

Summary
Transcriptional activation of the nuclear receptor RAR by retinoic acid (RA) often leads to inhibition of cell growth. However, in some tissues, RA promotes cell survival and hyperplasia, activities that are unlikely to be mediated by RAR. Here we show that, in addition to functioning through RAR, RA activates the ‘orphan’ nuclear receptor PPARβ/δ, which, in turn, induces the expression of pro-survival genes. Partitioning of RA between the two receptors is regulated by the intracellular lipid-binding proteins CRABP-II and FABP5. These proteins specifically deliver RA from the cytosol to nuclear RAR and PPARβ/δ, respectively, thereby selectively enhancing the transcriptional activity of their cognate receptors. Consequently, RA functions through RAR and is a pro-apoptotic agent in cells with high CRABP-II/FABP5 ratio, but it signals through PPARβ/δ and promotes survival in cells that highly express FABP5. Opposing effects of RA on cell growth thus emanate from alternate activation of two different nuclear receptors.
Introduction
The vitamin A metabolite retinoic acid (RA) regulates multiple biological processes and plays key roles in embryonic development and in tissue remodeling in the adult. It is well established that many of the activities of RA are mediated by retinoic acid receptors (RARα, β, and γ), ligand-inducible transcription factors that are members of the superfamily of nuclear hormone receptors (Laudet and Gronemeyer, 2002). RARs associate with the retinoid X receptor (RXR) to form heterodimers that bind to regulatory regions of specific target genes and modulate transcriptional rates in response to their cognate ligands (Chambon, 1996; Mangelsdorf et al., 1994). Transcriptional activation by RAR may trigger differentiation (Park et al., 1999; Rochette-Egly and Chambon, 2001), cell cycle arrest (Donato et al., 2007), and apoptosis (Altucci et al., 2001; Donato and Noy, 2005; Kitareewan et al., 2002), and thus often leads to inhibition of cell proliferation. Accordingly, RA displays distinct anticarcinogenic activities and is currently used in or is being tested as a therapeutic agent in several human cancers (Soprano et al., 2004).
However, in some tissues, RA appears to promote rather than inhibit cell survival. For example, RA is critical for neuronal survival (Henion and Weston, 1994; Jacobs et al., 2006; Plum et al., 2001; Rodriguez-Tebar and Rohrer, 1991), and it was reported that RA treatment can enhance skin tumor formation (Verma et al., 1982). RA is essential for normal keratinization in various epithelia (Wolbach and Howe, 1978), and topical administration of the hormone stimulates dermal repair and induces hyperproliferation of basal kertinocytes (Kang et al., 1995; Zouboulis, 2001). Interestingly, although RA plays critical roles in maintenance of skin integrity, mice lacking both RARα and RARγ as well as RARβ-null mice display normal keratinocyte proliferation (Chapellier et al., 2002). These observations indicate that RARs are dispensable for keratinocyte renewal, and suggest that some RA activities in the skin are mediated by an RAR-independent pathway.
We previously reported that RA binds with a high affinity to another nuclear receptor, namely PPARβ/δ, a member of a sub-class of receptors which also includes PPARα and PPARγ, and that, like RAR, functions as a heterodimer with RXR (Laudet and Gronemeyer, 2002). Selective association of RA with PPARβ/δ was suggested by the observations that the Kd for RA-binding by this receptor is ~15 nM, about an order of magnitude stronger than that displayed by PPARα and PPARγ Correspondly, in the context of a reporter gene construct, RA was found to efficiently activate PPARβ/δ but not PPARα or PPARγ (Shaw et al., 2003). Hence, an intriguing possibility is that, in some cells, RA may activate transcription not only through RAR but also through PPARβ/δ. It is noteworthy in regard to this that, in keratinocytes, PPARβ/δ induces differentiation and, importantly, displays pronounced anti-apoptotic activities mediated in part by direct transcriptional targeting of the PDK-1/Akt survival pathway (Di-Poi et al., 2002; Tan et al., 2004). Consequently, PPARβ/δ is central to maintenance of skin permeability-barrier integrity, and to keratinocyte survival during inflammation and wound healing (Di-Poi et al., 2003; Icre et al., 2006).
Ligands that activate RAR and the various PPAR isotypes also bind in cells to intracellular lipid binding proteins (iLBPs), a family of small proteins that share a remarkably similar 3-dimensional structure (Gutierrez-Gonzalez et al., 2002; Kleywegt et al., 1994; Veerkamp and Maatman, 1995), but bind lipophilic molecules with distinct selectivities. Some members of this family, termed cellular retinoic acid binding proteins (CRABP-I and II), specifically associate with retinoic acid with subnanomolar affinities (Dong et al., 1999). Other iLBPs, known as fatty acid binding proteins (FABPs), display broad selectivities and bind a variety of fatty acids and some fatty acid derivatives. In fact, the spectrum of ligands that bind to FABPs is reminiscent of that of PPARs (Gutierrez-Gonzalez et al., 2002; Hanhoff et al., 2002; Norris and Spector, 2002; Widstrom et al., 2001). The shared ligand selectivities of some iLBPs and some nuclear receptors suggest that specific members of the two classes of proteins may cooperate in regulating the biological activities of their common ligands. Such a cooperation is also suggested by overlapping tissue expression profiles and by involvement in similar biological functions.
Recent studies indeed demonstrated that three iLBPs, CRABP-II, FABP5 (K-FABP, eFABP, mal1), and FABP4 (A-FABP, aP2), selectively cooperate with the nuclear receptors RARα, PPARβ/δ and PPARγ, respectively. Specifically, these studies established that, upon association with particular ligands, these binding proteins translocate from the cytosol to the nucleus, that they engage in direct protein-protein interactions with their ‘cognate’ receptors, and that the resulting complex mediates ‘ligand-channeling’ from the binding protein to the receptor. Consequently, the binding proteins facilitate the ligation of the respective receptors and significantly augment their transcriptional activities (Budhu and Noy, 2002; Dong et al., 1999; Manor et al., 2003; Sessler and Noy, 2005; Tan et al., 2002). Interestingly, although FABP4 and FABP5 bind multiple ligands, only particular compounds trigger their nuclear translocation (Tan et al., 2002). FABP4 moves into the nucleus in response to ligands that activate PPARγ but not upon treatment with PPARβ/δ ligands. In contrast, FABP5 mobilizes to the nucleus only in response to ligands that activate PPARβ/δ.
Hence, available information suggests the possibility that, while RA inhibits cell-growth by signalling through RAR, the ‘non-traditional’ pro-proliferative activities of this hormone may be mediated by PPARβ/δ. Work described in this manuscript was undertaken in order to examine this hypothesis, and to investigate the possibility that CRABP-II and FABP5 control the partitioning of RA between the two receptors.
Results
RA facilitates tumor growth in the MMTV-neu transgenic mammary cancer model
In keratinocytes, RA exerts pro-proliferative activities that appear to be mediated by a pathway other than activation of RAR (Chapellier et al., 2002). In search for an additional model that will allow for exploring the mechanisms that underlie ‘non-traditional’ activities of RA, we considered the transgenic mammary cancer mouse model TgN(MMTVneu)202Mul (Akagi et al., 1997; Guy et al., 1992). The oncogenic hallmark of this model is a mammary-specific amplification of the growth factor receptor c-erb-B2/neu, which is often amplified in primary human breast cancers (King et al., 1985). This model, in which 100% of female mice develop mammary adenocarcinomas (Muller et al., 1988), was chosen because it has been reported that over-expression of neu in mammary carcinoma cells leads to RA-resistance, suggesting down-regulation of RAR signalling (Tari et al., 2002). To examine the effects of RA on tumor development, the rate of tumor growth was studied in untreated mice and in mice systemically treated with RA as of age 140 days. Measurements were initiated when tumors reached a volume of 0.065 cm3. The data (Fig. 1A) showed that RA treatment dramatically facilitated tumors formation with mean tumor volume on day 20 of 1.09 ± 0.22 cm3 in untreated mice, and 1.87 ± 0.25 cm3 in the RA-treated group (mean ± S.E.M., n = 12 in each group, P = 0.04). Assessment of mouse survival, defined as the time when tumor volume reached 0.524 cm3 (Fig. 1B), indicated that, although all mice eventually developed tumors of that size, the median survival of RA-treated vs untreated mice was 205 vs 240 days (P = 0.001). Hence, RA exerts pro-proliferative activities in tumors that arise in MMTVneu mice.
Figure 1. RA facilitates tumor growth in MMTV-neu mice.
