

Abstract
CD8+ cytotoxic T cells recognize their targets by the presence of unique peptide bound to a major histocompatibility complex (MHC) class I molecules on the cell surface. The MHC molecules normally display thousands of distinct peptides, making it difficult to identify individual antigenic peptides, their protein precursors, and their relative importance in the T-cell response. Here we used the EL-4 tumor-specific lacZ-inducible KZ30.6 T cell as a probe for detecting the peptide/MHC ligand that was generated in cells transfected with an EL-4 cDNA library. These expression screens allowed identification of a mouse mammary tumor virus (MMTV) transcript as the source of the antigenic peptide presented by the Kb MHC molecule. The antigenic activity was encoded within the MMTV env gene and was defined by the octapeptide ANYDFICV (AFV8). Synthetic AFV8 stimulated KZ30.6 T cells at picomolar concentrations and coeluted with one of two active peptides in HPLC-fractionated extracts of EL-4 cells. The AFV8/Kb complex was also recognized by two other EL-4-specific T cells. The results illustrate a novel strategy for identifying T-cell-stimulating antigens and suggest that the MMTV env gene and its naturally processed AFV8 peptide product can serve as a model for study of antigen processing and tumor immunotherapy.
Cytotoxic CD8+ T cells survey the target cell surface for presence of unique complexes between a peptide and a major histocompatibility complex (MHC) class I molecule and cause lysis of the target cell. These MHC-bound peptides are derived from endogenous proteins via the antigen-processing pathway and serve as the basis for immune surveillance of virally infected, transformed, allogeneic, or even self tissues (1, 2, 3). The pool of peptides displayed by any one MHC molecule depends upon the nature of its antigen binding groove and represents thousands of distinct peptides (4, 5). Identifying individual peptides from among this complex pool is nevertheless the key to defining the unique ligands recognized by the T cells, for determining the role of distinct peptide/MHC complexes in the T-cell response, and for the study of the antigen-processing pathway (6).
Notwithstanding the enormous complexity of the peptide pool displayed by MHC molecules, several CD8+ T-cell-stimulating peptides have been identified by two different strategies. In the biochemical strategy, the unique T-cell-stimulating peptide is purified from the target cell by several rounds of HPLC and its sequence is determined by Edman degradation (7) or by mass spectrometry (8). Alternatively, the antigen gene can be identified first by its ability to generate the peptide/MHC complex in transfected cells and the antigenic peptide can be defined subsequently (9, 10, 11, 12). Unlike, peptide purification, the expression cloning of the antigen gene does not depend upon the abundance of the peptide in the target tissue. Furthermore, the sensitivity of the assays can be significantly enhanced by using lacZ-inducible single-cell, rather than conventional bulk T-cell, assays and by screening cDNA libraries in transiently transfected cells (12, 13). Indeed, we recently showed that even rare processed peptides (<10 copies per cell) can be identified by using lacZ-inducible T cells as probes for detecting the expression of the relevant peptide/MHC complex (14, 15).
The identification of several T-cell-stimulating antigenic peptides and their donor proteins are providing novel insights into diverse immunological phenomena. For example, it is now clear that the high frequency of alloreactive T cells is due to recognition of a large number of (self) peptides that are normally presented by (foreign) MHC (7, 8, 15, 16, 17, 18). The pool of self peptides displayed by the MHC in the thymus is also the basis for the generation of the diverse T-cell repertoire in the thymus (19). Likewise, T-cell immunity to virus-infected or tumor cells is due to the presence of unique peptide/MHC complexes on their surface (10, 20, 21). Indeed several MHC-bound peptides have been identified in melanoma and other tumors that are derived from tissue-specific or mutant genes (11, 22, 23, 24). These T-cell-stimulating tumor peptides may serve as targets for immunological intervention of tumor growth (25).
The central element of any strategy for identifying unique peptide–MHC complexes is the T cells used to detect their presence. Often tumor cells are poor immunogens in syngeneic animals making it difficult to obtain high-affinity tumor-specific T cells (26). One strategy to obviate this problem is to elicit tumor-specific T-cell responses in allogeneic animals (17). Indeed, previous studies have shown that despite the complexity of potential ligands that can be recognized by alloreactive T cells, it is possible to generate T-cell clones that are specific for unique peptide/MHC complexes expressed by tumor cells (27). However, the molecular basis for their tumor-specificity and the extent of clonal diversity among these T cells remained unclear.
Herein we define the antigenic peptide recognized by one of these allogeneic tumor-specific T cells by a novel expression cloning strategy. We used the Kb30 cytotoxic T-cell clone that is specific for an unknown peptide presented by Kb MHC on the murine EL-4 thymoma cells. The Kb30 cells were used to generate the lacZ-inducible T-cell hybrid KZ30.6 (28) and used to probe Kb-COS cells transiently transfected with an EL-4 cDNA library (12, 15). The expression screens led to identification of a previously unknown mouse mammary tumor virus (MMTV) transcript as the source of the antigenic peptide. The antigenic peptide was identified as the octapeptide ANYDFICV (AFV8) encoded within the lumenal region of the endoplasmic reticulum (ER)-translocated MMTV env protein, but its presentation was strictly transporter of antigenic peptides (TAP)-dependent. The AFV8 peptide matched one of two HPLC peaks of antigenic activity among naturally processed peptides expressed in EL-4 cells and was recognized by two other independently derived EL-4-specific T cells.
