

Abstract
Presenilin-1 (PS-1) gene mutations are responsible for the majority of the early onset familial forms of Alzheimer disease (AD). Neither PS-1’s anatomic distribution in brain nor expression in AD have been reported. Using in situ hybridization in the rat forebrain, we show that PS-1 mRNA expression is primarily in cortical and hippocampal neurons, with less expression in subcortical structures, in a regional pattern similar to APP695. Excitotoxic lesions lead to loss of PS-1 signal. A neuronal pattern of expression of PS-1 mRNA was also observed in the human hippocampal formation. AD and control levels did not differ. PS-1 is expressed in brain areas vulnerable to AD changes more so than in areas spared in AD; however, PS-1 expression is not sufficient to mark vulnerable regions. Collectively, these data suggest that the neuropathogenic process consequent to PS-1 mutations begins in neuronal cell populations.
Keywords: hippocampal formation, excitotoxic lesion
Alzheimer disease (AD) is a widespread and devastating neurodegenerative disorder that manifests itself as a progressive and irreversible decline in cognitive abilities associated with the development of neurofibrillary tangles and senile plaques in a distinctive pattern in the brain. The underlying causes and pathophysiologic mechanisms of AD remain unknown. Molecular analysis in recent years has suggested that the causes may be heterogeneous. There are now four well established genes associated with AD. Inheritance of a common allele of the apolipoprotein E gene, apoE ɛ4, is a risk factor for late onset (>60 years old) AD (1). The other three genes are causative and lead to autosomal dominant forms of the disease with fairly early ages of onset (<60 years, frequently even in the 40s). Mutations in the amyloid precursor protein (APP) (2) account for a small percentage of early onset familial AD cases (3). The majority of early onset familial AD appears to be due to mutations in two recently discovered genes, presenilin 1 (PS-1) located on chromosome 14 (4), which is responsible for AD in multiple pedigrees, and PS-2, located on chromosome 1, which is responsible for AD in the well studied Volga German families (5, 6). Twenty-four mutations have already been discovered in PS-1 in 52 pedigrees, and two mutations in PS-2 have been described (4, 5, 6, 7, 8, 9, 10, 11, 12, 13). PS-1 and PS-2 are 67% identical to one another, and also share marked homology of approximately 50% identity with the Caenorhabditis elegans gene product Sel-12. (14). The predicted amino acid sequence of PS-1 suggests a protein structure that is serpentine, with multiple hydrophilic loops separated by transmembrane domains.
The normal role of the PS-1 gene product is unknown, and its role in the pathophysiology of AD remains speculative. PS-1 mRNA is ubiquitously expressed, but detailed knowledge of its expression and localization in the brain is not yet available. Our initial studies suggest a neuronal localization in normal human brain (15). We have now used oligonucleotide probes to examine AD and control brain expression of PS-1 mRNA. In addition, a mouse homologue of PS-1 has been cloned and shows strong homology to human PS-1, affording the opportunity to study PS-1 expression in experimental models. A loss of PS-1 mRNA signal was observed after excitotoxic lesion at a time when the lesion contained marked gliosis and neuronal loss. These data suggest that PS-1 is expressed in the central nervous system primarily in neurons in both rat and human brain. However, PS-1 expression is not sufficient to predispose neurons to Alzheimer-related degeneration.
MATERIALS AND METHODS
All human samples were obtained from the Brain Tissue Resource Center, McLean Hospital, Belmont, MA, or from the Massachusetts Alzheimer Disease Research Center, Massachusetts General Hospital. The diagnosis of AD was made from paraffin embedded sections of the contralateral hemisphere using Beilschowski silver stain and Khachaturian criteria (16). Six control (three males, three females; age range, 44–86 yr; postmortem interval range, 2–32.3 hr) and three AD (two males, one female; age range, 77–92 yr; postmortem interval range, 2.3–48.5 hr) were examined. Sections of the hippocampal formation were rapidly frozen and stored at −80°C until cryosectioning (17).
Stereotaxic Surgery.