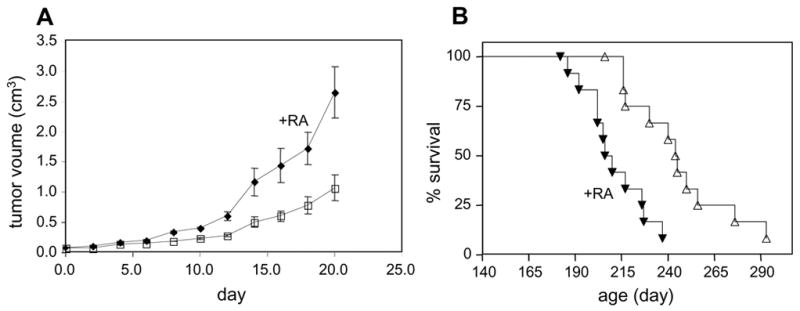
Tumor growth was monitored in untreated female MMTV-neu mice and in mice subjected to systemic RA as of age 140 d. A. Sizes of tumors were measured with calipers with a start point (day 0) of tumor volume = 0.065 cm3. Volumes of tumors in untreated (closed symbols) and RA-treated (open symbols) on day 20 were 1.09 ± 0.22 cm3 vs. 1.87 ± 0.25 cm3 (mean ± SEM; n=12 in each group, P = 0.04). B. Mice were treated as described in a., and survival, defined as the age in which tumor volume reached 0.524 cm3, was assessed. Median survivals were 240 and 205 days for untreated mice (open symbols) and RA-treated mice (closed symbols, n=12 in each group, P = 0.001).
Subsequent experiments thus utilized two cell lines in which RA displays ‘non-traditional’ activities, leading to cell survival and growth: HaCaT keratinocyte cells, and NaF cells, derived from tumors that arise in MMTVneu mice. For comparison, we employed MCF-7 mammary carcinoma cells, in which RA is known to function through RAR and to inhibit cell proliferation (Donato and Noy, 2005; Donato et al., 2007; Mangiarotti et al., 1998; Toma et al., 1998).
In HaCaT keratinocytes, RA activates PPARβ/δ in parallel to activation of RAR
The ability of RA to activate PPARβ/δ in the human keratinocyte cell line HaCaT was examined. Transcriptional activation assays conducted using a luciferase reporter driven by a consensus PPAR response element (PPRE) showed that the synthetic PPARβ/δ-selective ligand GW0742, but not the RAR-selective ligand 4-[(E)-2-(5,6,7,8-Tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)-1-propenyl]benzoic acid (TTNPB), induced transcription of the reporter (Fig. 2A). These observations attest to the expression and functionality of PPARβ/δ in these cells and demonstrate specificity of reporter response. RA also enhanced the expression of the PPRE-driven reporter and did so in a dose-responsive manner (Fig. 2B). The response was markedly suppressed when the expression of PPARβ/δ in the cells was decreased by about 80% by siRNA methodology (Fig. 2B), indicating that the ability of RA to induce reporter expression was indeed mediated by this receptor and not by RAR.
Figure 2. In HaCaT cells, RA activates both PPARβ/δ and RAR.
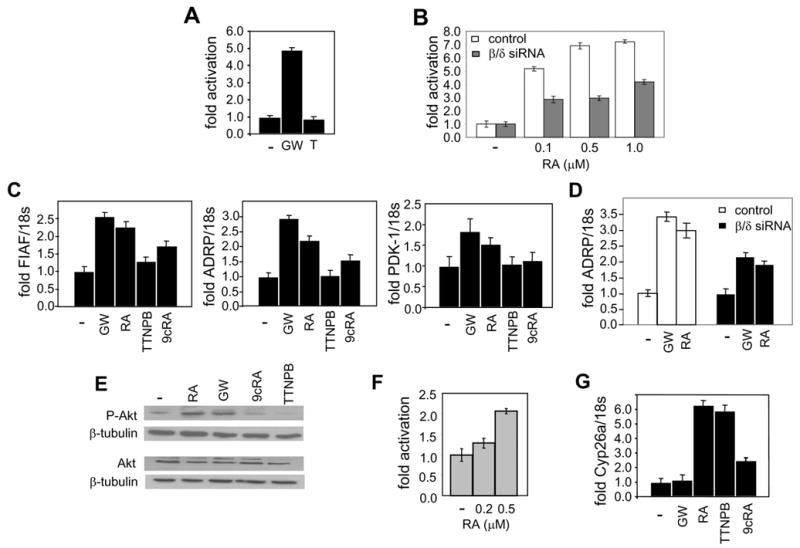
a. and b. Transactivation assays in HaCaT cells transfected with a PPRE-driven luciferase reporter. Cells were treated with the denoted ligands for 15 hr, lysed, and luciferase activity measured and corrected for β-galactosidase activity. Data were normalized to the basal activity. Data are mean ± SEM, n=3 A. Cells were treated with vehicle or GW0742 (GW) or TTNPB (T, 1 μM). B. Cells were cotransfected with either control siRNA or siRNA for PPARβ/δ, and than treated with RA at the denoted concentrations. C. HaCaT cells were treated with denoted ligands (0.1 μM, 4 hr) and RNA extracted. Levels of mRNA of the PPARβ/δ target genes FIAF, ADRP, and PDK-1 were analyzed by Q-PCR and normalized to 18s mRNA. Data are mean ± SEM, n=3. D. HaCaT cells were transfected with control siRNA or PPARβ/δ siRNA (24 hr.), and then treated with the denoted ligands (0.1 μM, 4 hr) and RNA extracted. ADRP mRNA was analyzed by Q-PCR and normalized to 18s mRNA. Data are mean ± SEM, n=3. E. Cells were treated with denoted ligands (0.1 μM, 12 hr) and lysed. Thr-307-phospho-Akt, total Akt, and β-tubulin were assessed by immunoblots. Data from a representative experiment which was repeated 4 times with similar results are shown. F. Transactivation assays in HaCaT cells transfected with a RARE-driven luciferase reporter. Cells were treated with RA at the denoted concentrations (15 hr), lysed, and luciferase activity measured and corrected for β-galactosidase activity. Data were normalized to the basal activity. Data are mean ± SEM, n=3. G. Cells were treated with the denoted ligands and expression of mRNA of the RAR target gene Cyp26a analyzed by Q-PCR and normalized to 18s mRNA. Data are mean ± SEM, n=3.
We then set out to examine the ability of RA to induce the expression of endogenous PPARβ/δ target genes in HaCaT cells. One of these, PDK-1, was previously shown to comprise a direct PPARβ/δ target in HaCaT cells (Di-Poi et al., 2002). Two other genes, fasting induced adipose factor (FIAF) (Kersten et al., 2000) and adipose differentiation-related protein (ADRP) (Schmuth et al., 2004), were reported to be targeted by PPARβ/δ in other cells. Chromatin immunoprecipitation assays demonstrated that PPARβ/δ associates with the PPAR response elements of both FIAF and ADRP in HaCaT cells (Fig. S1), verifying that both are direct targets for this receptor in the context of these cells. The PPARβ/δ ligand GW0742 as well as RA upregulated the expression of mRNA for all three of these endogenous PPARβ/δ target genes (Fig. 2C). The observations that the RAR-ligand TTNPB had little effect on the expression of these genes further confirm that RAR is not involved in this activity of RA. As a control, cells were treated with 9-cis-RA (9cRA), a ligand that activates RXR, the obligatory heterodimerization partner for both RAR and PPARs (Levin et al., 1992). This ligand induced a modest response, which likely emanated from activation of the RXR moiety of the RXR-PPARβ/δ heterodimer. The small magnitude of the response indicates that the induction of ADRP and FIAF by RA is not a result of conversion of RA to its 9-cis isomer within the cells. Further support for the conclusion that upregulation of these genes by RA is mediated by PPARβ/δ was provided by the observations that an 80% decrease in the expression of this receptor significantly hampered the induction of ADRP by both GW0747 and RA (Fig. 2D).
Notably, one of the PPARβ/δ targets found to be induced by RA, PDK-1, is an important component of the anti-apoptotic activities of this receptor in keratinocytes, where induction of this kinase leads to phosphorylation and activation of the downstream PDK-1-target survival factor Akt (Di-Poi et al., 2002). The effects of RA or GW0742 on the phosphorylation level of Akt were thus examined. Treatment with either of these ligands, but not with TTNPB or 9cRA, significantly increased the phosphorylation level of Akt (Fig. 2E).
In addition to activating PPARβ/δ RA also upregulated the expression of a reporter gene construct driven by an RAR response element (Fig. 2F), and it efficiently upregulated the expression of mRNA for CYP26a, a known direct RAR target gene (Loudig et al., 2000) (Fig. 2G). Hence, in HaCaT cells, RA treatment results in parallel activation of both RAR and PPARβ/δ.
