

Abstract
The first step of V(D)J recombination, specific cleavage at the recombination signal sequence (RSS), can be carried out by the recombination activating proteins RAG1 and RAG2. In vivo, the cleaved coding and signal ends must be rejoined to generate functional antigen receptors and maintain chromosomal integrity. We have investigated signal joint formation using deletion and inversion substrates in a cell free system. RAG1 and RAG2 alone or in combination were unable to generate signal joints. However, RAG1 and RAG2 complemented with nuclear extracts were able to recombine an extrachromosomal substrate and form precise signal joints. The in vitro reaction resembled authentic V(D)J recombination in being Ku-antigen-dependent.
Keywords: recombination activating protein RAG1, recombination activating protein RAG2
Lymphocyte antigen receptors are encoded by multiple copies of gene segments in germ-line DNA. During B- and T-lymphocyte development, these gene segments are assembled into functional transcription units by the mechanism of V(D)J recombination. The DNA sequence requirements for V(D)J recombination consist of highly conserved heptamer and nonamer DNA motifs separated by a spacer of 12 or 23 base pairs (12 RSS and 23 RSS, where RSS is recombination signal sequence). During the V(D)J recombination reaction, two types of DNA joints are formed: signal joints, generally involving precise head-to-head ligation of two heptamers, and coding joints, usually containing deletions or additions of a few nucleotides (1).
Several lymphoid-specific factors are known to be involved in V(D)J recombination. These include terminal deoxynucleotidyltransferase (2, 3, 4) and the recombination activating proteins RAG1 and RAG2 (5, 6, 7). RAG1 and RAG2 are sufficient for the formation of specific double-strand DNA breaks at RSSs (8, 9). Efficient recombination occurs almost exclusively between RSSs with different spacers. This restriction is known as the 12/23 rule and is evident at the initial cleavage event (10, 11, 12). The cleavage reaction involves the formation of hairpin loops at the coding ends and precise double-strand breaks at the signal ends (8, 9, 13, 14, 15, 16). RAG1 and RAG2 can be coimmunoprecipitated from cells, suggesting that they function as part of a complex during V(D)J recombination (17, 18).
Other proteins that are not restricted in their pattern of expression to lymphoid cells are also required for V(D)J recombination. These include two components of the DNA-dependent protein kinase, the p86 subunit of the p70/p86 Ku antigen (product of XRCC5), and the large catalytic subunit (product of SCID), as well as the XRCC4 protein. These proteins are involved in DNA repair as well as in V(D)J recombination (19, 20). In addition other as yet unidentified factors may be required for antigen receptor assembly.
Herein we report an in vitro system in which RAG1 and RAG2 complemented with a nuclear extract are able to catalyze signal joint formation. This system depends on Ku proteins and may provide the means for identification of the range of factors involved in generation of the signal joint.
METHODS
PCR Analysis.
Recombined DNA was amplified and labeled with α-32P by using 30 cycles of 94°C for 0.5 min, 65°C for 1 min, and 75°C for 1 min, followed by incubation at 72°C for 10 min. The PCR amplification mixture contained 10 mM Tris·HCl (pH 9.0), 50 mM KCl, 5.5 mM MgCl2, BSA (0.1 mg/ml), 100 ng of each appropriate primer (R5, CCAGTCTGTAGCACTGTGCAC; R14, TCCAGCTGAACGGTCTGGT), all four dNTPs (each at 20 μM), 1 mCi of [α-32P]dCTP (3000 Ci/mmol; 1 Ci = 37 GBq; Amersham), and 1 unit of Taq DNA polymerase (Boehringer Mannheim). When primer R14 was used in combination with primer R3 (TGTTCCAGTCTGTAGCACTG), PCR was for 30 cycles of 95°C for 10 sec, 55°C for 0.5 min, 72°C for 1 min, followed by incubation at 72°C for 10 min.
Purification of Truncated Glutathione S-Transferase Fusion RAG1 and RAG2 Proteins.