Eight rats were anesthetised using Avertin (1,1,1-tribromoethanol/tertiary amyl alcohol, 0.5 ml/100 g. i.p) and placed into a stereotaxic frame (David Kopf), and a burr hole was drilled at the following stereotaxic coordinates: Bregma = −6.7, −7.6 mm; Lateral = 4.6, 4.5 mm with the incisor bar set at −3.3 mm. A stainless steel 10-gauge cannula attached to a 5-μl Hamilton syringe was lowered 5.5 mm into the hippocampal formation, and 0.5 μl of 10 mM α-amino-3-hydroxy-5-methyl-4-propionic acid HBr (Research Biochemicals, Natick, MA) was injected over 5 min (0.1 μl per min), with a delay of 5 min preceding and following each injection. On completion of the surgical procedure, the scalp was sewn with surgical silk, and the rats were returned to their home cages where they were monitored hourly for the first 6 hr. The rats were sacrificed at either 3, 7, or 11 days postsurgery, and the brains were processed for in situ hybridization. Rats were sacrificed by cervical dislocation under ether anesthesia, and their brains were removed and snap frozen in isopentane chilled with dry ice. The brains were wrapped in aluminum foil and stored at −70°C prior to sectioning. Sections were cut in either coronal or saggital orientations at 16 μm and thaw mounted onto sterile Probe-On (Fisher) slides coated with a sterile solution of 0.01% poly-l-lysine. Prior to fixation, the mounted sections were stored with desiccant at −70°C in air-tight boxes.
Prehybridization procedures have been described in detail elsewhere (16). Oligonucleotide probes (45-mer) were synthesized on an Applied Biosystems DNA synthesizer and purified by polyacrylamide gel electrophoresis on a 12% urea gel. Sequences were chosen from both the 5′- and 3′-untranslated regions of the mRNA sequence submitted to GenBank (http://www.ncbi.nlm.nih.gov/GenBank/index).
In situ hybridizations were performed on adjacent sections using both sense and antisense 45-mer oligonucleotide probes designed using the published sequence of mouse or human PS-1 mRNA (4). The sequences and GenBank source reference IDs used in the probe design are presented in Table 1. As an additional control, a 45-mer oligonucleotide probe consisting of a mixture of up to 444 random sequences was synthesized and also utilized.
Table 1.
The design regions and sequences of oligonucleotide probes used for in situ hybridization detection of PS-1 mRNA in human and rat brain
| Probe | GenBank accession | Region | Base |
|---|---|---|---|
| PS-1 probe 1 (rat) | L42177.MUSS1PR. | 5′-UTR | 81-125 |
| PS-1 probe 2 (rat) | L42177.MUSS1PR. | 3′-UTR | 1896-1940 |
| PS-1 probe 1 (human) | HUMS182R | 5′-UTR | 330-374 |
| PS-1 probe 2 (human) | HUMS182R | 3′-UTR | 1273-1317 |
| APP695 probe (human) | A02759A02759 | Junctional | 987-1031 |
| APP695 probe (rat) | RNAG | Junctional | 856-900 |
Probes were designed from the untranslated 5′ and 3′ termini, the percentage GC:AT ratio was approximately 55:45% in each case.
In Situ Hybridization.
The probes were diluted in diethyl pyrocarbonate-pretreated water to a working concentration of 5 ng/μl and end-labeled by the repetitive addition of [35S]ATP (DuPont/NEN) by terminal d-transferase (TdT, Promega). Labeled 45-mer probes were purified on prespun Bio-Rad Sephadex columns and diluted to a final concentration of between 8500 and 10,000 cpm/μl in hybridization buffer. The components of the hybridization buffer have been described in detail elsewhere (16). The sections were hybridized overnight at 42°C in sealed chambers humidified with 50% formamide/diethyl pyrocarbonate water.
Posthybridization Treatments.