FABP5 translocates into the nucleus in response to RA, and it enhances RA-induced, PPARβ/δ-mediated transcriptional activation
The observations that RA can activate both RAR and PPARβ/δ raise the question of the factors that regulate the dual activity of this hormone. We previously showed that the iLBPs CRABP-II and FABP5 mobilize to the nucleus in response to ligands that activate RAR and PPARβ/δ, respectively, and that they bind to their cognate receptors to form a complex through which the ligand is directly ‘channeled’ to the receptor. Consequently, CRABP-II enhances the transcriptional activity of RAR, while FABP5 facilitates the activation of PPARβ/δ (Budhu and Noy, 2002; Donato and Noy, 2005; Dong et al., 1999; Manor et al., 2003; Tan et al., 2002). The observations that RA serves as a ligand for PPARβ/δ thus raise the possibility that RA may be delivered to this receptor by FABP5. A fluorescence-based binding assay (Fig. 3A) demonstrated that GW0742 and RA bind to FABP5 with Kds of 42.3 ± 4.5 nM, and 34.8 ± 6.6 nM, respectively (mean ± SD, n=3), in good agreement with binding affinities of this protein towards other ligands (Tan et al., 2002). The subcellular localization of FABP5 was then examined. COS-7 cells were transfected with FABP5 fused to green fluorescence protein (GFP), and confocal fluorescence microscopy was used to image GFP-FABP5 in live cells treated with various ligands (Fig. 3B). Similarly to the behavior of GFP when transfected alone, GFP-FABP5 in untreated cells distributed between the cytoplasm and the nucleus, most likely reflecting that over-expression of the protein leads to leakage into the nucleus even in the absence of a specific nuclear localization signal (data not shown and (Sessler and Noy, 2005). Treatment of cells with stearic acid, a long chain fatty acid that binds FABP5 but does not activate it, did not affect the subcellular distribution of the protein. In contrast, treatment with either GW0742 or RA resulted in a distinct shift of the protein into the nucleus (Fig. 3B). To monitor the effects of ligands on the localization of endogenous FABP5 in HaCaT cells, cells were treated with vehicle, RA, or stearate, subjected to subcellular fractionation, and the presence of FABP5 in cytosol and in nuclei examined by immunoblots (Fig. 3C). The data demonstrated that endogenous FABP5 in HaCaT cells is predominantly cytosolic in the absence of ligand, and that it accumulates in the nucleus in response to RA, but not upon treatment with stearic acid. Hence, like known PPARβ/δ-ligands, RA activates the nuclear localization of FABP5.
Figure 3. FABP5 binds RA, translocates to the nucleus in response to this ligand, and enhances RA-induced activation of PPARβ/δ.
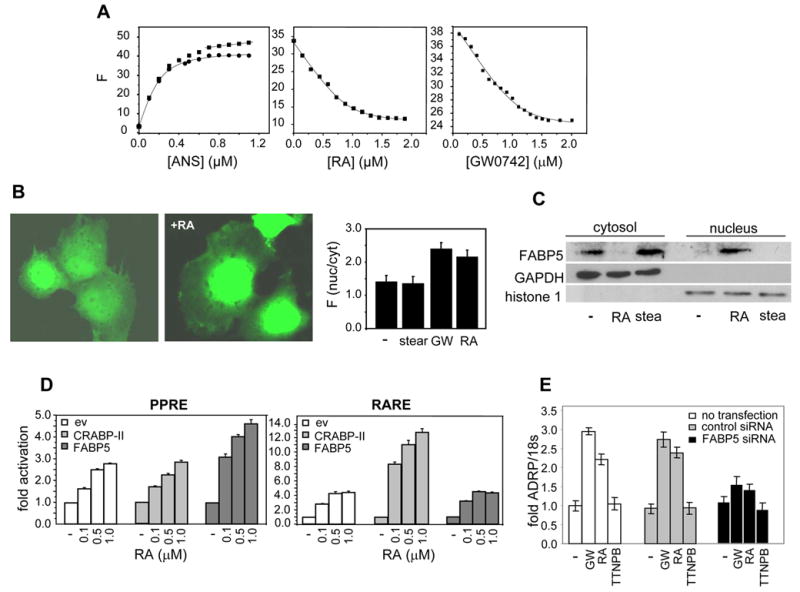
A. FABP5 was titrated with the fluorescence probe ANS. Titrations curves (left panel, filled squares) were corrected for linear non-specific fluorescence (solid line at end of titration curve), and corrected data (filled circles) analyzed to yield a Kd of 57 ± 7.3 nM (mean ± SD, n=3). Kds for the association of FABP5 with RA (middle panel) and with GW0742 (right panel) were determined by fluorescence competition titrations. B. COS-7 cells were transfected with an expression vector harboring GFP-FABP5 and imaged. Images were acquired from live cells before and after a 30 min. treatment with RA (1 μM). Right panel: quantitation of nuclear/cytoplsmic partitioning of FABP5 in cells treated with denoted ligands. Forty cells of each treatment group were analyzed (mean ± SEM) C. HaCaT cells were treated with denoted RA or with stearic acid (1μM, 30 min.). Nuclei were separated from cytosol by subcellular fractionation (Calbiochem ProtoExtract Subcellular Proteome Extraction kit) and anlyzed for the presence of FABP5 by immunoblots. Efficiency of fractionation was validated by immunoblotting for the cytosolic marker GAPDH, and the nuclear marker histone 1. D. Transactivation assays were carried out in COS-7 cells cotransfected with a luciferase reporter driven by a PPRE and an expression vector for PPARβ/δ (left panel) or with an RARE-driven reporter together with an expression vector for RARα (right panel) Cells were also transfected with an empty vector or with expression vectors for either FABP5 or CRABP-II, treated with RA, lysed, and luciferase activity determined. Data are mean ± SEM, n=3. E. HaCaT cells were not transfected, or transfected with either control siRNA or a construct harboring FABP5 siRNA (24 hr.). The ability of denoted ligands to induce ADRP expression was monitored by Q-PCR and normalized to 18s mRNA. Data are mean ± SEM, n=3.
The effects of FABP5 on the ability of RA to activate PPARβ/δ were examined by transactivation assay using COS-7 cells which express very low level of either FABP5 or CRABP-II. Cells were co-transfected with a luciferase reporter construct driven by a PPRE, an expression vector for PPARβ/δ, and a vector harboring cDNA for either FABP5 or CRABP-II. Cells were then treated with RA, and the expression of the reporter monitored (Fig. 3D, left panel). RA enhanced the expression of the PPRE-driven reporter in a dose-responsive manner. While expression of CRABP-II did not affect the activity, FABP5 significantly enhanced RA-induced, PPARβ/δ-mediated transactivation. To investigate the effect of the binding proteins on RA-induced activation of RAR, cells were transfected with a luciferase reporter under the control of an RAR response element (RARE), an expression construct for RARα, and a vector harboring cDNA for either FABP5 or CRABP-II. In agreement with previous reports, CRABP-II augmented RA-induced transactivation of RAR. On the other hand, FABP5 had little effect on this activity (Fig. 3D, right pane). Cells in which the receptors were not ectopically expressed displayed qualitatively similar behaviour but the magnitudes of the ligand-induced responses were significantly smaller (not shown).
The involvement of FABP5 in RA-induced activation of PPARβ/δ was further investigated by examining the effect of decreasing the expression level of this binding protein on the ability of RA to activate the receptor in HaCaT cells. Cells were transfected with FABP5 siRNA, resulting in an about 80% decrease in the level of the protein, and induction of the PPARβ/δ target gene ADRP was monitored (Fig. 3E). Decreasing the expression of FABP5 markedly attenuated the ability of both GW0742 and RA to upregulate the expression of the ADRP, further substantiating that the presence of FABP5 is necessary for efficient activation of PPARβ/δ by its ligands, including RA.
Decreasing the FABP5/CRABP-II ratio in HaCaT and NaF cells converts RA from a survival factor to a pro-apoptotic agent
The observations that RA is delivered to RAR by CRABP-II, and is shuttled to PPARβ/δ by FABP5 suggest that differential expression profiles of these iLBPs may regulate the partitioning of RA between the two receptors in different cells. The levels of the two binding proteins were thus examined in several tissues and cells. Of special interest are mammary tumors that arise in the transgenic mouse cancer model MMTVneu. RA treatment of MMTVneu mice facilitates mammary tumor development (Fig. 1). On the other hand, it was previously reported that ectopic expression of CRABP-II in these tumors induces carcinoma cell apoptosis and suppresses tumor growth (Manor et al., 2003), indicating that, under these conditions, RA exerts growth-inhibitory activities. Comparison between the expression levels of CRABP-II and FABP5 in tumors that arise in MMTVneu mice and in adjacent normal mammary tissue revealed that CRABP-II expression is markedly decreased, while the level of FABP5 is significantly higher in the tumors (Fig. 4A). Hence, tumor development in MMTVneu mice is accompanied by a dramatic decrease in the CRABP-II/FABP5 ratio. Corresponding to this expression profile in vivo, NaF cells, a cell line derived from MMTVneu tumors, display a high level of FABP5 and minimal expression of CRABP-II (Fig. 4B). Similarly, HaCaT cells, in which RA can function through PPARβ/δ, express a high level of FABP5 and an undetectable CRABP-II content (Fig. 4B). In contrast, the mammary carcinoma MCF-7 cells, in which RA activates RAR to inhibit cell growth (Donato and Noy, 2005; Donato et al., 2007; Mangiarotti et al., 1998; Toma et al., 1998), and F9 teratocarcinoma cells, which differentiate into primitive endoderm in response to RA-induced, RAR-mediated signalling (Rochette-Egly and Chambon, 2001; Strickland and Mahdavi, 1978; Strickland et al., 1980), express a markedly higher CRABP-II/FABP5 ratio (Fig. 4B).