Truncated versions of RAG1 (amino acids 330-1040) and RAG2 (amino acids 1–383) were expressed as glutathione S-transferase fusion proteins under the transcriptional control of the elongation factor 1α promoter (21). 293T cells [293 cells expressing the simian virus 40 large tumor (T) antigen] were transiently transfected with the RAG1 and RAG2 constructs by calcium phosphate precipitation (22). Two days after transfection, the cells were harvested and lysed for 5 min in RSB buffer (10 mM Tris·HCl, pH 7.4/10 mM NaCl/5 mM MgCl2/0.5% Nonidet P-40/protease inhibitors). Lysates were brought to 0.6 M NaCl with buffer LSB (20 mM Tris·HCl, pH 7.4/1.0 M NaCl/0.2 mM MgCl2/0.1% Nonidet P-40/protease inhibitors) and incubated for 30 min on ice. Extracts were centrifuged and the supernatant was incubated with glutathione-agarose beads. RAG proteins were eluted (50 mM Tris·HCl, pH 8.3/20 mM glutathione/1 M NaCl/10% glycerol/protease inhibitors) and dialyzed against buffer D (20 mM Hepes·NaOH, pH 7.5/1 mM DTT/10% glycerol/0.3 M NaCl/0.1 mM EDTA/protease inhibitors). Both proteins were expressed at levels corresponding to approximately 1 to 2 μg of each protein from one 100-mm tissue culture dish. Although RAG1 and RAG2 were highly purified (as determined by Coomassie blue staining, data not shown), we cannot rule out the possibility that our preparations contained additional factors that contributed to the biochemical activities described. All of the experiments described were done at least three times with two different preparations of extracts and proteins.
Preparation of Nuclear Extracts.
293 and CHO cells stably transfected with full-length RAG1 and RAG2 are indicated as 293-S and CHO-S, respectively. BASC6C2 is a pro-B-cell line and 22D6 is an Abelson virus-transformed pre-B-cell line. Nuclear extracts were prepared using a modification of the Dignam protocol (23). Briefly, cells were harvested and resuspended in 5 vol of buffer A (10 mM Hepes·KOH, pH 7.9/1.5 mM MgCl2/10 mM KCl/0.1% Nonidet P-40/protease inhibitors). After 10 min of incubation on ice, samples were centrifuged at 1000 × g for 10 min. The pellet was resuspended in 1.5 vol of buffer C (20 mM Hepes·KOH, pH 7.9/20% glycerol/0.6 M NaCl/1.5 mM MgCl2/0.2 mM EDTA/0.5 mM DTT/0.1% Nonidet P-40/protease inhibitors) and incubated on ice for 30 min. Cellular debris was removed by high-speed centrifugation, and the supernatant was dialyzed against low salt buffer C (100 mM NaCl) as described by Dignam et al. (23).
In Vitro Recombination.
Fifty nanograms of pJH200 or 10 ng of pJH288 was incubated with RAG proteins and nuclear extracts in 20 μl in the presence of 12.5 mM Hepes·KOH, pH 7.5/100 mM KCl/1 mM MnCl2/0.05 mM EDTA/5% glycerol/0.5 mM ATP/all four dNTPs (each at 50 mM). RAG1 (200 ng), RAG2 (200 ng), or nuclear extracts from 293-S (8 mg/ml), BASC6C2 (11 mg/ml), HeLa (10 mg/ml), CHO-S (9 mg/ml), or 22D6 (3 mg/ml) were added to the reaction mixtures. After a 5-hr incubation at 30°C, the samples were treated with proteinase K for 2 hr. The samples were then extracted once with phenol/chloroform and twice with chloroform. The DNA was recovered by ethanol precipitation, using 3 μg of poly(dI·dC) as carrier, and resuspended in 20 μl of 1 mM Tris·HCl, pH 7.5/0.1 mM EDTA. Five percent of the recovered DNA was used as template in the PCR detection assay.
In Vivo Recombination.
293T cells (293 cells expressing the simian virus 40 large tumor antigen) were transiently transfected with the recombination substrate alone or together with the RAG1 and RAG2 constructs by a calcium phosphate precipitation (22). Plasmid DNA was recovered from the cells 2 days later by performing a standard alkaline lysis protocol.
Southern Blot Analysis.