Nonspecific hybridizations were removed by washing the sections for 30 min in 1× SSC at 55°C repeated once and then subsequently in 1× SSC at room temperature (rt; 10 sec), 0.1× SSC (10 sec, rt), double-distilled water (10 sec, rt), 50% ethanol (10 sec, rt), 70% ethanol (10 sec, rt) 95% ethanol (10 sec, rt). The slides were dried for 2 hr in a stream of warm air and then apposed to Amersham β-max autoradiography film for 15–30 days. Films were developed using Kodak D-19 (100%), stopped in 1% acetic acid, and fixed in Kodak Rapid Fixer. To achieve cellular resolution of the distribution of PS-1 mRNA, the randomer-, sense-, and antisense-hybridized slides were dipped in a 50% suspension of photographic emulsion in 600 mM ammonium acetate (LM-1; Amersham), dried, and stored in lightproof boxes at 4°C for 5–15 weeks. Emulsion-dipped slides were then developed in Kodak D-19 (50% in double distilled water), stopped in 0.1% acetic acid, and fixed in Kodak Fixer prior to counter-staining for Nissl material. Autoradiograms were analyzed using an Imaging Densitometer (Bio-Rad GS-700 series). Autoradiographic images of sections were scanned at maximum resolution (1200 dpi, pixel depth 12) using molecular analyst software (Bio-Rad) on a Power Macintosh 7100/80. One slide of brain sections from each subject (three sections) per target probe experiment was measured. Outlines of brain areas were drawn in each section. Relative optical densities (RODs) for each brain area from each section were measured directly within these drawn measurement frames. These ROD measurements were then corrected for background, and average RODs for each brain area from each subject per experiment were calculated.
RESULTS
Expression of PS-1 mRNA in the Adult Rat Brain.
PS-1 mRNA was expressed throughout the cortical gray matter and in the choroid plexus of the adult rat central nervous system (Fig. 1). PS-1 mRNA was preferentially expressed in the dentate gyrus of the hippocampal formation, neo- and allocortical laminae, and in the cerebellum. PS-1 mRNA was present in diencephalic nuclei but at a lower level than that found in cortical and hippocampal layers; it was nearly undetectable in other subcortical and brain stem areas.
Figure 1.
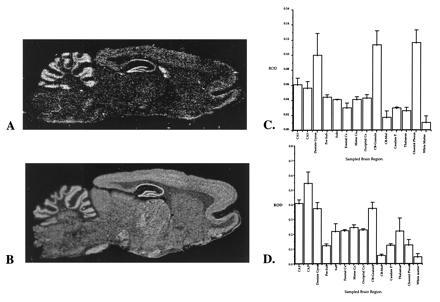
(A–D) Comparison between the expression of PS-1 (A and C) and APP(695) (B and D) in rat brain. The relative optical densities of PS-1 and APP(695) expression were measured in a range of brain regions and are illustrated as the mean adjusted optical density ± SD (n = 3). PS-1 is expressed maximally in the dentate gyrus, in cerebellar granule cells, and in choroid plexus. APP(695) is maximally expressed in the dentate gyrus, hippocampal pyramidal fields, and cerebellar granule cells. These observations suggest that PS-1 and APP(695) share a fairly common regional distribution in brain. CA1–CA3, CA subfields of the hippocampus; Pre-Sub, presubiculum; Sub, subiculum; CB-granule, cerebellar granule cell layer; CB-Mol, molecular layer of the cerebellum.
We compared this pattern of expression to that seen for the APP. APP695 mRNA is much more robustly expressed, giving a signal that is approximately 5– to 10-fold more intense than PS-1 for comparable labeling of probes. However, the regional distribution of APP matches closely with that of PS-1. Fig. 1 illustrates the regional distribution in the brain of both PS-1 and APP. The histograms provide a semiquantitative analysis of the pattern of expression in different brain regions. The similar pattern of the histograms highlights the conclusion that PS-1 and APP are expressed in overlapping brain regions.