Figure 4. In HaCaT cells, decreasing the FABP5/CRABP-II ratio diverts RA away from PPARβ/δ.
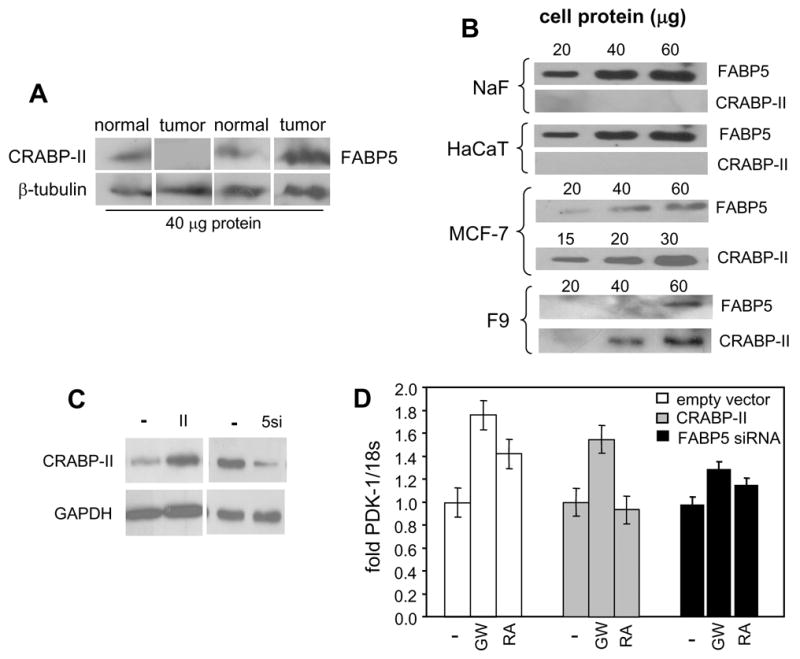
A. Expression of CRABP-II and FABP5 in mammary tumors that arise in MMTVneu mice and in adjacent normal mammary tissue estimated by immunoblots. B. Expression levels of CRABP-II and FABP5 in HaCaT, NaF, MCF-7, and F9 cells were estimated by immunoblots. C. HaCaT cells were transfected with an expression vector for CRABP-II or with a vector harboring FABP5 siRNA. Effectiveness of transfection in modulating the proteins’ expression levels was monitored by immunoblotting. D. Parental HaCaT cells and HaCaT cells over-expressing CRABP-II or under-expressing FABP5 were treated with denoted ligands (1 μM, 4 hr) and expression of PDK-1 mRNA was measured by Q- PCR. Data are mean ± SEM, n=3.
The effect of reversing the CRABP-II/FABP5 ratio in HaCaT cells on the ability of RA to activate PPARβ/δ was then examined. Cells were transfected with either an expression vector for CRABP-II, or with a construct harboring FABP5 siRNA (Fig. 4C), treated with RA or GW0742, and induction of the PPARβ/δ target gene PDK-1 was monitored (Fig. 4D). Over-expression of CRABP-II had little effect on GW0742-induced expression of PDK-1 mRNA, indicating that this protein does not directly affect PPARβ/δ activity. Either over-expression of CRABP-II or under-expression of FABP5 abolished the ability of RA to induce PDK-1 expression. Additionally, in accordance with the observations that GW0742 tightly binds to FABP5 and mobilizes it to the nucleus (Fig. 3), decreasing FABP5 expression augmented the transcriptional activity of this ligand. The data thus show that increasing the CRABP-II/FABP5 ratio in HaCaT cells diverts RA away from PPARβ/δ. The involvement of FABP5 in induction of PDK-1 by both RA and GW0742 also supports the notion that this binding protein mediates the nuclear targeting of both of these PPARβ/δ ligands.
Upregulation of PDK-1 expression upon activation of PPARβ/δ by RA in HaCaT cells results in the phosphorylation and thus the activation of the survival factor Akt (Fig. 2E). It can thus be predicted that RA treatment will protect these cells against apoptosis. To examine this notion, HaCaT cells were treated with the apoptosis inducer tumor necrosis factor α (TNFα), and the effects of RA, the PPARβ/δ ligand GW0742, and the RAR ligand TTNPB on TNFα-induced apoptosis were examined. To this end, we monitored the ability of TNFα to trigger the cleavage of poly(ADP-ribose) polymerases (PARP), one of the earliest proteins targeted for a specific cleavage during apoptosis (Fig. 5A, 5B). TNFα treatment significantly enhanced PARP cleavage, demonstrating the efficacy of the cytokine as an apoptosis-inducing agent in these cells. In agreement with a role for PPARβ/δ in enabling keratinocyte survival during inflammation (Di-Poi et al., 2002; Tan et al., 2001), activation of the receptor by GW0742 inhibited TNFα-induced apoptosis. Similarly to GW0742, RA protected the cells from TNFα-induced apoptosis. In contrast, the RAR-ligand TTNPB induced apoptosis when administered alone, and augmented TNFα-induced PARP cleavage, demonstrating that RAR displays pro-apoptotic activities in these cells. Hence, in HaCaT cells, RA inhibits apoptosis like a bona-fide PPARβ/δ ligand, an activity that diametrically contrasts with the pro-apoptotic activities of RAR.
Figure 5. In HaCaT and NaF cells decreasing the FABP5/CRABP-II ratio converts RA from an anti-apoptotic to a pro-apoptotic agent.
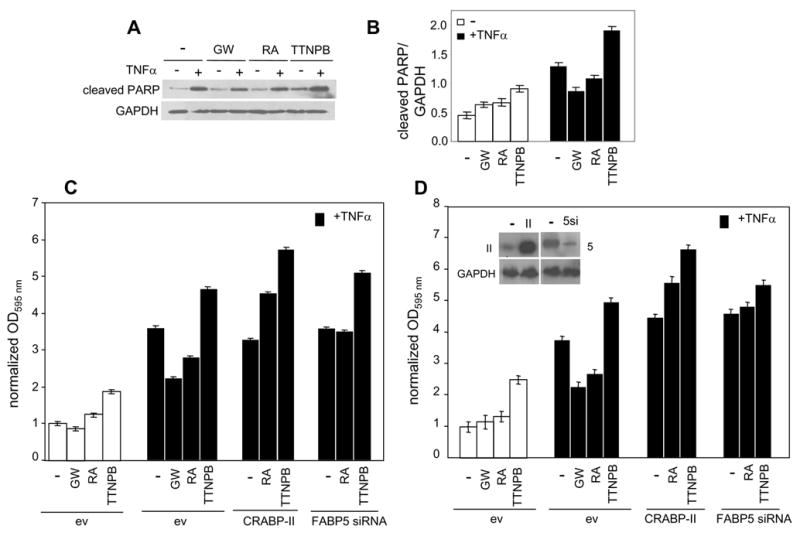
A. HaCaT cells were treated with denoted ligands (2 μM, 2 hr) prior to addition of TNFα (20 ng/ml, 14 hr.). Cells were lysed and the level of cleaved PARP assessed by immunoblots. A representative experiment is shown. B. Quantitation of data described in panel a. Band intensities were normalized to the loading control GAPDH. Data are mean ± SEM, n=3. C. and D. Parental HaCaT (C.) or NaF (D.) cells and corresponding cells over-expressing CRABP-II or under-expressing FABP5 were treated with denoted ligands (1 μM, 16 hr.), or pretreated with ligands for 2 hr. prior to addition of TNFα (20 ng/ml, 14 hr., solid bars). Apoptosis was evaluated using the APOPercentage Apoptosis Assay kit. Data are mean ± SEM, n=3.
