

Abstract
The multiple genetic hit model of cancer predicts that normal individuals should have stable populations of cancer-prone, but noncancerous, mutant cells awaiting further genetic hits. We report that whole-mount preparations of human skin contain clonal patches of p53-mutated keratinocytes, arising from the dermal–epidermal junction and from hair follicles. These clones, 60–3000 cells in size, are present at frequencies exceeding 40 cells per cm2 and together involve as much as 4% of the epidermis. In sun-exposed skin, clones are both more frequent and larger than in sun-shielded skin. We conclude that, in addition to being a tumorigenic mutagen, sunlight acts as a tumor promoter by favoring the clonal expansion of p53-mutated cells. These combined actions of sunlight result in normal individuals carrying a substantial burden of keratinocytes predisposed to cancer.
Keywords: sunlight, ultraviolet, carcinogenesis, tumor promotion, clonal expansion
Although skin cancers typically arise in patients aged 50–70, epidemiologic evidence indicates that much of the critical sunlight exposure is received before the age of 18 (1, 2). This early role of sunlight is supported by the finding of sunlight-induced mutations in the p53 tumor suppressor gene in actinic keratosis, the precancerous lesion for squamous cell carcinoma of the skin. In addition, p53-mutated cells are present in skin flanking human tumors and in UV-irradiated mouse skin (3, 4, 5, 6, 7, 8). Mutations at particular p53 codons are present in sun-exposed normal human skin at frequencies of 10−6 to 10−2 (5, 9). Other human tumors for which mutation of p53 appears to be an early event include head and neck cancer and hepatocellular carcinoma (10, 11).
Because keratinocytes are continuously lost through squamous differentiation and desquamation, it seems likely that the cell targeted by sunlight decades before a tumor’s appearance is a stem cell. If so, the keratinocytes containing the mutations measured in normal skin would not be randomly dispersed but instead would reside in clonal patches arising from mutated stem cells. The frequency of p53 mutations measured in a biopsy of normal skin would then depend on whether the biopsy included a clone. The spatial arrangement of the cells in the clone might give clues to the geometry of early carcinogenesis, including the site of the stem cells from which skin tumors originate in humans.
We therefore devised a whole-mount preparation method for human epidermis that permitted immunohistochemical analysis for stabilized p53 protein. A p53 mutation usually leads to nuclear immunopositivity (12). A large enough sample of skin might contain rare patches staining intensely for p53. We report here that such patches are not only present and contain mutations but are also frequent, indicating the existence of a large population of cells in normal skin that are presumably predisposed to skin cancer. In addition, we present evidence that sunlight can act both as a tumor initiator and as a tumor promoter for p53-mutated cells.
MATERIALS AND METHODS
Epidermal Whole Mounts.
Fresh skin samples were obtained from discarded cosmetic surgery tissue or from volunteers. No individuals had skin cancer. Protocols were approved by an institutional human subjects committee; informed consent was obtained from volunteers after explaining the nature and possible consequences of the studies. Whole mounts were prepared by a modification of previous procedures (13, 14). Tissue was scraped of excess fat, immersed 30–60 sec in 55°C phosphate-buffered saline (PBS)/20 mM EDTA, and immediately transferred to 4°C PBS for at least 2 min. The sample was then placed epidermis side up on a 4°C Petri dish, and an incision through the epidermis was made in one corner. Using curved no. 7 forceps, the epidermis was grasped from the point of the incision and gently separated from the dermis. Separated epidermis was fixed overnight in 10% neutral buffered formalin and rinsed in PBS.
Immunohistochemistry.
Endogenous peroxidase activity was quenched with 0.15% H2O2/PBS, followed by PBS rinse. Epitope was unmasked with 0.05% saponin (Sigma) in H2O, followed by PBS. Nonspecific antibody binding was blocked with normal goat serum (Vector) in 3% BSA (Sigma)/PBS. Excess blocking solution was blotted, and samples were incubated 1 hr at room temperature with rabbit polyclonal antibody CM-1 (NovoCastra, Newcastle, U.K.) at 1:5000 dilution, followed by PBS wash. Samples were incubated 45 min with 5 μl of biotinylated goat anti-rabbit antiserum (Vector) in 1000 μl of PBS/3% bovine serum albumin and washed in PBS. Diaminobenzamide staining used ABC and diaminobenzidine (DAB) reagents (Vector) for 30 min. Samples were dehydrated through ethanol, xylene-cleared, and mounted with Cytoseal nonaqueous mounting medium (Stephens Scientific, Riverdale, NJ). Fluorescent immunostaining of tissue was performed as above, except ABC and DAB reactions were replaced by incubation with Cy-3 avidin (1:500 dilution in PBS/3% BSA; generous gift of D. Ward, Yale University), washed in PBS, and mounted with Vectashield aqueous mounting medium (Vector). Samples were stored at 4°C in the dark until imaged.
Light and Confocal Microscopy.