Examination of emulsion-dipped sections reinforce the conclusion that PS-1 is expressed primarily in neurons; many but not all cortical and hippocampal neurons appear to contain PS-1 mRNA (Fig. 2), whereas glial populations do not. Neurons of various morphologies, including large and small pyramidal neurons in cortex and CA fields, and dentate gyrus granule neurons all expressed PS-1 mRNA. Because PS-1 mRNA is expressed at low levels, we were concerned that glial expression was not detected because it was below the threshold of detection on the emulsion-dipped sections. We re-examined the question of which cell types express PS-1 using an experimental manipulation designed to kill neurons and result in a predictable time course of reactive gliosis. Following excitotoxic lesion of the hippocampus, there was a significant reduction in the optical densities of PS-1 mRNA expression within the lesion site (Fig. 3). At each of the time points studied (from 4 to 11 days postinjection), there was a loss of neurons and an increase in astrocytes as assessed histologically and by analysis of adjacent section hybridized with oligonucleotide probes against glial fibrillary acidic protein mRNA. There was no effect on PS-1 mRNA expression in the contralateral, unlesioned side relative to unoperated controls. Examination of the emulsion-dipped sections confirmed lack of PS-1 mRNA expression in glia in the lesion bed. These results support the conclusion that PS-1 mRNA expression is primarily neuronal rather than glial and further suggest that PS-1 mRNA levels do not increase following lesion.
Figure 2.
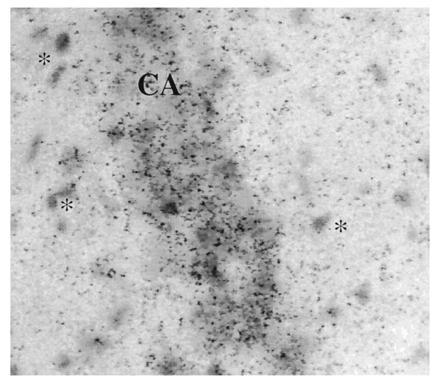
Photomicrograph of a rat hippocampal emulsion-dipped section after hybridization for PS-1 mRNA. The CA1 pyramidal cells (center of field) are clearly labeled, whereas glia in the molecular layers (∗) are not labeled.
Figure 3.
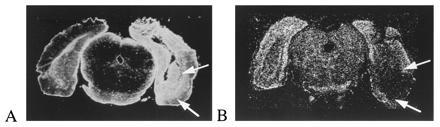
Dark field autoradiograms comparing the effects of an excitotoxic lesions of the caudal hippocampus/entorhinal cortical area on the expression of glial fibrillary acidic protein (A) and PS-1 mRNA (B). Animals were sacrificed at either 3, 7, or 11 days postlesion. There was a marked loss of PS-1 mRNA within the bed of the lesion (arrows) after 7 days. This contrasts with the massive up-regulation of glial fibrillary acidic protein mRNA expression in adjacent sections.
Expression of PS-1 mRNA in AD and Control Brain.
PS-1 mRNA expression (Fig. 4) appears to be present at very low levels in the control elderly human brain (n = 6). Expression was, however, detected in the hippocampal dentate gyrus, the CA fields, and all lamina of temporal neocortex, whereas it was not above background level in the white matter. The pattern of expression, and even the degree to which various hippocampal subfields contained PS-1 mRNA, was quite similar to that observed in the rat. In AD (n = 3), essentially the same pattern of very low expression throughout the hippocampal formation and temporal neocortex was observed. Thus there was no overall qualitative difference in the pattern of PS-1 mRNA expression in AD compared with control brain.
Figure 4.