The hypothesis that CRABP-II and FABP5 control the partitioning of RA between RAR and PPARβ/δ and thus the biological activities of this hormone, was further investigated by examining the outcome of changing the CRABP-II/FABP5 ratio on the ability of RA to modulate TNFα-induced apoptosis. In these experiments, apoptosis was followed by monitoring the transfer of phosphatidylserine to the outer leaflet of the cell membrane, a process linked to the execution phase of apoptosis. In agreement with observations obtained by monitoring PARP cleavage as a ‘readout’ for apoptosis, treatment of HaCaT cells with GW0742 or RA had little effect, while the RAR-ligand TTNPB increased the fraction of apoptotic cells (Fig. 5C). Also in agreement, TNFα efficiently induced apoptosis, both GW0742 and RA inhibited, and TTNPB enhanced the response. Strikingly, over-expression of CRABP-II not only abolished the protective activity of RA, but converted it into an agent which, similarly to TTNPB, enhanced TNFα-induced apoptosis. Reduced expression of FABP5 also negated the anti-apoptotic activity of RA. The observation that the decreased expression of FABP5 did not quite lead to an enhancement of the apoptotic response likely reflects that transfection of FABP5 siRNA reduced but did not completely inhibit the expression of the protein (Fig. 4C). Similar effects were observed in NaF cells, derived from mammary tumors of the MMTVneu mouse model; RA ‘rescued’ NaF cells from TNFα-induced apoptosis, and either over-expression of CRABP-II or under-expression of FABP5 abolished the protective activity of RA and enhanced the apoptotic response (Fig. 5D).
In MCF-7 cells, increasing the FABP5/CRABP-II ratio converts RA from a pro-apoptotic to an anti-apoptotic agent
Unlike HaCaT and NaF cells, the mammary carcinoma MCF-7 cells display a high CRABP-II/FABP5 ratio (Fig. 4B). To reverse this ratio, cell were co-transfected with an expression vector for FABP5 and a construct harboring CRABP-II siRNA (Fig. 6A). The ensuing concomitant increase in FABP5 and decrease in CRABP-II levels did not affect the expression of either RAR or PPARβ/δ (Fig. S2). Nevertheless, the reversal hampered the ability of RA to upregulate the expression of the cell cycle regulator BTG2, a gene that was recently shown to comprise a direct target for RAR and to be involved in mediating RA-induced cell cycle arrest in MCF-7 cells (Donato et al., 2007)(Fig. 6B). Correspondingly, the reversal abolished the ability of RA to down-regulate the expression of cyclin D1, a known downstream target for BTG2 (Guardavaccaro et al., 2000; Kawakubo et al., 2006; Kawakubo et al., 2004; Lim et al., 1989) (Fig. 6C). Reversing the FABP5/CRABP-II also inhibited RA-induced induction of the RAR target cyp26a (Fig. 6D). Remarkably, while RA had little effect the level of the direct PPARβ/δ target gene PDK-1 in parental MCF-7 cells, the ligand significantly upregulated the expression of this survival factor upon the reversal of the ratio of the binding proteins (Fig. 6E). Hence, increasing the FABP5/CRABP-II ratio in MCF-7 cells directed RA away from RAR and towards PPARβ/δ. Considering the disparate nature of the RAR and PPARβ/δ target genes, the observations suggest that the reversal of the CRABP-II/FABP5 ratio hampered growth-inhibitory activities and triggered a survival response.
Figure 6. In MCF-7 cells, increasing the FABP5/CRABP-II ratio converts RA from a pro-apoptotic agent to a survival factor.
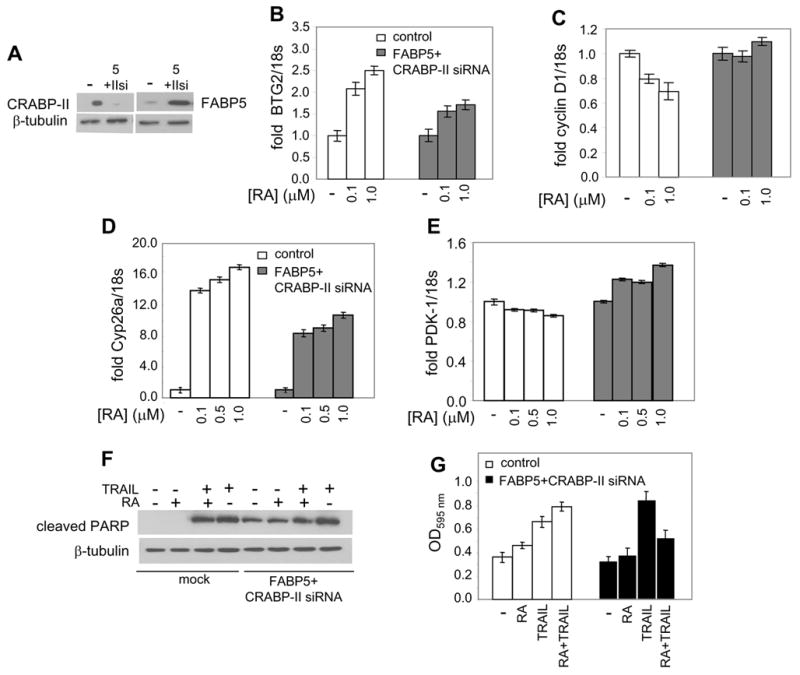
A. MCF-7 cells were co-transfected with an expression vector for FABP5 together with CRABP-II siRNA, and protein expression levels monitored by immunoblotting. B.-E. MCF-7 cells were mock transfected or co-transfected with an expression vector for FABP5 and with CRABP-II siRNA, and treated with RA. Levels of mRNA for BTG2 (B.), cyclin D1 (C.), cyp26a (D.), and PDK-1 (E.) were measured by Q-PCR and normalized to 18s. F. and G. MCF-7 cells were mock transfected or co-tranfected with an expression vector for FABP5 and with CRABP-II siRNA. Cells were treated with TRAIL (4 ng/ml), or RA (0.1 μM), or both, and apoptosis was assessed by assessing PARP cleavage (F.) and by monitoring transfer of phosphatidylserine to the plasma membrane outer leaflet using the APOPercentage Apoptosis Assay kit (G.).
To further examine the consequences of the ‘switch’ in binding protein profile, parental MCF-7 cells and cells in which the CRABP-II/FABP5 ratio was reversed were treated with the apoptosis-inducing agent tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). Apoptosis was monitored by following PARP cleavage (Fig. 6F) as well as by monitoring transfer of phosphatidylserine to the outer leaflet of the cell membrane (Fig. 6G). Transfection of constructs encoding FABP5 and CRABP-II siRNA resulted in PARP cleavage even in the absence of TRAIL. The effect is likely to stem from a degree of apoptosis induced by the forced over-expression, and was not observed upon monitoring later apoptosis events, represented by phosphatidylserine membrane “flip”. Treatment with TRAIL induced marked apoptosis as judged by both PARP cleavage and by plasma membrane events. Addition of TRAIL in conjunction with RA had little effect (PARP cleavage) or somewhat enhanced apoptosis (phosphatidylserine translocation). Remarkably, both ‘read-outs’ clearly demonstrated that, in cells in which the CRABP-II/FABP5 ratio was reversed, RA treatment markedly inhibited TRAIL-induced apoptosis. Hence, increasing the FABP5/CRABP-II ratio in MCF-7 cells results in conversion of RA from a growth-inhibitory to a pro-survival factor.
Discussion
RA activates the nuclear receptor RAR and, in many embryonic and adult tissues, the biological activities of this hormone can be traced to induction of expression of RAR target genes. However, in some tissues, such as skin, important functions of RA appear to be mediated by an RAR-independent pathway, the nature of which remained unclear. The observations presented here indicate that, in addition to activating RAR, RA can also activate the nuclear receptor PPARβ/δ, and thus that the repertoire of genes and cellular responses that can be controlled by this hormone include both RAR and PPARβ/δ-targets. The data show further that partitioning of RA between its two receptors is regulated by cognate iLBPs; CRABP-II delivers RA to RAR, while FABP5 shuttles the hormone to PPARβ/δ (Fig. 7). It should be noted that the binding affinity of the CRABP-II/RAR pathway for RA exceeds that of the FABP5/PPARβ/δ path. The interactions of RA with both CRABP-II and RAR are characterized by Kds in the 0.1–0.2 nM range (Dong et al., 1999; Sussman and de Lera, 2005), while both FABP5 and PPARβ/δ associate with this compound at a Kd of 10–50 nM ((Tan et al., 2002) and Fig. 3A). It can thus be predicted that, in most cells, RA signalling through RAR will predominate, and that activation of PPARβ/δ will become apparent only in cells that exhibit a high FABP5/CRABP-II ratio. This indeed appears to be the case. In MCF-7 cells, which express a low FABP5/CRABP-II ratio, RA activates RAR, while keratinocytes and NaF cells, which display a high FABP5/CRABP-II ratio, respond to RA by activation of PPARβ/δ.
