Sci USA. 1986;83:9684–9688. doi: 10.1073/pnas.83.24.9684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haddada H, Sogn J A, Coligan J E, Carbone M, Dixon K, Levine A S, Lewis A M., Jr Cell. 1988;61:2755–2761. doi: 10.1128/jvi.62.8.2755-2761.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Routes J, Metz B, Cook J. J Virol. 1993;67:3176–3181. doi: 10.1128/jvi.67.6.3176-3181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Arase H, Arase N, Saito T. J Exp Med. 1995;181:1235–1238. doi: 10.1084/jem.181.3.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Berke G. Annu Rev Immunol. 1994;12:735–773. doi: 10.1146/annurev.iy.12.040194.003511. [DOI] [PubMed] [Google Scholar]
- 57.Nagata S, Golstein P. Science. 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 58.Lowe S W, Ruley H E, Jacks T, Housman D E. Cell. 1993;74:957–967. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- 59.Arany Z, Sellers W, Livingston D, Eckner R. Cell. 1994;77:799–800. doi: 10.1016/0092-8674(94)90127-9. [DOI] [PubMed] [Google Scholar]
- 60.Eckner R, Ewen M, Newson D, Gerdes M, DeCaprio J, Lawrence J, Livingston D. Genes Dev. 1994;8:869–884. doi: 10.1101/gad.8.8.869. [DOI] [PubMed] [Google Scholar]
- 61.Lundblad J, Kwok R, Laurance M, Harter M, Goodman R. Nature (London) 1995;374:85–88. doi: 10.1038/374085a0. [DOI] [PubMed] [Google Scholar]
- 62.Gedrich R, Bayley S, Engel D. J Virol. 1992;66:5849–5859. doi: 10.1128/jvi.66.10.5849-5859.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hagmeyer B, König H, Herr I, Offringa R, Zantema A, van der Eb A, Herrlich P, Angel P. EMBO J. 1993;12:3559–3572. doi: 10.1002/j.1460-2075.1993.tb06030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kitabayashi I, Eckner R, Arany Z, Chiu R, Gachelin G, Livingston D, Yokoyama K. EMBO J. 1995;14:3496–3509. doi: 10.1002/j.1460-2075.1995.tb07356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stein R, Corrigan M, Yaciuk P, Whelan J, Moran E. J Virol. 1990;64:4421–4427. doi: 10.1128/jvi.64.9.4421-4427.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Song C-Z, Lowenstein P, Green M. J Virol. 1995;69:2907–2911. doi: 10.1128/jvi.69.5.2907-2911.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dorsman J, Hagmeyer B, Veenstra J, Elfferich P, Nabben N, Zantema A, van der Eb A. J Virol. 1995;69:2962–2967. doi: 10.1128/jvi.69.5.2962-2967.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Smits P, de Wit L, van der Eb A, Zantema A. Oncogene. 1996;12:1529–1535. [PubMed] [Google Scholar]