MATERIALS AND METHODS
Cell Lines.
Cell lines were maintained in RPMI 1640 medium (Cellgro, Mediatech, Washington, DC) supplemented with 2 mM glutamine, 1 mM pyruvate, 50 μM 2-mercaptoethanol, penicillin (100 units/ml), streptomycin (100 μg/ml), and 10% fetal bovine serum (HyClone), at 37°C in a 5% CO2/95% air atmosphere. Kb and Db-COS cells transfectants have been described (12, 29). T2-Kb cell line was a gift of P. Cresswell (Yale University School of Medicine). EL4-B7 (H-2b) and RMA (H-2b) and its TAP derivative RMA/S cell lines were obtained from J. Allison and D. Raulet (University of California, Berkeley). Kb30 CTL clone was obtained by immunization of a B10.D2 mouse with EL-4 cells. CTL clones were obtained by limiting dilution cloning from a B10.D2 mouse immunized 10 days earlier with 20 × 106 EL-4 tumor cells. Kb35 was from a similarly immunized (B10.BR × B10.D2)F1 mouse (16), and AB-1 was derived from a mixed lymphocyte culture using irradiated C57BL/6 stimulators and BALB/c responders (30). Kb30 cytotoxic T lymphocyte (CTL) clone was fused with the BWZ36/CD8α fusion partner as described (28). The resulting lacZ-inducible hybrid was designated KZ30.6.
cDNA Libraries, Expression Screens, and Constructs.
A unidirectional cDNA library using poly(A)+ mRNA from EL4 cells was constructed in the BstXI–NotI sites of the mammalian expression vector pcDNAI (Invitrogen). The cDNA inserts were ligated to the vector using the BstXI linker (5′-CTTTCCAGCACA-3′) at the 5′ end and primer–linker oligo(dT)/NotI (5′-CAACCGGCTCGAGCGGCCGCT21-3′) at the 3′ end. Recombinant plasmids were selected by transforming competent bacteria and were cultured in pools of 30–100 colony-forming units in U-bottom 96-well culture plates. For expression screens, aliquots of DNA (≈100 ng/ml), prepared directly in the 96-well plate (31), were transiently transfected into 3 × 104 Kb COS cells per well of 96-well plates (29). Two days later, 3 × 104 KZ30.6 T cells were added to each well, and after overnight incubation, plates were developed with the chlorophenol red β-galactoside (CPRG) substrate (28). Positive pools were scored by increased absorbance over other wells and vector alone. The single plasmid responsible for the positive signal was isolated by repeating the screen with individual bacterial colonies obtained using DNA from the positive pool.
Deletion constructs of the cDNA clone were prepared using the BAL-31 nuclease (see Fig. 2A). The 39G3.9 plasmid DNA was cut with XbaI at the 3′ end and digested with BAL-31. Aliquots were removed at 5-min intervals, and the ends were blunted with the Klenow fragment of DNA polymerase I. The insert fragments were removed by excision with the 5′ flanking BamHI site and subcloned into BamHI/EcoRV-cut pcDNAI vector. The deletions were confirmed by restriction digests and by nucleotide sequencing. The functional activity of only the smallest construct, 25.4, is shown in Fig. 2B. The Kb1 (CYI8), Kb2 (AFV8), and AFV8-H constructs were prepared with complementary oligonucleotides corresponding to the indicated sequences in Fig. 2A with an additional ATG codon for translation initiation (29).
Figure 2.
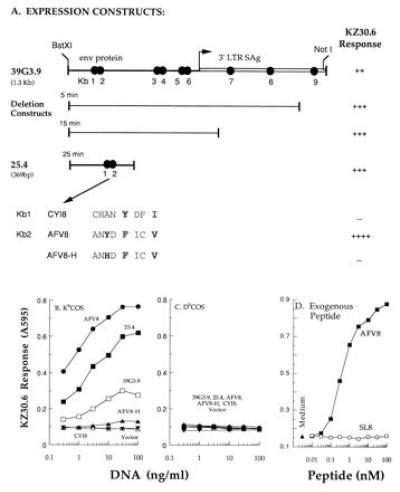
KZ30.6 T cells recognize the 39G3.9 encoded octapeptide AFV8/Kb MHC complex. (A) Schematic representation of the cDNA 39G3.9 and its derivative expression constructs and their ability to stimulate the KZ30.6 T-cell response. The ORFs of cDNA clone 39G3.9 corresponding to the env protein and 3′ LTR superantigen (SAg) are indicated as well as the potential Kb peptide motifs, Kb1-9 (solid circles). The indicated deletion constructs were prepared by digestion of 39G3.9 with the Bal-31 exonuclease for 5, 15, or 25 min. Kb1 and Kb2 represent the two Kb binding octapeptide motifs [XXXX(F,Y)XX(I,L,M,V)] and their amino acid sequence is shown. The p5 and p8 anchor residues are outlined. The peptide AFV8-H has 1 aa substitution at p3 position (boldface type) of AFV8 sequence. The relative KZ30.6 response to each construct is indicated by + or −. KZ30.6 response to either Kb-COS (B) or Db-COS (C) cells transfected with various concentrations of DNA constructs shown in A. (D) KZ30.6 response to APCs in presence of various concentrations of synthetic peptides AFV8 or OVA-(257–264) (SL8) or medium alone. KZ30.6 response was measured by the lacZ assay using CPRG. Data points represent average absorbance of replicate wells.