Figure 7.
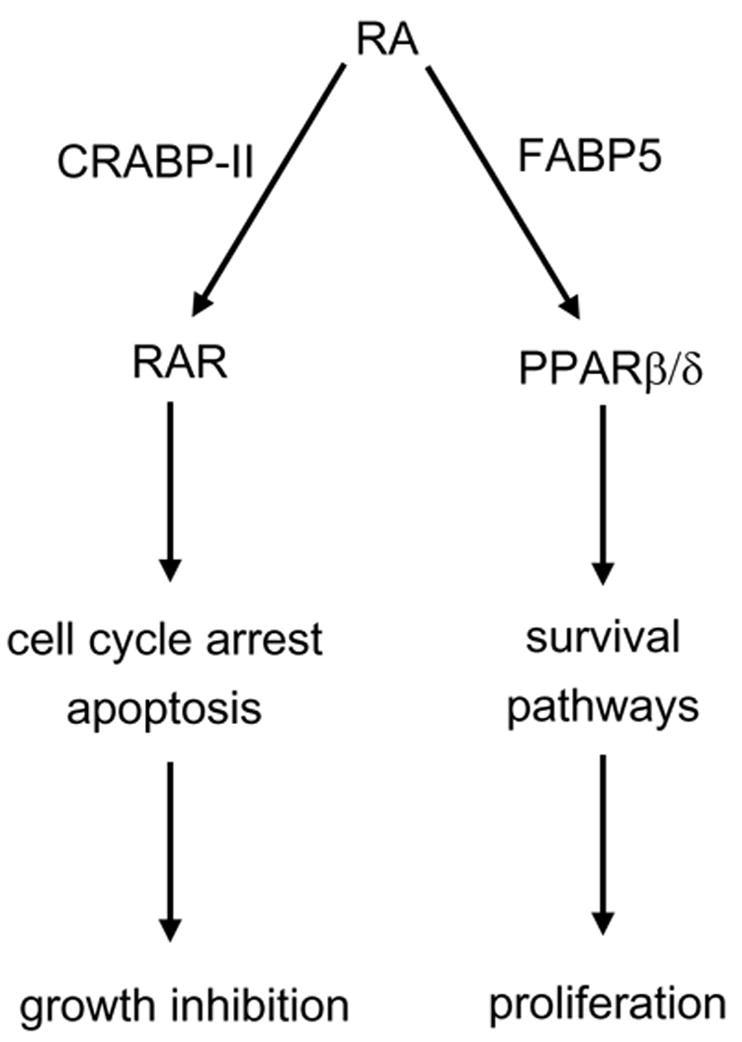
A model outlining the dual transcriptional activity of RA. CRABP-II and FABP5 target RA to RAR and PPARβ/δ, respectively. In cells that express a high CRABP-II/FABP5 ratio, RA is ‘channeled’ to RAR, often resulting in growth inhibition. Conversely, in the presence of a low CRABP-II/FABP5 expression ratio, RA is targeted to PPARβ/δ, thereby upregulating survival pathways.
A high FABP5/CRABP-II ratio abolishes RA-triggered upregulation of RAR target genes that mediate important biological responses, such as cell cycle arrest (Fig. 6B, 6C). It is worth noting however that cells that display such a ratio retain the ability to induce the expression of very efficient RAR targets, such as the RA-degrading enzyme Cyp26a, albeit at a muted response (Fig. 2G and 6D). Hence, critical functions of RA that are mediated by RAR, such as the ability to trigger its own degradation by upregulting the expression of cyp26 (White et al., 1996), can proceed in the background of the predominant action of RA-evoked PPARβ/δsignalling.
The present work demonstrates that RA serves as a physiological ligand for PPARβ/δunder some but not all circumstances. However, this receptor displays near-ubiquitous tissue expression (Kliewer et al., 1994), raising the question of the nature of the ligand(s) that activate it in tissues that do not support activation by RA. The ligand-binding pocket of PPARβ/δis much larger than the pockets of other nuclear receptors (Xu et al., 1999). It may thus accommodate multiple ligands, and it has been suggested that various long chain fatty acids and eicosanoids may serve as effective PPARβ/δ activators (Bucco et al., 1997). Whether some of these ligands function as true physiological ligands for the receptor remains to be clarified, but the present work and the similar nature of ligands that bind to FABPs and PPARs (Hanhoff et al., 2002; Widstrom et al., 2001) raise the possibility that FABPs other than FABP5 may act to deliver ligands other than RA to PPARβ/δ, and thus that they may regulate the functionality of distinct ligands in specific tissues.
When enabled, RA signalling through PPARβ/δ has profound functional consequences. One consequence, explored here, is that such signalling evokes potent anti-apoptotic activities that overcome the growth-inhibitory activities of RAR, and allow cells to survive in the face of powerful apoptotic agents. Hence, RA-dependent maintenance of skin integrity, proliferation of basal keratinocytes, and survival of these cells during wound repair likely stem from a high expression level of FABP5, enabling RA to activate PPARβ/δ. Activation of PPARβ/δ by RA also appears to underlie the facilitation of mammary tumor progression in MMTVneu mice by this hormone. In support of this conclusion are the observations that RA functions as anti-apoptotic agent in cells derived from MMTVneu tumors, that a high FABP5/CRABP-II ratio is required for this activity, that tumor progression in MMTVneu mice is accompanied by marked increase in the FABP5/CRABP-II ratio, and that decreasing this ratio by ectopic over-expression of CRABP-II triggers apoptosis and inhibits tumor development in vivo (Manor et al., 2003). It is worth noting in regard to this that it has been reported FABP5 is expressed in melanocytic tumors, but not in normal human melanocytes (Brouard et al., 2002), indicating that elevation in the expression of this protein, and thus activation of PPARβ/δ accompanies tumorigenesis in various cancers. While not directly addressed here, the observations that RA is critical for neuronal survival (Henion and Weston, 1994; Jacobs et al., 2006; Plum et al., 2001; Rodriguez-Tebar and Rohrer, 1991; Wuarin et al., 1990) raise the possibility that the brain may comprise another organ in which RA signals through PPARβ/δ. It is interesting to note regarding this possibility that the expression level FABP5 is high during neurogenesis in the developing rat brain (Liu et al., 2000), that this protein is required for neuronal outgrowth in PC12 neuronal cells (Allen et al., 2000), and that its expression is induced following nerve injury (De Leon et al., 1996), suggesting an involvement in survival/repair processes.