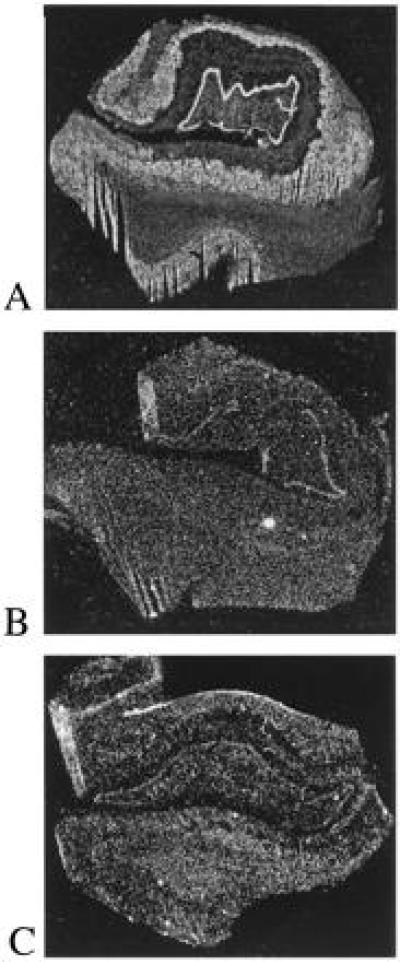
Dark field autoradiograms demonstrating the expression of APP(695) and PS-1 mRNA (A) in human temporal lobe. This figure, and Figs. 2 and 4, were prepared using molecular analyst (Bio-Rad) and adobe photoshop image handling applications. (A) APP(695) mRNA expression in a neuropathologically normal case; (B) PS-1 mRNA expression in the same case as A; (C) expression of PS-1 mRNA in an 88-year-old female AD patient. There were no major differences in the pattern of expression of PS-1 mRNA between control and AD cases. Comparison between the level of expression of APP(695) and PS-1 in the control case reflects the relative abundance of the two mRNAs in human brain.
The pattern of expression of PS-1 mRNA was also compared with the pattern of AD neuropathological changes noted on thioflavine S-stained adjacent sections. The CA1/subiculum area contained many neurofibrillary tangles and senile plaques, whereas in general CA3 and CA4 hippocampal fields contained few. Despite the presence of these pathological changes in CA1/subiculum, no clear alteration in the pattern of PS-1 mRNA expression was observed.
DISCUSSION
Mutations in the PS-1 gene lead to autosomal dominant forms of AD. Very little is known about the expression of this gene product in the brain. Our studies demonstrate that PS-1 mRNA is detectable primarily within neuronal populations within the parenchyma, and in nonneuronal support tissues such as the choroid plexus. To explore the possibility that PS-1 would be expressed in other cell types, or in astrocytes or microglia upon activation, we also examined the patterns of PS-1 expression following excitotoxic lesion. This experiment provided further data that PS-1 is expressed primarily, if not exclusively, in neurons because hybridization signal was lost from the bed of the lesion at times when neuronal loss was observed but the lesion bed was invaded by reactive gliosis.
The pathogenic mechanism of the PS-1 mutations is unknown, but two recent observations implicate PS-1 in the formation of Aβ deposits in senile plaques. Younkin and coworkers (17) showed that mutations in PS-1 and PS-2 lead to increased production of Aβ 42 and an elevation in the Aβ 42/40 ratio in fibroblasts and plasma from affected and at-risk carriers of the disease genes (17). Because Aβ 42 is also elevated in AD caused by APP717 mutations (18), this leads to the possibility that the presenilin mutations lead to enhanced Aβ production and increased numbers of senile plaques. This hypothesis implies an interaction between APP and PS-1. Our data support the idea that such an interaction is plausible, at least in neurons, since PS-1 and APP mRNA are present in very similar patterns in brain. However, APP mRNA is present in much higher amounts.
Recent immunohistochemical data suggest that PS-1 immunoreactivity can be localized to senile plaques (19). Therefore we also compared the distribution of PS-1 expression with the known patterns of distribution of pathological changes in the AD brain. No clear difference in expression patterns were observed between AD and control brains, although our in situ hybridization data do not rule out the possibility of small quantitative changes in AD. In the rat, PS-1 expression is strongest in the hippocampal formation and cortical regions, with relatively less expression in the basal ganglia, thalamus, or brain stem, although the cerebellum contains high levels of signal. We observed a similar pattern in the human temporal lobe, with strongest expression in the parahippocampal gyrus and hippocampal formation. In general AD neurofibrillary tangles and senile plaques affect the hippocampal formation and neocortex much more than subcortical structures (20, 21). Thus areas that are vulnerable in AD express PS-1, whereas regions that are generally spared appear to have lower levels of expression. However, an exact match between PS-1 expression and senile plaques or neurofibrillary tangles is not seen. For example, senile plaques are present in greater numbers in the temporal neocortex than in hippocampal fields, and they are sparse in hippocampal fields CA2 and CA3 (20, 21). Similarly, neurons in entorhinal cortex and the CA1/subiculum area are most vulnerable for neurofibrillary tangles (22); by contrast, although there is PS-1 expression in these vulnerable areas, expression is equally high in relatively spared areas such as CA2 and CA3. Thus PS-1 expression by itself does not specifically identify vulnerable sets of neurons.