T-Cell-Activation Assays.
Peptide/MHC ligand-specific T-cell responses were measured by the LacZ activity induced in the T cells (13, 28). About 3–10 × 104 T cells were cocultured overnight with 2–6 × 104 appropriate normal cells or transfected antigen-presenting cells (APCs) with or without exogenous peptides in 96-well plates. The ligand-induced T-cell response was determined using the LacZ substrate chlorophenol red β-galactoside (CPRG) as described (28). The conversion of CPRG to chlorophenol red, in each well of the 96-well plate, was measured at 595 nm and 655 nm as reference wavelength. Data show the mean absorbance of replicate cultures and are representative of at least three experiments.
Peptides, Extracts, and HPLC Analysis.
The peptide NH2-Ala-Asn-Tyr-Asp-Phe-Ile-Cys-Val-OH (abbreviated as AFV8) was prepared using solid-phase F-Moc chemistry and purified by HPLC, and synthesis was confirmed by mass spectrometry as described (15). Total acid-soluble peptide pool from EL4 cells was extracted by trifluoroacetic acid (TFA) as described (32, 33). Briefly, 5 × 108 cells were washed with PBS, lysed in 6 ml of 0.1% TFA in water, and homogenized by ultrasonication using Sonic Dismembrator (Fisher Scientific). The homogenate was centrifuged at 12,000 × g for 30 min. The supernatant was passed through a 10-kDa Ultra Free-MC filter (Millipore). The filtrate was dried in a vacuum centrifuge, resuspended in 0.1% TFA, and fractionated by HPLC (Hewlett–Packard 1050 HPLC symtem controlled by HP Chemstation software). Reverse-phase C18 columns (Vydac, 4.6 × 250 mm, 5 μm, 300 Å) were run in 0.1% TFA in water (solvent A) and 0.1% TFA in acetonitrile (solvent B). The gradient used for separations was as follows: 0–5 min, 23% B; 5–35 min, a linear increase of B to 38%; 35–40 min, a rapid linear increse of B to 100%; 40–45 min, 100% B; 55–60 min, decrease of B to 23%. Flow rate was maintained at 1 ml/min and fractions were collected using the synchronized Frac100 fraction collector (Pharmacia). Mock runs were performed prior to each experimental sample and assayed in parallel to ensure absence of cross-contamination between samples. Fractions were dried in a vacuum centrifuge (Savant) and resuspended in 100 μl of PBS, and aliquots were assayed at 1:7.5 dilution for stimulating KZ30.6 T cell as described above using Kb L cells as APCs. Synthetic AFV8 peptide was used as a standard in parallel to estimate the quantitative recovery of the naturally processed peptide.
RESULTS AND DISCUSSION
Expression Cloning of the KZ30.6-Stimulating Antigen.
The Kb30 CTL clone was chosen for this study because it was specific for a unique peptide/Kb complex expressed by the EL-4 tumor cells, that was not detected on normal spleen cells. The Kb30 CTL clone was fused with BWZ36/CD8α fusion partner to obtain the LacZ-inducible T-cell hybrid, designated as KZ30.6 (12, 28). To identify the antigenic peptide component, KZ30.6 T cells were used as a probe to detect the peptide/Kb ligand generated in Kb-COS cells transfected with an EL-4 cDNA expression library as described (12, 15). A cDNA pool, 39G3, was identified by its ability to stimulate the lacZ response in KZ30.6 T cells (Fig. 1A). This positive pool was then further subdivided and the screen was repeated to obtain the individual cDNA clones (Fig. 1B). The cDNA clone 39G3.9 was selected for further analysis. This clone encoded the antigen gene because DNA transfection caused expression of the KZ30.6-stimulating ligand in Kb-COS but not in Db cells (Fig. 2B). The antigen clone obtained by fractionating the second postive pool (Fig. 1A) was identical to 39G3.9, suggesting that this gene was well represented in the cDNA library.
Figure 1.
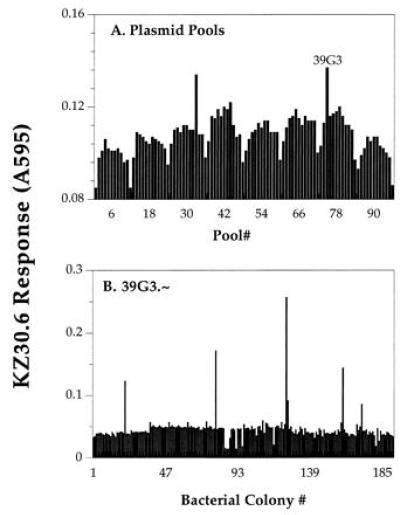
Isolation of KZ30.6 T-cell-stimulating antigen gene, 39G3.9, by expression cloning. (A) Pools of an EL-4 cDNA library were transiently transfected into Kb-COS cells in 96-well plates. Two days later lacZ-inducible KZ30.6 T cells were added to the wells and after an overnight incubation, assayed for the induction of lacZ activity measured by the color change due to conversion of the CPRG substrate. Representative absorbance data is shown for a single 96-well plate containing the postive pool 39G3 that was selected for further fractionation. (B) DNA from individual bacterial colonies of the pool 39G3 were assayed for their ability to stimulate KZ30.6 T-cell response. Plasmid 39G3.9 was active in generating the KZ30.6-stimulating ligand and was selected for further analysis.