Supplementary Material
Acknowledgments
We thank Lee Kraus’ laboratory for help with ChiP assays, P. Leder for NaF cells, A. Senderowicz for HaCaT cells, and P. Chambon for CRABP-II constructs and antibodies. This work was supported by grants R01 CA68150 and RO1 DK60684 from the NIH. TTS was supported by NIH grant 5T32CA009682.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akagi K, Sandig V, Vooijs M, Van der Valk M, Giovannini M, Strauss M, Berns A. Cre-mediated somatic site-specific recombination in mice. Nucleic Acids Res. 1997;25:1766–1773. doi: 10.1093/nar/25.9.1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen GW, Liu JW, De Leon M. Depletion of a fatty acid-binding protein impairs neurite outgrowth in PC12 cells. Brain Res Mol Brain Res. 2000;76:315–324. doi: 10.1016/s0169-328x(00)00014-0. [DOI] [PubMed] [Google Scholar]
- Altucci L, Rossin A, Raffelsberger W, Reitmair A, Chomienne C, Gronemeyer H. Retinoic acid-induced apoptosis in leukemia cells is mediated by paracrine action of tumor-selective death ligand TRAIL. Nat Med. 2001;7:680–686. doi: 10.1038/89050. [DOI] [PubMed] [Google Scholar]
- Brouard MC, Saurat JH, Ghanem G, Siegenthaler G. Urinary excretion of epidermal-type fatty acid-binding protein and S100A7 protein in patients with cutaneous melanoma. Melanoma Res. 2002;12:627–631. doi: 10.1097/00008390-200212000-00013. [DOI] [PubMed] [Google Scholar]
- Bucco RA, Zheng WL, Davis JT, Sierra-Rivera E, Osteen KG, Chaudhary AK, Ong DE. Cellular retinoic acid-binding protein(II) presence in rat uterine epithelial cells correlates with their synthesis of retinoic acid. Biochemistry. 1997;36:4009–4014. doi: 10.1021/bi962094o. [DOI] [PubMed] [Google Scholar]
- Budhu AS, Noy N. Direct channeling of retinoic acid between cellular retinoic acid-binding protein II and retinoic acid receptor sensitizes mammary carcinoma cells to retinoic acid-induced growth arrest. Mol Cell Biol. 2002;22:2632–2641. doi: 10.1128/MCB.22.8.2632-2641.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambon P. A decade of molecular biology of retinoic acid receptors. Faseb J. 1996;10:940–954. [PubMed] [Google Scholar]
- Chapellier B, Mark M, Messaddeq N, Calleja C, Warot X, Brocard J, Gerard C, Li M, Metzger D, Ghyselinck NB, Chambon P. Physiological and retinoid-induced proliferations of epidermis basal keratinocytes are differently controlled. Embo J. 2002;21:3402–3413. doi: 10.1093/emboj/cdf331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Leon M, Welcher AA, Nahin RH, Liu Y, Ruda MA, Shooter EM, Molina CA. Fatty acid binding protein is induced in neurons of the dorsal root ganglia after peripheral nerve injury. J Neurosci Res. 1996;44:283–292. doi: 10.1002/(SICI)1097-4547(19960501)44:3<283::AID-JNR9>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Di-Poi N, Michalik L, Tan NS, Desvergne B, Wahli W. The anti-apoptotic role of PPARbeta contributes to efficient skin wound healing. J Steroid Biochem Mol Biol. 2003;85:257–265. doi: 10.1016/s0960-0760(03)00215-2. [DOI] [PubMed] [Google Scholar]
- Di-Poi N, Tan NS, Michalik L, Wahli W, Desvergne B. Antiapoptotic role of PPARbeta in keratinocytes via transcriptional control of the Akt1 signaling pathway. Mol Cell. 2002;10:721–733. doi: 10.1016/s1097-2765(02)00646-9. [DOI] [PubMed] [Google Scholar]
- Donato LJ, Noy N. Suppression of mammary carcinoma growth by retinoic acid: proapoptotic genes are targets for retinoic acid receptor and cellular retinoic acid-binding protein II signaling. Cancer Res. 2005;65:8193–8199. doi: 10.1158/0008-5472.CAN-05-1177. [DOI] [PubMed] [Google Scholar]
- Donato LJ, Suh JH, Noy N. Suppression of mammary carcinoma cell growth by retinoic acid: the cell cycle control gene Btg2 is a direct target for retinoic acid receptor signaling. Cancer Res. 2007;67:609–615. doi: 10.1158/0008-5472.CAN-06-0989. [DOI] [PubMed] [Google Scholar]
- Dong D, Ruuska SE, Levinthal DJ, Noy N. Distinct roles for cellular retinoic acid-binding proteins I and II in regulating signaling by retinoic acid. J Biol Chem. 1999;274:23695–23698. doi: 10.1074/jbc.274.34.23695. [DOI] [PubMed] [Google Scholar]
- Guardavaccaro D, Corrente G, Covone F, Micheli L, D’Agnano I, Starace G, Caruso M, Tirone F. Arrest of G(1)-S progression by the p53-inducible gene PC3 is Rb dependent and relies on the inhibition of cyclin D1 transcription. Mol Cell Biol. 2000;20:1797–1815. doi: 10.1128/mcb.20.5.1797-1815.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-Gonzalez LH, Ludwig C, Hohoff C, Rademacher M, Hanhoff T, Ruterjans H, Spener F, Lucke C. Solution structure and backbone dynamics of human epidermal-type fatty acid-binding protein (E-FABP) Biochem J. 2002;364:725–737. doi: 10.1042/BJ20020039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, Muller WJ. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc Natl Acad Sci U S A. 1992;89:10578–10582. doi: 10.1073/pnas.89.22.10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanhoff T, Lucke C, Spener F. Insights into binding of fatty acids by fatty acid binding proteins. Mol Cell Biochem. 2002;239:45–54. [PubMed] [Google Scholar]
- Henion PD, Weston JA. Retinoic acid selectively promotes the survival and proliferation of neurogenic precursors in cultured neural crest cell populations. Dev Biol. 1994;161:243–250. doi: 10.1006/dbio.1994.1024. [DOI] [PubMed] [Google Scholar]
- Icre G, Wahli W, Michalik L. Functions of the Peroxisome Proliferator-Activated Receptor (PPAR) alpha and beta in Skin Homeostasis, Epithelial Repair, and Morphogenesis. J Invest Dermatol. 2006;126(Suppl):30–35. doi: 10.1038/sj.jidsymp.5650007. [DOI] [PubMed] [Google Scholar]
- Jacobs S, Lie DC, DeCicco KL, Shi Y, DeLuca LM, Gage FH, Evans RM. Retinoic acid is required early during adult neurogenesis in the dentate gyrus. Proc Natl Acad Sci U S A. 2006;103:3902–3907. doi: 10.1073/pnas.0511294103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S, Duell EA, Fisher GJ, Datta SC, Wang ZQ, Reddy AP, Tavakkol A, Yi JY, Griffiths CE, Elder JT, et al. Application of retinol to human skin in vivo induces epidermal hyperplasia and cellular retinoid binding proteins characteristic of retinoic acid but without measurable retinoic acid levels or irritation. J Invest Dermatol. 1995;105:549–556. doi: 10.1111/1523-1747.ep12323445. [DOI] [PubMed] [Google Scholar]
- Kawakubo H, Brachtel E, Hayashida T, Yeo G, Kish J, Muzikansky A, Walden PD, Maheswaran S. Loss of B-cell translocation gene-2 in estrogen receptor-positive breast carcinoma is associated with tumor grade and overexpression of cyclin d1 protein. Cancer Res. 2006;66:7075–7082. doi: 10.1158/0008-5472.CAN-06-0379. [DOI] [PubMed] [Google Scholar]
- Kawakubo H, Carey JL, Brachtel E, Gupta V, Green JE, Walden PD, Maheswaran S. Expression of the NF-kappaB-responsive gene BTG2 is aberrantly regulated in breast cancer. Oncogene. 2004;23:8310–8319. doi: 10.1038/sj.onc.1208008. [DOI] [PubMed] [Google Scholar]
- Kersten S, Mandard S, Tan NS, Escher P, Metzger D, Chambon P, Gonzalez FJ, Desvergne B, Wahli W. Characterization of the fasting-induced adipose factor FIAF, a novel peroxisome proliferator-activated receptor target gene. J Biol Chem. 2000;275:28488–28493. doi: 10.1074/jbc.M004029200. [DOI] [PubMed] [Google Scholar]
- King CR, Kraus MH, Aaronson SA. Amplification of a novel v-erbB-related gene in a human mammary carcinoma. Science. 1985;229:974–976. doi: 10.1126/science.2992089. [DOI] [PubMed] [Google Scholar]
- Kitareewan S, Pitha-Rowe I, Sekula D, Lowrey CH, Nemeth MJ, Golub TR, Freemantle SJ, Dmitrovsky E. UBE1L is a retinoid target that triggers PML/RARalpha degradation and apoptosis in acute promyelocytic leukemia. Proc Natl Acad Sci U S A. 2002;99:3806–3811. doi: 10.1073/pnas.052011299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleywegt GJ, Bergfors T, Senn H, Le Motte P, Gsell B, Shudo K, Jones TA. Crystal structures of cellular retinoic acid binding proteins I and II in complex with all-trans-retinoic acid and a synthetic retinoid. Structure. 1994;2:1241–1258. doi: 10.1016/s0969-2126(94)00125-1. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Forman BM, Blumberg B, Ong ES, Borgmeyer U, Mangelsdorf DJ, Umesono K, Evans RM. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc Natl Acad Sci U S A. 1994;91:7355–7359. doi: 10.1073/pnas.91.15.7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laudet V, Gronemeyer H. The Nuclear Receptor FactsBook. London and San Diego: Academic Press; 2002. [Google Scholar]