PCR products were resolved by electrophoresis through a 2% agarose gel, denatured, renatured within the gel, and then transferred onto Biotrans membrane (ICN). The DNA was UV-crosslinked to the membrane with a Stratalinker (Stratagene) and probed with an oligonucleotide which encompasses a correct signal joint (CTGTGCACAGTGGTA) according to the protocol described by Oettinger et al. (7).
RESULTS AND DISCUSSION
As a further step toward defining the biochemical requirements for the V(D)J recombination reaction, we have developed an in vitro system to study signal joint formation. The system uses the active core regions of RAG1 (amino acid 330-1040) and RAG2 (amino acid 1–383), which were purified as fusion proteins with glutathione S-transferase (24, 25, 26, 27). Truncated RAG1 and RAG2 fusion proteins mediated V(D)J recombination in vivo and specific cleavage of the RSSs in vitro (data not shown). The truncated fusion proteins were used for all of the experiments shown herein and will be referred to as RAG1 and RAG2.
A sensitive PCR assay was used to test for signal joint formation in a cell-free system (Fig. 1). The substrate used for the joining reaction was pJH200, a plasmid that undergoes deletional V(D)J recombination (28). Recombined signal joints were detected by amplification with primers R5 and R14 to yield a 252-bp PCR fragment (Fig. 1A). Hybridization of the R5 primer to recombined DNA is precise, whereas hybridization to the unrecombined substrate results in a mismatch at the 3′ end of R5 (Fig. 1B). Despite the mismatch, some amplification of a 456-bp PCR fragment was observed with R5 and R14 on the unrecombined substrate (Fig. 1C, lane 5). However, there was a preference for the recombined product even when the unrecombined plasmid was present in vast excess (Fig. 1C). Furthermore, the presence of the unrecombined substrate did not significantly inhibit detection of the recombined product (Fig. 1C). As a further control for the in vitro signal joining reaction, pJH200 was cotransfected with or without RAG1 and RAG2 into 293T cells. The 252-bp product was only amplified from DNA recovered from cells transfected with the combination of pJH200, RAG1, and RAG2 (Fig. 2A).
Figure 1.
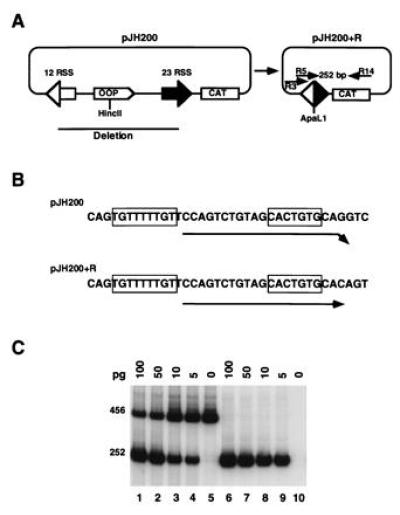
PCR assay for signal joint formation. (A) Schematic representation of the pJH200 substrate (28). The unrecombined plasmid and the recombined product are shown on the left and right, respectively. The relative positions of the primers (R5 and R14) used to amplify the 252-bp region of the recombined product are indicated by arrows (not to scale). (B) Nucleotide sequence to which primer R5 hybridizes in the unrecombined (pJH200) and recombined (pJH200+R) plasmids. Note that in the unrecombined substrate there is a 1-bp mismatch at the 3′ end of R5 (arrowhead). (C) Evaluation of primer combination R5/R14 on recombined and unrecombined sequences. The PCR assay was performed using the indicated amounts of recombined plasmid as template in the presence (lanes 1–5) or absence (lanes 6–10) of 2.5 ng of unrecombined substrate. The 456- and 252-bp fragments are the amplified products from pJH200 and pJH200+R, respectively.
Figure 2.