These data begin to define the locale and possible roles for PS-1 in the brain and in AD suggest that the initial neuropathogenic events in familial AD due to PS-1 mutations occur in neuronal populations. There are four proteins known on the basis of genetic data to be involved in the cascade of events that lead to AD pathlogical changes. Both PS-1 and PS-2 are expressed primarily in neurons. In PS-1-transfected cells, the majority of the protein is associated with the Golgi and endoplasmic reticulum (15). APP is expressed in all cell types in the brain, but APP695 appears to be restricted to neurons (23). APP is a cell surface molecule but is also highly expressed in the Golgi and endoplasmic reticulum. Apolipoprotein E is expressed only in astrocytes from which it is secreted (24, 25), but may influence neurons through neuronal apoE receptors such as the low density lipoprotein receptor-related protein (LRP) (26). Expression of both APP and apoE are elevated by lesion (24, 25, 27, 28). PS-1 expression is not. Taken together with previous findings, these observations lead us to believe that the PS-1 mutations in familial AD most likely alter endoplasmic reticulum/Golgi functions specifically within neurons. Future studies should elucidate whether familial AD associated PS-1 mutations affect neuronal metabolism of APP.
Acknowledgments
We thank the Brain Tissue Resource Center, McLean Hospital (Grant MH/NS 31862) and the Massachusetts Alzheimer Disease Research Center (AG05134) for brain tissue samples and diagnostic evaluations. This work was supported by National Institutes of Health Grants AG05134 and AG11337.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: AD, Alzheimer disease; APP, amyloid precursor protein; PS-1, presenilin 1.
References
- 1.Strittmatter W J, Saunders A M, Schmechel D, Pericak-Vance M, Enghild J, Salvesen G S, Roses A D. Proc Natl Acad Sci USA. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goate A, Chartier-Harlin M-C, Mullan M. Nature (London) 1991;349:704–707. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 3.Tanzi R E, Vaula G, Romano D M, Mortilla M, Huang T L, et al. Am J Hum Genet. 1992;51:273–282. [PMC free article] [PubMed] [Google Scholar]
- 4.Sherrington R, Rogaev E I, Liang Y, Rogaeva E A, Levesque G, et al. Nature (London) 1995;375:754–760. [Google Scholar]
- 5.Levy-Lahad E, Wasco W, Poorkaj P, Romano D, Oshima J, Pettingell W, Yu C, Jondro P, Schmidt S, Wang K, Crowley A, Fu Y-H, Guenette S, Galas D, Nemens E, Wijsman E, Bird T, Schellenberg G, Tanzi R. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 6.Rogaev E I, Sherrington R, Rogaeva E A, Levesque G, Ikeda M, et al. Nature (London) 1995;376:775–778. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 7.Alzheimer’s Disease Collaboration Group. Nat Genet. 1995;11:219–222. [Google Scholar]
- 8.Cruts M, Backhovens H, Wang S-Y, Van Gassen G, Theuns J, De Jonghe C, Wehnert A, De Voecht J, De Winter G, Cras P, Bruyland M, Datson N, Weissenbach J, den Dunnen J, Martin J, Hendriks L, Van Broeckhoven C. Hum Mol Genet. 1995;4:2363–2372. doi: 10.1093/hmg/4.12.2363. [DOI] [PubMed] [Google Scholar]