The plasmid clone 39G3.9 contained a 1.3-kb cDNA insert. A perfect match for the 39G3.9 nucleotide sequence was not found among the sequences in the databases, but it was closely related to MMTV-derived transcripts (Fig. 2A). The closest match to both the the 5′ (361 nt) and the 3′ (426 nt) ends of 39G3.9 sequence was the MMTV env gene (GenBank accession no. M11024M11024) (34), with, respectively, 98% and 99% identities. Because of the large number of MMTV integrants carried by laboratory mouse strains, as well as those known to be amplified in murine lymphomas, the exact origin of 39G3.9 gene is difficult to assign (35, 36). The MMTV env gene is expressed as a uniquely spliced mRNA that encodes the env gp73 as well as the 3′ long terminal repeat (LTR) ORF proteins (37). The gp73 protein is post-translationally processed into the gp52 and the gp36 polypeptides (38), while the 3′ LTR encodes the ≈320-aa endogenous superantigen vSAg that associates with MHC class II molecules (39). The 39G3.9 clone contained the 244 C-terminal residues of gp73 (aa 464–688) that included the gp36 as well as SAg residues (Fig. 2A). The KZ30.6 T-cell-stimulating antigenic peptide could, therefore, be encoded within either the env or the vSAg regions.
AFV8 Is the Antigenic Peptide Encoded Within the MMTV env Gene Product.
The antigenic peptide within the isolated cDNA was defined by testing a panel of deletion constructs. Nucleotides were systematically deleted from the 3′ end of the parental 39G3.9 clone by treatment with BAL-31 exonuclease for various time periods. The resulting DNA fragments were subcloned into the pcDNAI expression vector and tested for their antigenic activity in transiently transfected Kb-COS cells. All the deletion constructs were active in stimulating the KZ30.6 T cells as exemplified by the smallest deletion construct, 25.4 (Fig. 2 A and B). The antigenic activity was, therefore, located within the env, rather than the 3′ LTR region, and was encoded within the 369 bp of the smallest deletion construct 25.4.
Examination of the predicted amino acid sequence of clone 25.4 showed the presence of two potential Kb MHC binding octapeptide motifs XXXX(F,Y)XX(I,L,V,M) (4, 40) (Kb1 and Kb2, Fig. 2A). Expression constructs encoding these two peptides (designated as CYI8 and AFV8 with an additional translational initiation codon ATG) were prepared and tested for activity. Expression of only the AFV8 construct (ANYDFICV) in Kb-COS but not Db-COS cells was sufficient for inducing the KZ30.6 T-cell response (Fig. 2 B and C). Furthermore, the AFV8 construct was significantly more active than the parental 39G3.9 clone or the 25.4 deletion fragment. The designation of the antigenic activity to the AFV8 sequence was further confirmed by testing the corresponding synthetic peptide. Again KZ30.6 T cells specifically recognized the AFV8 peptide but not SL8 (OVA257-264) that stimulates Kb-restricted B3Z T cells, when it was added exogenously to T2-Kb cells expressing empty Kb MHC (Fig. 2D) or to other cells expressing Kb but not Db MHC molecules (data not shown). Furthermore, the AFV8 peptide, as is characteristic of most naturally processed peptides recognized by CD8+ T cells, was active at picomolar concentrations (29). We conclude that the AFV8 peptide is presented by Kb MHC in EL-4 tumor as an unique ligand for the KZ30.6 T cells.
Relationship of AFV8 to the Naturally Processed Peptides in EL-4 Cell Extracts.
The relationship of AFV8 to its naturally processed counterpart(s) in EL-4 target cells was determined by HPLC analysis of peptides in trifluoracetic acid (TFA) extracts (41). The MHC-bound peptides in EL-4 cells were extracted in TFA and fractionated by reverse-phase HPLC. The fractions were tested for their ability to stimulate KZ30.6 T cells using the Kb-L cells as APCs. Two peaks of KZ30.6 stimulating activity were found in the HPLC fractions (Fig. 3 Upper). Although the relative amounts of activity in the two peaks varied in different experiments, the second HPLC peak had the same retention time as synthetic AFV8 peptide run under identical conditions (Fig. 3 Lower). This result strongly suggests that AFV8 represents one of the two naturally processed peptides in EL-4 cells. Assuming the two peaks contain peptides of similar activity as AFV8, and are recovered quantitatively, we estimate that EL-4 cells express 200–500 copies of these peptides per cell. Given that the abundance of naturally processed peptides can range between >10,000 and <10 copies per cell (5, 20, 42), the AFV8 peptide appears to be about average.
Figure 3.