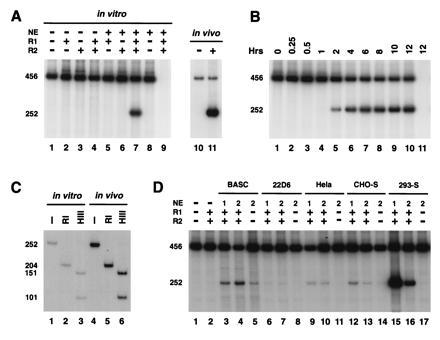
In vitro signal joint formation by deletion. (A) In vitro signal joint formation by deletion requires RAG1, RAG2, and nuclear extracts. pJH200 was incubated at 30°C with the indicated combinations of purified RAG1 (R1), RAG2 (R2), and nuclear extracts (NE) from 293-S cells under the conditions describe. The DNA was then purified and analyzed by PCR assay as described in Fig. 1. The autoradiograph from one such experiment is presented on the left (lanes 1–9), while the right (lanes 10 and 11) shows the 32P-labeled PCR products amplified from in vivo-recombined DNA purified from 293T cells transfected with pJH200 alone (−) or cotransfected with RAG1 and RAG2 (+). Lane 9 correspond to a control reaction, RAG1, RAG2, and NE but not template DNA. (B) Time course of in vitro signal joint formation. The reaction conditions were identical to those described in A, lane 7 (with RAGs and nuclear extract) and the products were analyzed as explained in Fig. 1. Lane 11 is identical to lane 10 except that template DNA was not included in the reaction. (C) Digestion pattern of the in vivo- and in vitro-recombined products. Purified 252-bp 32P-labeled PCR products were incubated with no enzyme (−), RsaI (RI), or HindIII (HIII) and separated on an 8% polyacrylamide/1× TBE gel. The 252-bp fragment contains a unique HindIII site 151 bp from the R14 oligonucleotide. This fragment also contains two RsaI sites, one in the 23 RSS and the other 25 bp from oligonucleotide R14. (D) Different nuclear extracts can complement RAG proteins in signal joint formation. RAG1 (R1) and RAG2 (R2) were supplemented with extracts prepared from BASC6C2 (BASC), 22D6, HeLa, CHO-S, or 293-S cells, as indicated. Each sample was processed as in A and the bands were visualized by autoradiography.
Using this PCR assay, we found that RAG1 and RAG2 or a combination of both were not sufficient to carry out signal joint formation in vitro. However, when RAG1 and RAG2 proteins were complemented with nuclear extracts, signal joints were generated as evidenced by the presence of the 252-bp PCR-amplified fragment (Fig. 2 A, B, and D). Signal joints were detected after approximately 2 hr and the amount of product increased for up to 12 hr (Fig. 2B). In contrast, cleavage was observed after a few minutes under the same reaction conditions (data not shown). The time lag between cleavage and joining may reflect the time required for the two signals ends to find one another and be joined. A significant time lag between cleavage and signal joint formation was observed in vivo (29).
To estimate the number of signal joints formed in vitro, we constructed a standard curve with different amounts of prerecombined molecules diluted in 2.5 ng of the unrecombined substrate (Fig. 1C). We found that a typical in vitro reaction resulted in formation of approximately 107 recombined molecules, corresponding to an average of 0.2% of the pJH200 substrate being transformed into recombined product.
Nuclear extracts from BASC6C2, 22D6, HeLa, CHO-S, or 293-S cells complemented the RAG proteins in signal joint formation (Fig. 2D). However, nuclear extracts prepared from 293 cells stably expressing RAG1 and RAG2 (293-S) were at least 5 times more active than all other extracts tested for signal joint formation. Low levels of recombination were seen in unsupplemented extracts both from BASC6C2 pro-B cells, which constituitively express RAG1 and RAG2 and actively recombine their immunoglobulin genes, and CHO-S cells, which stably express low levels of RAG1 and RAG2. This recombination activity was enhanced by the addition of recombinant RAG1 and RAG2 (Fig. 2D).
To determine whether the amplified products from in vivo and in vitro reactions were molecularly identical, we purified the 252-bp fragment from both reaction mixtures and performed restriction digests with RsaI and HindIII. We found that the digestion pattern of the PCR products of the in vitro V(D)J joining reaction was identical to that displayed by authentic in vivo recombination reactions (Fig. 2C).