The area of DAB-visualized patches was measured with a glass reticule calibrated in microns at 100× magnification and was rounded to the nearest 0.005 mm2. Only patches having clearly recognizable borders and a condensed morphology were scored, with condensed morphology defined as patches in which immunopositive cells were separated by no more than one nonstaining cell. To minimize seasonal variation, samples measured were those collected from late spring to early fall. Confocal images of fluorescence-stained samples were captured using a Bio-Rad MRC 600 laser scanning confocal microscope with 20× objective. Images were reassembled using voxelview imaging software (Vital Images, Fairfield, IA) running on a Silicon Graphics IRIS 4D/240 GTX workstation.
DNA Amplification and Sequencing.
DAB-stained patches were microdissected with 30-gauge steel needles. Total genomic DNA was amplified by a variation of the method of ref. 15 to a copy number sufficient for multiple p53-specific PCRs. Semirandom nonamers of sequence 5′-NNNNNN(G/C)(G/C)(G/C)-3′ (Tag-It kit; Bios, New Haven, CT) were used as primers with a reaction mix of 2.5 μl of 10× Tag-It buffer [500 mM KCl/100 mM Tris·HCl, pH 8.4 at 25°C/15 mM MgCl2/0.1% (wt/vol) gelatin/1% Triton X-100], 5 μl of H2O, 4 μl of 1.25 mM dNTPs, 2.5 μl of Tag-It primers, 1 μl of Taq (Roche), and 10 μl of template DNA (2 ng), overlaid with 50 μl of mineral oil. Cycling was performed in a Perkin–Elmer/Cetus thermocycler as follows: one step-file cycle (60°C for 3 min/94°C for 5 min); 30 thermo-file cycles (2.75 min ramp to 94°C/94°C for 10 sec/2 min ramp to 24°C/24°C for 10 sec). Reamplification for exons of the p53 gene was performed with primers, amplification conditions, and controls for contamination and polymorphisms as previously described, as were direct DNA sequencing of uncloned PCR products and sequence confirmation (5, 16, 17).
Statistical Analysis.
Statistical analyses were performed with patients as the units of analysis. Comparisons of patch frequency and patch size between two levels of sun exposure were performed using the Wilcoxon rank sum test; tests of dose–response were performed using the Jonckheere–Terpstra test (18). All P values are two-sided.
RESULTS
p53-Mutated Clones.
Clinically normal skin from cosmetic surgery patients and from volunteers contained many p53-immunopositive patches visible by light microscopy. These were present in both interfollicular epidermis (Fig. 1A) and at hair follicles (Fig. 1B). The frequency of patches ranged from an average of 3 patches per cm2 in sun-shielded skin to 33 patches per cm2 in chronically sun-exposed skin (Table 1 and Fig. 2). Intermittently exposed skin had an intermediate patch frequency. The dose–response of patch frequency was statistically significant (P < 0.0001), as was the difference between each pair of categories (P = 0.0001 to 0.01). Patch frequency was independent of age. Two patch morphologies were apparent, compact and diffuse, the latter having nonstaining cells interspersed with immunopositive cells. Samples taken in summer also contained a background level of widely dispersed immunopositive single cells. These cells resemble those observed by others in UV-irradiated human and murine skin, resulting from transient UV-induction of wild-type p53 protein (19, 20).
Figure 1.
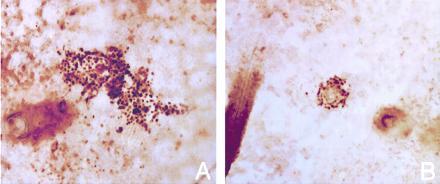
p53-immunopositive clones in whole-mount preparations of human epidermis. Clones arising from (A) the dermal–epidermal junction and (B) a hair follicle. In both cases, view is from the basal surface to show follicles. (×100.)
Table 1.