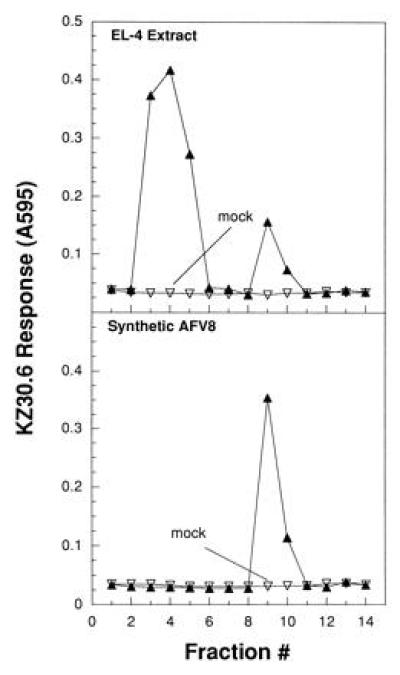
Naturally processed peptide in EL-4 cell extract coelutes with synthetic AFV8 in HPLC analysis. (A) Small molecular weight material in a 0.1% TFA extract of 6 × 107 EL-4 cell equivalents was fractionated on a C18 reverse-phase column by HPLC using an acetonitrile gradient. Fractions were collected at 1-min intervals, dried, resuspended, and tested for activity using Kb-L cells as APCs. KZ30.6 response was measured by the lacZ activity as described in Fig. 1. (B) HPLC fractions from synthetic AFV8 peptide run under identical conditions were analyzed similarly. Mock runs following injection of 0.1% TFA alone were performed prior to each sample and analyzed in parallel to ensure absence of cross-contamination between sample runs.
What is the identity and origin of the peptide in the first HPLC peak? By contrast to naturally processed peptides presented by MHC class II that invariably consist of a mixture of nested peptide sequences (43, 44), those presented by MHC class I molecules predominantly elute as single peaks in HPLC-fractionated extracts (45, 46). Yet a few examples are known where MHC class I-restricted T-cell-stimulating peptides elute as multiple peaks in HPLC analysis of cell extracts, raising questions of their origin (32, 47, 48). The best characterized examples of these is the CD8+ 2C CTL clone specific for the octapeptide (p2Ca) that is derived from α-ketoglutarate dehydrogenase (αKGDH) and is presented by the Ld MHC class I molecule (7, 49). Interestingly, in addition to the p2Ca peptide peak, most tissues contain two additional peaks, and the liver contains three peaks of 2C-stimulating activity (42). In remarkable feats of purification, the active peptides in two of these peaks were identified and found to represent analogues of the p2Ca peptide; one with eight additional N-terminal residues and the other with a single amino acid substitution within the p2Ca peptide (42, 49). Thus multiple T-cell-stimulating peaks in HPLC-fractionated cell extracts can correspond to either proteolytic intermediates or to substituted peptide analogues.
Given that AFV8 is derived from the MMTV env gene and that mice carry several endogenous MMTV proviral sequences (35), it was possible that additional peaks might correspond to AFV8 analogues derived from other MMTV integrants. Indeed the peptide ANHDFICV (AFV8-H) was homologous to the AFV8 (ANYDFICV) sequence in the closest database match M11024M11024, to the 39G3 cDNA clone. Yet, transfection of the AFV8-H construct encoding these residues into Kb-COS cells did not yield any detectable KZ30.6-stimulating activity (Fig. 2 A and B). Thus despite the fact that the M11024 MMTV env gene is expressed in the same EL-4 cells (34) and that the AFV8-H peptide differs from AFV8 in only a single amino acid residue, it was unable to stimulate KZ30.6 T cells and thus cannot account for the KZ30.6-stimulating activity in the first HPLC peak. The identity and the origin of the first HPLC peak thus remains to be determined.
The Expression of AFV8/Kb Complex Requires TAP and Is Not Detected on Normal Spleen Cells.
Most naturally processed peptides presented by MHC class I molecules are generated in the cytoplasm via the proteasome (50, 51) and transported into the ER by the TAP transporter (52, 53). Yet some proteins that are translocated into the ER can yield peptide/MHC I complexes despite the absence of TAP (54, 55, 56), while others exhibit a strict requirement for TAP transport (18, 57). Because the AFV8 peptide is located within the lumenal region (aa 544–551) of the gp73 precursor that is cotranslationally translocated into the ER, we asked whether the generation of the AFV8/Kb complex required the TAP transporter. Both EL-4 and the RMA cells strongly stimulated KZ30.6 T cells, indicating that the KZ30.6 ligand was expressed on both tumor cells (Fig. 4). However, the TAP-negative RMA/S cells as well as normal syngeneic B6 spleen failed to stimulate KZ30.6 T cells. Because the RMA/S and normal spleen cells could stimulate KZ30.6 T cells in presence of exogenously added AFV8 peptide demonstrates that they were capable of presenting the AFV8/Kb ligand. Thus the generation of AFV8/Kb ligand is TAP-dependent and is consistent with the notion that processed peptides from ER-translocated proteins are nevertheless generated in the cytoplasm (57). Interesting mechanisms for this phenomenon may involve either retention of a fraction of nascent translated products in the cytoplasm or retrieval of translocated proteins/peptides from the ER (58, 59).
Figure 4.