Because the R5 primer crosses the recombination border and could theoretically give an artifactual result, we used an alternative pair of flanking primers, R14 and R3 (Fig. 1A), to confirm in vitro signal joint formation. The recombined product was detected by Southern blot analysis with a probe that covered the recombination border. The results obtained with the flanking primers were identical to those obtained with the R5 and R14 primers. Using these primers, we found in vitro signal joint formation only when the RAG proteins and nuclear extract were present (Fig. 3A). Since the heptamer–heptamer junction generates an ApaL1 site, digestion with this enzyme was used as to define the precision of the signals joints amplified with the flanking primers. PCR was performed with R3 and R14 flanking primers, on in vitro and in vivo recombination reactions, in the presence of [32P]dCTP (Fig. 3B) and the 256-bp recombined product was analyzed by restriction digestion (Fig. 3C). Almost 100% of the amplified products from the in vitro and in vivo reaction were ApaL1-sensitive (Fig. 3C), indicating that the majority of the signal joints produced in vitro were precise.
Figure 3.
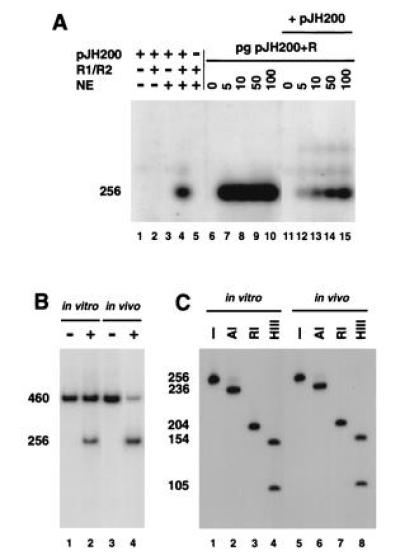
Signal joints generated in vitro and detected by PCR are precise. (A) Southern blot analysis of pJH200 recombined in vitro and PCR-amplified with flanking oligonucleotides R3 and R14. Because primers R3 and R14 hybridize equally well to the recombined and unrecombined pJH200, the DNA recovered from in vitro recombination reactions was first digested with HincII before PCR amplification, to enrich for the recombined DNA. The in vitro recombination assay was performed with the indicated combinations of RAGs, 293-S nuclear extracts, and 50 ng of pJH200 (lanes 1–5). The HincII-digested DNA from these recombination reactions, as well as a titration (0–100 pg) of pJH200+R without (lanes 6–10) or with (lanes 11–15) 2.5 ng of pJH200, was then subjected to PCR with the following modifications: no [α32P]dCTP was added to the mixture and the primer pair used was R3/R14 (which generates a 256-bp fragment). Unlike R5, which overlaps the signal joint, the 3′ end of R3 stops 3 bp before the signal joint and is, therefore, considered a flanking primer. The PCR products were separated on an agarose gel, transferred to nylon membrane, subjected to Southern blot analysis using a 32P-labeled oligonucleotide that overlaps the signal joint and visualized by autoradiography. (B) pJH200 recombined in vitro and PCR-amplified with flanking oligonucleotides R3 and R14, in the presence of [32P]dCTP. pJH200 was subjected to the in vitro recombination assay in the absence (lane 1) or presence of RAGs and 293-S nuclear extract (lane 2). As a control, pJH200 was transiently transfected into 293T cells alone (lane 3) or along with RAGs (lane 4) to generate in vivo-recombined plasmid. DNA recovered from recombination reactions was digested with HincII and PCR-amplified using flanking oligonucleotides R3 and R14. [α32P]dCTP was used during PCR amplification. The 32P-labeled PCR products were fractionated on an 8% native polyacrylamide/1× TBE gel and detected by autoradiography. (C) Restriction enzyme digestion of signal joints formed in vivo and in vitro. The 256-bp product from in vivo and in vitro reactions was extracted from a gel and submitted to restriction enzyme digestion with ApaL1 (A1), RsaI (RI), and HindIII (HIII). Undigested and digested aliquots were separated on an 12% native polyacrylamide gel and the 32P-labeled PCR products were detected by autoradiography. ApaL1 cuts at the correct signal joint, located 20 bp from the end of the PCR product.