- 9.Campion D, Flaman J-M, Brice A, Hannequin D, Dubois B, et al. Hum Mol Genet. 1995;4:2373–2377. doi: 10.1093/hmg/4.12.2373. [DOI] [PubMed] [Google Scholar]
- 10.Wasco W, Pettingell W P, Jondro P D, Schmidt S D, Gurubhagavatula S, Rodes L, DiBlasi T, Romano D M, Guenette S Y, Kovacs D M, Growdon J T, Tanzi R E. Nat Med. 1995;1:848. doi: 10.1038/nm0995-848a. [DOI] [PubMed] [Google Scholar]
- 11.Van Broeckhoven C. Nat Genet. 1995;11:230–232. doi: 10.1038/ng1195-230. [DOI] [PubMed] [Google Scholar]
- 12.Tanahashi H, Mitsunaga Y, Takahashi K, Tasaki H, Watanabe S, Tabira T. Lancet. 1995;346:440. doi: 10.1016/s0140-6736(95)92810-3. [DOI] [PubMed] [Google Scholar]
- 13.Sorbi S, Nacmias B, Forleo P, Piacentini S, Sherrington R, Rogaev E, St. George-Hyslop P, Amaducci L. Lancet. 1995;346:439–440. doi: 10.1016/s0140-6736(95)92809-x. [DOI] [PubMed] [Google Scholar]
- 14.Levitan D, Greenwald I. Nature (London) 1995;377:351–355. doi: 10.1038/377351a0. [DOI] [PubMed] [Google Scholar]
- 15.Kovacs D M, Fausett H J, Page K J, Kim T-W, Moir R D, Merriam D E, Hollister R D, Hallmark O G, Mancini R, Felsenstein K M, Hyman B T, Tanzi R E, Wasco W. Nat Med. 1996;2:224–229. doi: 10.1038/nm0296-224. [DOI] [PubMed] [Google Scholar]
- 16.Sirinathsinghji D J S, Dunnett S B. In: Imaging Gene Expression in Neural Grafts. Sharif N A, editor. Oxford: Oxford Univ. Press; 1993. pp. 43–70. [Google Scholar]
- 17.Scheuner D, Bird T, Citron M, Lannfelt L, Schellenberg G, Selkoe D, Viitanen M, Younkin S G. Soc Neurosci Abstr. 1995;21:1500. [Google Scholar]
- 18.Suzuki N, Cheung T T, Cai X-D, Odaka A, Otvos L, Jr, Eckman C, Golde T E, Younkin S G. Science. 1994;264:1336–1340. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 19.Wisniewski T, Palha J A, Ghiso J. Lancet. 1995;346:1366. doi: 10.1016/s0140-6736(95)92379-9. [DOI] [PubMed] [Google Scholar]
- 20.Arnold S E, Hyman B T, Flory J, Damasio A R, Van Hoesen G W. Cereb Cortex. 1991;1:103–116. doi: 10.1093/cercor/1.1.103. [DOI] [PubMed] [Google Scholar]
- 21.Arriagada P V, Growdon J H, Hedley-Whyte E T, Hyman B T. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 22.Hyman B T, Van Hoesen G W, Damasio A R, Barnes C L. Science. 1984;225:1168–1170. doi: 10.1126/science.6474172. [DOI] [PubMed] [Google Scholar]
- 23.Johnson S, McNeil T, Cordel B, Finch C. Science. 1990;248:854–857. doi: 10.1126/science.2111579. [DOI] [PubMed] [Google Scholar]
- 24.Poirier J, Hess M, May P C, Finch C E. Mol Brain Res. 1991;11:97–106. doi: 10.1016/0169-328x(91)90111-a. [DOI] [PubMed] [Google Scholar]
- 25.Page K, Hyman B T. Soc Neurosci Abstr. 1995;21:1484. [Google Scholar]
- 26.Rebeck G W, Reiter J S, Strickland D K, Hyman B T. Neuron. 1993;11:575–580. doi: 10.1016/0896-6273(93)90070-8. [DOI] [PubMed] [Google Scholar]
- 27.Abe K, St. George-Hyslop P H, Tanzi R E, Kogure K. Neurosci Lett. 1991;125:169–171. doi: 10.1016/0304-3940(91)90019-p. [DOI] [PubMed] [Google Scholar]
- 28.Brugg B, Lemaigre-Dubreuil Y, Huber G, Wollman E E, Delhaye-Bouchaud N, Mariani J. Proc Natl Acad Sci USA. 1995;92:3032–3035. doi: 10.1073/pnas.92.7.3032. [DOI] [PMC free article] [PubMed] [Google Scholar]