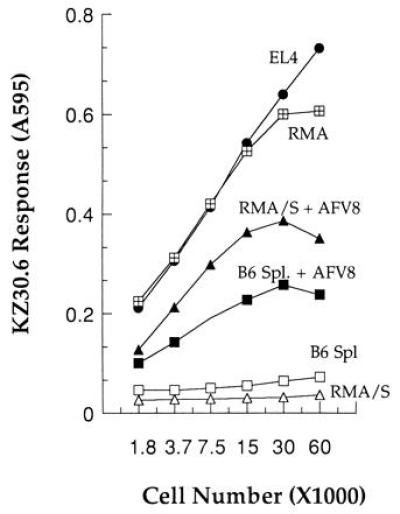
Generation of the KZ30.6 stimulating ligand is TAP-dependent. Various numbers of indicated cell lines or normal B6 spleen cells were used as APCs to stimulate KZ30.6 T cells in absence or presence of 10 nM AFV8 peptide. RMA is a tumor cell line from B6 mice, and RMA/S is its TAP− derivative. KZ30.6 response was measured by the lacZ assay as described in Fig. 1.
Why normal spleen cells fail to stimulate KZ30.6 T cells is intriguing. Given the origin of the AFV8 peptide in the MMTV env gene, and that B6 mice contain four endogenously integrated MMTVs (Mtv-8, -9, -17, and -30) suggests that the env precursors should be expressed and presented as a peptide/MHC I complex (60). Furthermore, with the exception of M11024 discussed above, the known MMTV env sequences do contain the AFV8 peptide. Thus the explanation for why normal spleen cells fail to generate the AFV8/Kb complex could lie in differential regulation of the MMTV transcription as noted in the mammary gland and in T-cell tumors (36, 61). It is also possible that the processing of the env protein to the AFV8 peptide may not occur efficiently in splenic APCs. Which of these possibilities account for the difference in AFV8/Kb expression in normal spleen versus the EL-4 or RMA tumor cells is presently unknown but can be distinguished by analysis of MMTV env gene expression and processing of the env gene product(s) in different cell types. Note that the lack of detectable AFV8/Kb complex in syngeneic B6 mice could allow the generation of EL-4 (or RMA) tumor-specific T-cell responses in immunized B6 mice.
The Kb30 CTL clone was derived 6 years ago by immunizing a B10.D2 mouse with EL-4 tumor cells. Of the 41 clones obtained from this mouse, 3 were EL-4-specific and did not recognize normal spleen cells. Although the other two clones were no longer available, screening other EL-4-specific CTLs revealed that clones Kb35 and AB-1 were also specific for the AFV8/Kb ligand (data not shown). Because these CTL clones were independently derived from the (B10.D2 × B10.BR)F1 and BALB/c mice, respectively, strongly suggests that the AFV8/Kb complex is frequently recognized by EL-4-specific CTL clones.
In conclusion, we have demonstrated a novel expression cloning strategy by defining a specific T-cell-stimulating antigenic peptide/MHC complex expressed in EL-4 tumor cells. The identity of the peptide, the precursor MMTV env gene, and its relationship to naturally processed peptides in normal and in EL-4 and other tumor cells provide models for study of antigen processing, as well as immunological approaches to intervention of tumor growth.
Acknowledgments
We acknowledge F. Gonzalea for excellent technical assistance in the construction and screening of the EL-4 cDNA library. This research was supported by grants to N.S. from the National Institutes of Health and the University of California Tobacco Disease Related Research Program. S.M. and T.S. were supported in part by a postdoctoral fellowship from the American Cancer Society, California Division, and by a predoctoral training grant from the National Institutes of Health.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: APC, antigen-presenting cell; CPRG, chlorophenol red β-galactoside; CTL, cytotoxic T cell; MHC, major histocompatibility complex; MMTV, mouse mammary tumor virus; ER, endoplasmic reticulum; TAP, transporter of antigenic peptides; TFA, trifluoroacetic acid; LTR, long terminal repeat.
References
- 1.Townsend A, Bodmer H. Annu Rev Immunol. 1989;7:601–624. doi: 10.1146/annurev.iy.07.040189.003125. [DOI] [PubMed] [Google Scholar]
- 2.Yewdell J W, Bennink J R. Adv Immunol. 1992;52:1–123. doi: 10.1016/s0065-2776(08)60875-5. [DOI] [PubMed] [Google Scholar]
- 3.Germain R N. Cell. 1994;76:287–299. doi: 10.1016/0092-8674(94)90336-0. [DOI] [PubMed] [Google Scholar]
- 4.Falk K, Rotzschke O, Stevanovic S, Jung G, Rammensee H-G. Nature (London) 1991;351:290–296. doi: 10.1038/351290a0. [DOI] [PubMed] [Google Scholar]
- 5.Hunt D F, Henderson R A, Shabanowitz J, Sakaguchi K, Michel H, Sevilir N, Cox A L, Appella E, Engelhard V H. Science. 1992;255:1261–1263. doi: 10.1126/science.1546328. [DOI] [PubMed] [Google Scholar]