Inversion V(D)J reactions are characteristic of antigen receptor gene rearrangement in lymphoid cells. To examine signal joint formation in an inversion substrate, we used pJH288 (30) and the PCR assay with oligonucleotides R5 and R14. In this assay, the recombined plasmid should generate a PCR product of 231 bp. Neither RAG1 or RAG2 alone nor the combination was sufficient to generate an inversion product. However, extracts from 293-S cells were able to complement the purified RAG proteins and produce signal joints by inversion (Fig. 4A). To verify the accuracy of the inversion-mediated signal joints, the products of in vivo and in vitro reactions were compared by restriction enzyme digestion (Fig. 4B). Just as with deletional joining, the joints mediated by inversion in vitro were indistinguishable from their counterparts produced in vivo. Signal joints in this inversion substrate were detected after approximately 2 hr (Fig. 4C), whereas cleavage could be observed after a few minutes (data not shown).
Figure 4.
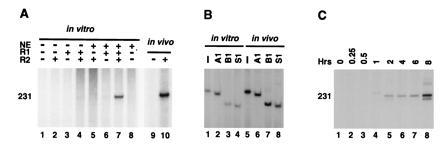
In vitro signal joint formation by inversion. (A) Autoradiogram of the 32P-labeled PCR products amplified from the recombination reaction using oligonucleotides R5 and R14 and separated in an 8% polyacrylamide/1× TBE gel. pJH288 inversion substrate was incubated with RAG1 (R1), RAG2 (R2), and nuclear extract (NE) as indicated. The in vivo results, to the right, show the 32P-labeled PCR products from 293T cells transfected with pJH288 alone (−) or cotransfected with RAG1 and RAG2 (+). The 231-bp 32P-labeled PCR product is indicated. (B) Digestion pattern of the in vivo and in vitro products, recombined by inversion. The 231-bp 32P-labeled PCR products were incubated with no enzyme (−), ApaLI (A1), BamHI (BI), and SmaI (SI) as indicated and separated in an 8% polyacrylamide/1× TBE gel. The sizes of the fragments are indicated. (C) Time course of in vitro signal joint formation by invertion. The reaction conditions were identical to those described in A, lane 7 (with RAGs and nuclear extract).
During in vivo V(D)J recombination, a DNA sequence with a 12 RSS recombines with a sequence containing a 23 RSS. This restriction is known as the 12/23 rule and has been shown in vitro to regulate cleavage when Mg2+ is included in the reaction (10, 12). Under our reaction conditions, we were unable to observe the 12/23 rule in cleavage and, therefore, signal joint formation could not obey the 12/23 rule. We are pursuing further studies to attempt reconstitution of signal joint formation under conditions that allow observation of the 12/23 rule. Reproduction of this restriction is key to producing an in vitro system that completely mimics the in vivo reaction.
Cleavage of the RSS at the heptamer results in blunt-ended 5′-phosphorylated linear DNA molecules (8, 9, 13, 14, 15, 16). These ends could potentially be joined to one another nonspecifically by DNA ligases to complete the signal joining reaction. In fact, complementation of RAG1 and RAG2 with purified T4 ligase will cause signal joint formation (data not shown). However, genetic experiments indicate that several factors other than RAG1 and RAG2 are required for efficient V(D)J recombination in vivo including the p86 subunit of Ku (19). To determine whether in vitro signal joint formation has similar requirements, we performed immunodepletion experiments with antibodies against the Ku antigen. We found that polyclonal antibodies to p70, one of the Ku subunits, and monoclonal antibodies that recognize the p70/p86 Ku heterodimer (31) specifically inhibited signal joint formation (Fig. 5). Preimmune serum and monoclonal isotype controls had no effect on the reaction. Thus, the in vitro reaction resembles authentic V(D)J recombination in that it involves genetically defined factors other than RAG1, RAG2, and ligase.
Figure 5.
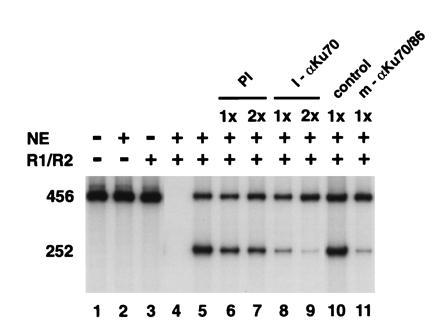
Anti- (α) Ku70 antibodies specifically inhibit in vitro signal joint formation. On the left (lanes 1–5), the in vitro recombination assay was performed with the indicated combinations of RAGs, 293-S nuclear extracts, and 50 ng of pJH200. Lane 4 corresponds to a control reaction: RAG1, RAG2, and NE but no template DNA. Lanes 6–11 show complete reactions in which the RAGs and nuclear extracts were preincubated for 15 min at 4°C with a preimmune sera (PI), a polyclonal αKu70 (I-αKu70) raised against rat Ku-70, αKu monoclonal (m-αKu70/80) raised to the human p70/p86 heterodimer (ref. 31, monoclonal 162), or an isotype-matched monoclonal antibody (HB95; American Type Culture Collection). The 1× and 2× refer to the relative amounts of sera used. Each in vitro recombination reaction was then analyzed by PCR as described in Fig. 1.