- Levin AA, Sturzenbecker LJ, Kazmer S, Bosakowski T, Huselton C, Allenby G, Speck J, Kratzeisen C, Rosenberger M, Lovey A, et al. 9-cis retinoic acid stereoisomer binds and activates the nuclear receptor RXR alpha. Nature. 1992;355:359–361. doi: 10.1038/355359a0. [DOI] [PubMed] [Google Scholar]
- Lim RW, Varnum BC, O’Brien TG, Herschman HR. Induction of tumor promotor-inducible genes in murine 3T3 cell lines and tetradecanoyl phorbol acetate-nonproliferative 3T3 variants can occur through protein kinase C-dependent and -independent pathways. Mol Cell Biol. 1989;9:1790–1793. doi: 10.1128/mcb.9.4.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Longo LD, De Leon M. In situ and immunocytochemical localization of E-FABP mRNA and protein during neuronal migration and differentiation in the rat brain. Brain Res. 2000;852:16–27. doi: 10.1016/s0006-8993(99)02158-7. [DOI] [PubMed] [Google Scholar]
- Loudig O, Babichuk C, White J, Abu-Abed S, Mueller C, Petkovich M. Cytochrome P450RAI(CYP26) promoter: a distinct composite retinoic acid response element underlies the complex regulation of retinoic acid metabolism. Mol Endocrinol. 2000;14:1483–1497. doi: 10.1210/mend.14.9.0518. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf D, Umesono K, Evans RM. The retinoid receptors. In: Sporn MB, Roberts AB, Goodman DS, editors. The Retinoids, Biology, Chemistry and Medicine. New York: Raven Press; 1994. pp. 319–350. [Google Scholar]
- Mangiarotti R, Danova M, Alberici R, Pellicciari C. All-trans retinoic acid (ATRA)-induced apoptosis is preceded by G1 arrest in human MCF-7 breast cancer cells. Br J Cancer. 1998;77:186–191. doi: 10.1038/bjc.1998.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manor D, Shmidt EN, Budhu A, Flesken-Nikitin A, Zgola M, Page R, Nikitin AY, Noy N. Mammary carcinoma suppression by cellular retinoic acid binding protein-II. Cancer Res. 2003;63:4426–4433. [PubMed] [Google Scholar]
- Muller WJ, Sinn E, Pattengale PK, Wallace R, Leder P. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell. 1988;54:105–115. doi: 10.1016/0092-8674(88)90184-5. [DOI] [PubMed] [Google Scholar]
- Norris AW, Spector AA. Very long chain n-3 and n-6 polyunsaturated fatty acids bind strongly to liver fatty acid-binding protein. J Lipid Res. 2002;43:646–653. [PubMed] [Google Scholar]
- Park DJ, Chumakov AM, Vuong PT, Chih DY, Gombart AF, Miller WH, Jr, Koeffler HP. CCAAT/enhancer binding protein epsilon is a potential retinoid target gene in acute promyelocytic leukemia treatment. J Clin Invest. 1999;103:1399–1408. doi: 10.1172/JCI2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plum LA, Parada LF, Tsoulfas P, Clagett-Dame M. Retinoic acid combined with neurotrophin-3 enhances the survival and neurite outgrowth of embryonic sympathetic neurons. Exp Biol Med (Maywood) 2001;226:766–775. doi: 10.1177/153537020222600809. [DOI] [PubMed] [Google Scholar]
- Rochette-Egly C, Chambon P. F9 embryocarcinoma cells: a cell autonomous model to study the functional selectivity of RARs and RXRs in retinoid signaling. Histol Histopathol. 2001;16:909–922. doi: 10.14670/HH-16.909. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Tebar A, Rohrer H. Retinoic acid induces NGF-dependent survival response and high-affinity NGF receptors in immature chick sympathetic neurons. Development. 1991;112:813–820. doi: 10.1242/dev.112.3.813. [DOI] [PubMed] [Google Scholar]
- Schmuth M, Haqq CM, Cairns WJ, Holder JC, Dorsam S, Chang S, Lau P, Fowler AJ, Chuang G, Moser AH, et al. Peroxisome proliferator-activated receptor (PPAR)-beta/delta stimulates differentiation and lipid accumulation in keratinocytes. J Invest Dermatol. 2004;122:971–983. doi: 10.1111/j.0022-202X.2004.22412.x. [DOI] [PubMed] [Google Scholar]
- Sessler RJ, Noy N. A ligand-activated nuclear localization signal in cellular retinoic acid binding protein-II. Mol Cell. 2005;18:343–353. doi: 10.1016/j.molcel.2005.03.026. [DOI] [PubMed] [Google Scholar]
- Shaw N, Elholm M, Noy N. Retinoic acid is a high affinity selective ligand for the peroxisome proliferator-activated receptor beta/delta. J Biol Chem. 2003;278:41589–41592. doi: 10.1074/jbc.C300368200. [DOI] [PubMed] [Google Scholar]
- Soprano DR, Qin P, Soprano KJ. Retinoic Acid Receptors and Cancers. Annu Rev Nutr. 2004;24:201–221. doi: 10.1146/annurev.nutr.24.012003.132407. [DOI] [PubMed] [Google Scholar]
- Strickland S, Mahdavi V. The induction of differentiation in teratocarcinoma stem cells by retinoic acid. Cell. 1978;15:393–403. doi: 10.1016/0092-8674(78)90008-9. [DOI] [PubMed] [Google Scholar]
- Strickland S, Smith KK, Marotti KR. Hormonal induction of differentiation in teratocarcinoma stem cells: generation of parietal endoderm by retinoic acid and dibutyryl cAMP. Cell. 1980;21:347–355. doi: 10.1016/0092-8674(80)90471-7. [DOI] [PubMed] [Google Scholar]
- Sussman F, de Lera AR. Ligand recognition by RAR and RXR receptors: binding and selectivity. J Med Chem. 2005;48:6212–6219. doi: 10.1021/jm050285w. [DOI] [PubMed] [Google Scholar]
- Tan NS, Michalik L, Desvergne B, Wahli W. Peroxisome proliferator-activated receptor-beta as a target for wound healing drugs. Expert Opin Ther Targets. 2004;8:39–48. doi: 10.1517/14728222.8.1.39. [DOI] [PubMed] [Google Scholar]
- Tan NS, Michalik L, Noy N, Yasmin R, Pacot C, Heim M, Fluhmann B, Desvergne B, Wahli W. Critical roles of PPAR beta/delta in keratinocyte response to inflammation. Genes Dev. 2001;15:3263–3277. doi: 10.1101/gad.207501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan NS, Shaw NS, Vinckenbosch N, Liu P, Yasmin R, Desvergne B, Wahli W, Noy N. Selective cooperation between fatty acid binding proteins and peroxisome proliferator-activated receptors in regulating transcription. Mol Cell Biol. 2002;22:5114–5127. doi: 10.1128/MCB.22.14.5114-5127.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tari AM, Lim SJ, Hung MC, Esteva FJ, Lopez-Berestein G. Her2/neu induces all-trans retinoic acid (ATRA) resistance in breast cancer cells. Oncogene. 2002;21:5224–5232. doi: 10.1038/sj.onc.1205660. [DOI] [PubMed] [Google Scholar]
- Toma S, Isnardi L, Riccardi L, Bollag W. Induction of apoptosis in MCF-7 breast carcinoma cell line by RAR and RXR selective retinoids. Anticancer Res. 1998;18:935–942. [PubMed] [Google Scholar]
- Veerkamp JH, Maatman RG. Cytoplasmic fatty acid-binding proteins: their structure and genes. Prog Lipid Res. 1995;34:17–52. doi: 10.1016/0163-7827(94)00005-7. [DOI] [PubMed] [Google Scholar]
- Verma AK, Conrad EA, Boutwell RK. Differential effects of retinoic acid and 7,8-benzoflavone on the induction of mouse skin tumors by the complete carcinogenesis process and by the initiation-promotion regimen. Cancer Res. 1982;42:3519–3525. [PubMed] [Google Scholar]
- White JA, Guo YD, Baetz K, Beckett-Jones B, Bonasoro J, Hsu KE, Dilworth FJ, Jones G, Petkovich M. Identification of the retinoic acid-inducible all-trans-retinoic acid 4- hydroxylase. J Biol Chem. 1996;271:29922–29927. doi: 10.1074/jbc.271.47.29922. [DOI] [PubMed] [Google Scholar]
- Widstrom RL, Norris AW, Spector AA. Binding of cytochrome P450 monooxygenase and lipoxygenase pathway products by heart fatty acid-binding protein. Biochemistry. 2001;40:1070–1076. doi: 10.1021/bi001602y. [DOI] [PubMed] [Google Scholar]
- Wolbach SB, Howe PR. Nutrition Classics. The Journal of Experimental Medicine 42: 753–77, 1925. Tissue changes following deprivation of fat-soluble A vitamin. S. Burt Wolbach and Percy R. Howe. Nutr Rev. 1978;36:16–19. doi: 10.1111/j.1753-4887.1978.tb03675.x. [DOI] [PubMed] [Google Scholar]
- Wuarin L, Sidell N, de Vellis J. Retinoids increase perinatal spinal cord neuronal survival and astroglial differentiation. Int J Dev Neurosci. 1990;8:317–326. doi: 10.1016/0736-5748(90)90038-4. [DOI] [PubMed] [Google Scholar]
- Xu HE, Lambert MH, Montana VG, Parks DJ, Blanchard SG, Brown PJ, Sternbach DD, Lehmann JM, Wisely GB, Willson TM, et al. Molecular recognition of fatty acids by peroxisome proliferator- activated receptors. Mol Cell. 1999;3:397–403. doi: 10.1016/s1097-2765(00)80467-0. [DOI] [PubMed] [Google Scholar]
- Zouboulis CC. Retinoids--which dermatological indications will benefit in the near future? Skin Pharmacol Appl Skin Physiol. 2001;14:303–315. doi: 10.1159/000056361. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.