- 6.Shastri N. Curr Opin Immunol. 1996;8:271–277. doi: 10.1016/s0952-7915(96)80067-7. [DOI] [PubMed] [Google Scholar]
- 7.Udaka K, Tsomides T J, Eisen H N. Cell. 1992;69:989–998. doi: 10.1016/0092-8674(92)90617-l. [DOI] [PubMed] [Google Scholar]
- 8.Henderson R A, Cox A L, Sakaguchi K, Appella E, Shabanowitz J, Hunt D F, Engelhard V H. Proc Natl Acad Sci USA. 1993;90:10275–10279. doi: 10.1073/pnas.90.21.10275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boon T, Van Pel A, De Plaen E, Chomez P, Lurquin C, Szikora J-P, Sibille C, Mariame B, Van Den Eynde B, Lethe B, Brichard V. Cold Spring Harbor Symp Quant Biol. 1989;54:587–596. doi: 10.1101/sqb.1989.054.01.070. [DOI] [PubMed] [Google Scholar]
- 10.Boon T, Cerottini J-C, Van Den Eynde B, can der Bruggeni P, Van Pel A. Annu Rev Immunol. 1994;12:337–365. doi: 10.1146/annurev.iy.12.040194.002005. [DOI] [PubMed] [Google Scholar]
- 11.Kawakami Y, Eliyahu S, Delgado C H, Robbins P F, Sakaguchi K, Appella E, Yannelli J R, Adema G J, Miki T, Rosenberg S A. Proc Natl Acad Sci USA. 1994;91:6458–6462. doi: 10.1073/pnas.91.14.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karttunen J, Sanderson S, Shastri N. Proc Natl Acad Sci USA. 1992;89:6020–6024. doi: 10.1073/pnas.89.13.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karttunen J, Shastri N. Proc Natl Acad Sci USA. 1991;88:3972–3976. doi: 10.1073/pnas.88.9.3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shastri N. Curr Opin Immunol. 1995;7:258–262. doi: 10.1016/0952-7915(95)80012-3. [DOI] [PubMed] [Google Scholar]
- 15.Malarkannan S, Afkarian M, Shastri N. J Exp Med. 1995;182:1739–1750. doi: 10.1084/jem.182.6.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heath W R, Hurd M E, Carbone F R, Sherman L A. Nature (London) 1989;341:749–752. doi: 10.1038/341749a0. [DOI] [PubMed] [Google Scholar]
- 17.Sherman L A, Chattopadhyay S. Annu Rev Immunol. 1993;11:385–402. doi: 10.1146/annurev.iy.11.040193.002125. [DOI] [PubMed] [Google Scholar]
- 18.Aldrich C J, DeCloux A, Woods A S, Cotter R J, Soloski M J, Forman J. Cell. 1994;79:649–658. doi: 10.1016/0092-8674(94)90550-9. [DOI] [PubMed] [Google Scholar]
- 19.Jameson S C, Hogquist K A, Bevan M J. Annu Rev Immunol. 1995;13:93–126. doi: 10.1146/annurev.iy.13.040195.000521. [DOI] [PubMed] [Google Scholar]
- 20.Rammensee H-G, Falk K, Rotzschke O. Annu Rev Immunol. 1993;11:213–268. doi: 10.1146/annurev.iy.11.040193.001241. [DOI] [PubMed] [Google Scholar]
- 21.Engelhard V H. Annu Rev Immunol. 1994;12:181–207. doi: 10.1146/annurev.iy.12.040194.001145. [DOI] [PubMed] [Google Scholar]
- 22.van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van Den Eynde B, Knuth A, Boon T. Science. 1991;254:1643–1647. doi: 10.1126/science.1840703. [DOI] [PubMed] [Google Scholar]
- 23.Cox A L, Skipper J, Chen Y, Henderson R A, Darrow T L, Shabanowitz J, Engelhard V H, Hunt D F. Science. 1994;264:716–719. doi: 10.1126/science.7513441. [DOI] [PubMed] [Google Scholar]
- 24.Wolfel T, Hauer M, Schneider J, Serrano M, Wolfel C, Klehmann-Hieb E, De Plaen E, Hankeln T, Meyer zum Buschenfelde K-H, Beach D. Science. 1995;269:1281–1284. doi: 10.1126/science.7652577. [DOI] [PubMed] [Google Scholar]
- 25.Boon T, Gajewski T F, Coulie P G. Immunol Today. 1995;16:334–336. doi: 10.1016/0167-5699(95)80149-9. [DOI] [PubMed] [Google Scholar]
- 26.Townsend S E, Allison J P. Science. 1993;259:368–370. doi: 10.1126/science.7678351. [DOI] [PubMed] [Google Scholar]
- 27.Heath W R, Sherman L A. Eur J Immunol. 1991;21:153–159. doi: 10.1002/eji.1830210123. [DOI] [PubMed] [Google Scholar]
- 28.Sanderson S, Shastri N. Int Immunol. 1994;6:369–376. doi: 10.1093/intimm/6.3.369. [DOI] [PubMed] [Google Scholar]
- 29.Shastri N, Gonzalez F. J Immunol. 1993;150:2724–2736. [PubMed] [Google Scholar]
- 30.Kane K, Sherman L A, Mescher M F. J Immunol. 1989;142:4153–4160. [PubMed] [Google Scholar]
- 31.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K. Current Protocols in Molecular Biology. New York: Wiley; 1994. [Google Scholar]
- 32.Rotzschke O, Falk K, Faath S, Rammensee H-G. J Exp Med. 1991;174:1059–1071. doi: 10.1084/jem.174.5.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malarkannan S, Goth S, Buchholz D R, Shastri N. J Immunol. 1995;154:585–598. [PubMed] [Google Scholar]