In summary, we have developed a cell-free system that mediates signal joint formation. This in vitro reaction recapitulates key aspects of signal joint formation as observed during in vivo V(D)J recombination. The joints are precise head to head heptamer fusions and their formation involves factors beyond RAG1 and RAG2. Because RAG1 and RAG2 are known to be sufficient for cleavage at RSSs (ref. 8 and data not shown), the other cellular proteins must be needed for the joining reaction. Our antibodies identify Ku-70 as one component that we assume acts in complex with Ku-86 and the DNA protein kinase. Other factors could well be necessary. The availability of this system to study signal joint formation should allow for rapid progress in identifying all of the factors required to mediate this reaction.
Acknowledgments
We thank Dr. David Schatz for critical review of this manuscript and also for providing the DNA substrate to test the 12/23 rule, Bruce Meyer for providing the pEBG plasmid, and Juan Cárcamo and Dennis Sawchuk for comments on the manuscript and very helpful discussions. P.C. was supported by a fellowship from the Irvington Institute and is now supported by the Lymphoma Research Foundation of America. J.-C.L. is supported by a fellowship from the Irvington Institute. This work was supported by grants from the National Institutes of Health to D.B. and M.C.N. M.C.N. is an associate investigator in the Howard Hughes Medical Institute and D.B. is an American Cancer Society Research Professor.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: RAG, recombination activating protein; RSS, recombination signal sequence.
References
- 1.Lewis S. Adv Immunol. 1994;56:27–151. doi: 10.1016/s0065-2776(08)60450-2. [DOI] [PubMed] [Google Scholar]
- 2.Landau N R, Schatz D G, Rosa M, Baltimore D. Mol Cell Biol. 1987;7:3237–3243. doi: 10.1128/mcb.7.9.3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gilfillan S, Dierich A, Lemeur M, Benoist C, Mathis D. Science. 1993;261:1175–1178. doi: 10.1126/science.8356452. [DOI] [PubMed] [Google Scholar]
- 4.Komori T, Okada A, Stewart V, Alt F W. Science. 1993;261:1171–1175. doi: 10.1126/science.8356451. [DOI] [PubMed] [Google Scholar]
- 5.Schatz D, Baltimore D. Cell. 1988;53:107–115. doi: 10.1016/0092-8674(88)90492-8. [DOI] [PubMed] [Google Scholar]
- 6.Schatz D, Oettinger M A, Baltimore D. Cell. 1989;59:1035–1048. doi: 10.1016/0092-8674(89)90760-5. [DOI] [PubMed] [Google Scholar]
- 7.Oettinger M A, Schatz D G, Gorka C, Baltimore D. Science. 1990;248:1517–1523. doi: 10.1126/science.2360047. [DOI] [PubMed] [Google Scholar]
- 8.McBlane F J, van Gent D C, Ramsden D A, Romeo C, Cuomo C A, Gellert M, Oettinger M A. Cell. 1995;83:387–395. doi: 10.1016/0092-8674(95)90116-7. [DOI] [PubMed] [Google Scholar]
- 9.van Gent D C, McBlane J F, Ramsdem D A, Sadofsky M J, Hesse J A, Gellert M. Cell. 1995;81:825–934. doi: 10.1016/0092-8674(95)90012-8. [DOI] [PubMed] [Google Scholar]
- 10.Eastman Q M, Leu T M J, Schatz D G. Nature (London) 1996;380:85–88. doi: 10.1038/380085a0. [DOI] [PubMed] [Google Scholar]