- 34.Kwon B S, Weissman S M. J Virol. 1984;52:1000–1004. doi: 10.1128/jvi.52.3.1000-1004.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scherer M T, Ignatowicz L, Winslow G M, Kappler J W, Marrack P. Annu Rev Cell Biol. 1993;9:101–128. doi: 10.1146/annurev.cb.09.110193.000533. [DOI] [PubMed] [Google Scholar]
- 36.Racevskis J. J Virol. 1990;64:4043–4050. doi: 10.1128/jvi.64.9.4043-4050.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dickson C, Peters G. Curr Top Microbiol Immunol. 1983;106:1–34. doi: 10.1007/978-3-642-69357-1_1. [DOI] [PubMed] [Google Scholar]
- 38.Dickson C, Atterwill M. J Virol. 1980;35:349–361. doi: 10.1128/jvi.35.2.349-361.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choi Y, Kappler J W, Marrack P. Nature (London) 1991;350:203–207. doi: 10.1038/350203a0. [DOI] [PubMed] [Google Scholar]
- 40.Joyce S, Kuzushima K, Kepecs G, Angeletti R H, Nathenson S G. Proc Natl Acad Sci USA. 1994;91:4145–4149. doi: 10.1073/pnas.91.10.4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Falk K, Rötzschke O, Rammensee H-G. Nature (London) 1990;348:248–251. doi: 10.1038/348248a0. [DOI] [PubMed] [Google Scholar]
- 42.Wu M X, Tsomides T J, Eisen H N. J Immunol. 1995;154:4495–4502. [PubMed] [Google Scholar]
- 43.Rudensky A Y, Preston-Hurlburt P, Hong S-C, Barlow A, Janeway C A., Jr Nature (London) 1991;353:622–627. doi: 10.1038/353622a0. [DOI] [PubMed] [Google Scholar]
- 44.Hunt D F, Michel H, Dickinson T A, Shabanowitz J, Cox A L, Sakaguchi K, Appella E, Grey H M, Sette A. Science. 1992;256:1817–1820. doi: 10.1126/science.1319610. [DOI] [PubMed] [Google Scholar]
- 45.Wallny H-J, Rammensee H-G. Nature (London) 1990;343:275–278. doi: 10.1038/343275a0. [DOI] [PubMed] [Google Scholar]
- 46.Van Bleek G M, Nathenson S G. Nature (London) 1990;348:213–216. doi: 10.1038/348213a0. [DOI] [PubMed] [Google Scholar]
- 47.Griem P, Wallny H-J, Falk K, Rotschke O, Arnold B, Schonrich G, Hammerling G, Rammensee H-G. Cell. 1991;65:633–640. doi: 10.1016/0092-8674(91)90095-g. [DOI] [PubMed] [Google Scholar]
- 48.Kuzushima K, Sun R, Van Bleek G M, Vegh Z, Nathenson S G. J Immunol. 1995;155:594–601. [PubMed] [Google Scholar]
- 49.Udaka K, Tsomides T J, Walden P, Fukusen N, Eisen H N. Proc Natl Acad Sci USA. 1993;90:11272–11276. doi: 10.1073/pnas.90.23.11272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Monaco J J. Immunol Today. 1992;13:173–179. doi: 10.1016/0167-5699(92)90122-N. [DOI] [PubMed] [Google Scholar]
- 51.Rock K L, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, Goldberg A L. Cell. 1994;78:761–771. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 52.Shepherd J C, Schumacker T N, Ashton-Rickardt P G, Imaeda S, Ploegh H L, Janeway C A, Jr, Tonegawa S. Cell. 1993;74:577–584. doi: 10.1016/0092-8674(93)80058-m. [DOI] [PubMed] [Google Scholar]
- 53.Van Kaer L, Ashton-Rickardt P G, Ploegh H L, Tonegawa S. Cell. 1992;71:1205–1214. doi: 10.1016/s0092-8674(05)80068-6. [DOI] [PubMed] [Google Scholar]
- 54.Anderson K, Cresswell P, Gammon M, Hermes J, Williamson A, Zweerink H. J Exp Med. 1991;174:489–492. doi: 10.1084/jem.174.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Snyder H L, Yewdell J W, Bennink J R. J Exp Med. 1994;180:2389–2393. doi: 10.1084/jem.180.6.2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Elliott T, Willis A, Cerundolo V, Townsend A. J Exp Med. 1995;181:1481–1491. doi: 10.1084/jem.181.4.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Siliciano R F, Soloski M J. J Immunol. 1995;155:1–5. [PubMed] [Google Scholar]
- 58.Wiertz E J H J, Jones T R, Sun L, Bogyo M, Geuze H J, Ploegh H L. Cell. 1996;84:769–779. doi: 10.1016/s0092-8674(00)81054-5. [DOI] [PubMed] [Google Scholar]
- 59.Skipper J C A, Hendrickson R C, Gulden P H, Brichard V, Van Pel A, Chen Y, Shabanowitz J, Wolfel T, Slingluff C L, Boon T, Hunt D F, Engelhard V H. J Exp Med. 1996;183:527–534. doi: 10.1084/jem.183.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kozak C, Peters G, Pauley R, Morris V, Michaelides R, et al. J Virol. 1987;61:1651–1654. doi: 10.1128/jvi.61.5.1651-1654.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Knepper J E, Medina D, Butel J S. J Virol. 1986;59:518–521. doi: 10.1128/jvi.59.2.518-521.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]