- 11.Steen S B, Gomelsky L, Roth D. Genes Cell. 1996;1:543–553. doi: 10.1046/j.1365-2443.1996.d01-259.x. [DOI] [PubMed] [Google Scholar]
- 12.van Gent D C, Ramsden D R, Gellert M. Cell. 1996;85:107–113. doi: 10.1016/s0092-8674(00)81086-7. [DOI] [PubMed] [Google Scholar]
- 13.Roth D B, Nakajima P B, Menetski J P, Bosma M J, Gellert M. Cell. 1992;69:41–53. doi: 10.1016/0092-8674(92)90117-u. [DOI] [PubMed] [Google Scholar]
- 14.Roth D B, Menetski J P, Nakajima P B, Bosma M J, Gellert M. Cell. 1992;70:983–991. doi: 10.1016/0092-8674(92)90248-b. [DOI] [PubMed] [Google Scholar]
- 15.Roth D B, Zhu C, Gellert M. Proc Natl Acad Sci USA. 1993;90:10788–10792. doi: 10.1073/pnas.90.22.10788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schlissel M, Constantinescu A, Morrow T, Baxter M, Peng A. Genes Dev. 1993;7:2520–2532. doi: 10.1101/gad.7.12b.2520. [DOI] [PubMed] [Google Scholar]
- 17.Spanopoulou E, Cortes P, Shih C, Huang E, Silver D P, Svec P, Baltimore D. Immunity. 1995;3:715–727. doi: 10.1016/1074-7613(95)90061-6. [DOI] [PubMed] [Google Scholar]
- 18.Leu T M, Schatz D G. Mol Cell Biol. 1995;15:5657–5670. doi: 10.1128/mcb.15.10.5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roth D B, Lindahl T, Gellert M. Curr Biol. 1995;5:496–499. doi: 10.1016/s0960-9822(95)00101-1. [DOI] [PubMed] [Google Scholar]
- 20.Li Z, Otevrel T, Gao Y, Cheng H-L, Seed B, Stamoto T D, Taccioli G E, Alt F. Cell. 1996;83:1079–1089. doi: 10.1016/0092-8674(95)90135-3. [DOI] [PubMed] [Google Scholar]
- 21.Mizushima S, Nagata S. Nucleic Acids Res. 1990;18:5322–5794. doi: 10.1093/nar/18.17.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pear W, Nolan G, Scott M, Baltimore D. Proc Natl Acad Sci USA. 1993;90:8392–8395. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dignam D J, Lebovitz R M, Roeder R G. Nucleic Acids Res. 1983;11:1475–1485. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Silver D P, Spanopoulou E, Mulligan R C, Baltimore D. Proc Natl Acad Sci USA. 1993;90:6100–6104. doi: 10.1073/pnas.90.13.6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sadofsky M J, Hesse J E, McBlane J F, Gellert M. Nucleic Acids Res. 1993;21:5644–5650. doi: 10.1093/nar/21.24.5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cuomo C A, Oettinger M A. Nucleic Acids Res. 1994;22:1810–1814. doi: 10.1093/nar/22.10.1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sadofsky M J, Hesse J E, Gellert M. Nucleic Acids Res. 1994;22:1805–1809. doi: 10.1093/nar/22.10.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lieber M R, Hesse J E, Mizuuchi K, Gellert M. Genes Dev. 1987;1:751–761. doi: 10.1101/gad.1.8.751. [DOI] [PubMed] [Google Scholar]
- 29.Ramsden D A, Gellert M. Genes Dev. 1995;9:2409–2420. doi: 10.1101/gad.9.19.2409. [DOI] [PubMed] [Google Scholar]
- 30.Lieber M R, Hesse J E, Lewis S, Bosma G C, Rosenberg N, Mizuuchi K, Bosma M J, Gellert M. Cell. 1988;55:7–16. doi: 10.1016/0092-8674(88)90004-9. [DOI] [PubMed] [Google Scholar]
- 31.Wang J, Satoh M, Pierani A, Schmitt J, Chou C H, Stunnenberg R, Roeder G, Reeves W H. J Cell Sci. 1994;107:3223–3233. doi: 10.1242/jcs.107.11.3223. [DOI] [PubMed] [Google Scholar]