Patches of p53-mutated keratinocytes in human epidermis
| Sample | Age | Location | Patch frequency, cm−2 | Normal sequence | Base change | Codon | Amino acid change |
|---|---|---|---|---|---|---|---|
| Sun-shielded | |||||||
| YC19A | 24 | Inner arm | 1 | ||||
| YC17A | 28 | Abdomen | 4 | ||||
| YC7A | 29 | Breast | 1 | ||||
| YC8A | 32 | Breast | 4 | ||||
| YC4A | 40 | Lower abdomen | 3 | ||||
| YC13A | 41 | Breast | 7 | ||||
| YC18A | 44 | Inner arm | 2 | ||||
| YC14A | 47 | Lower abdomen | 0 | ||||
| YC6A | 50 | Breast | 8 | ||||
| Intermittent | |||||||
| YC19B.1 | 24 | Upper back | 11 | tCCg | CC → TT/wt | 248 | Arg → Gln |
| YC19B.2 | tCCg | CC → TT/wt | 248 | Arg → Gln | |||
| tcCca | C → T/wt | 279 | Gly → Glu | ||||
| YC19B.3 | wt | ||||||
| YC9A | 31 | Left thigh | 6 | ||||
| YC18B | 44 | Upper back | 7 | ||||
| YC5A | 47 | Post-auricular | 8 | ||||
| YC2A | 48 | Post-auricular | 9 | ||||
| YC2B.1 | 48 | Post-auricular | 38 | gtCcc | C → T/wt | 266 | Gly → Glu |
| YC10A | 70 | Right thigh | 6 | ||||
| Chronic exposure | |||||||
| YC12A | 9 | Lip | 46 | wt | |||
| YC24A | 43 | Pre-auricular | 23 | ||||
| YC5B.1 | 47 | Pre-auricular (left) | 29 | gcTcc | ΔT/wt | 362 | … Stop |
| YC5B.2 | wt | ||||||
| YC5C | 47 | Pre-auricular (right) | 45 | ||||
| YC2C.1 | 48 | Lower eyelid | 32 | wt | |||
| YC2D.1 | 48 | Pre-auricular | 23 | wt | |||
| YC2D.2 | wt | ||||||
| YC2D.3 | wt | ||||||
| YC2E | 48 | Pre-auricular | 33 | ||||
| YC23A | 51 | Upper eyelid (right) | 24 | ||||
| YC23B | 51 | Upper eyelid (left) | 25 | ||||
| YC15A | 53 | Lower eyelid | 44 | ||||
| YC15B.1 | 53 | Lower eyelid | 41 | taCac | C → T/wt | 81 | Thr → Ile |
| YC15B.2 | aCg | C → T/wt | 273 | Arg → His | |||
| YC1A | 65 | Lower eyelid (right) | 36 | ||||
| YC1B.1 | 65 | Lower eyelid (left) | 37 | tCCg | CC → TT/wt | 248 | Arg → Gln |
| YC1B.2 | wt | ||||||
| YC1B.3 | wt | ||||||
| YC1B.4 | wt | ||||||
| YC22A | 66 | Upper eyelid (right) | 33 | ||||
| YC22B | 66 | Upper eyelid (left) | 25 | ||||
| YC11A | 74 | Pre-auricular | 39 | ||||
| YC3A | 79 | Upper eyelid | 27 | wt |
Each YC number denotes a different individual, letters denote different tissues, and decimal numbers denote sequenced patches. Both compact and diffuse patches were counted. Nucleotide sequences are for the strand containing the mutating pyrimidine. wt denotes the presence of a wild-type allele, alone or with a mutant allele.
Figure 2.
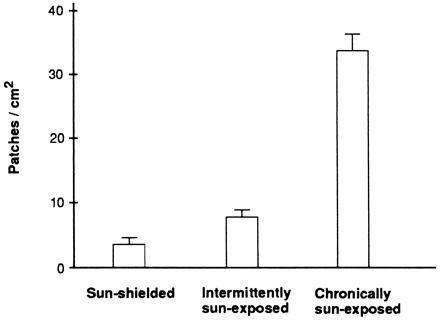
Increase of clone frequency with sun exposure. Histogram shows the average frequency of p53-mutated patches from sun-shielded, intermittently exposed, and chronically sun-exposed skin. Mean patch frequency for intermittently exposed skin excludes the outlier. Bars represent SEM.
To determine whether the patches were in fact composed of p53-mutated cells, we microdissected 16 stained compact patches from sun-exposed skin, randomly amplified the entire genome to increase the available template, and then amplified specific exons of the p53 gene. Upon direct DNA sequencing of uncloned PCR products, 50% of the patches were found to contain a mutation in the p53 structural gene (Table 1). Repeating the random and specific amplifications gave the same mutation, as did sequencing the opposite DNA strand. Nonstaining skin flanking the patches was wild-type, indicating that each p53 mutation was limited to a patch. The intensities of the mutant and wild-type DNA sequencing bands were approximately equal, suggesting that the entire patch was occupied by a heterozygous mutation; that is, each patch was a clone. The 50% figure may reflect technical limitations associated with microdissection, such as obscuring of the signal by contaminating normal cells.
Mutagenesis by Sunlight.
Mutations found in clones were predominantly C → T substitutions at sites of adjacent pyrimidines; several were CC → TT (Table 1). This pattern indicates that the mutagen is ultraviolet radiation (16, 21). Several of the mutated sites were the same as those previously seen in actinic keratoses, squamous cell carcinomas, or basal cell carcinomas (5, 16, 17, 22). All mutations seen in the clones changed the amino acid and so had been selected for. Consequently, it is likely that the mutations observed were required for the presence of the clone. For a stem cell, even a silent mutation—such as a C → T change at the third position of a codon—would be inherited by daughter cells even without selection pressure. However, no silent mutations were seen.
Three-Dimensional Geometry.
In three dimensions, the >1200 p53-mutated clones examined showed a gradation of morphologies. Patches encircling hair follicles often continued downward to the base of the follicle. Interfollicular clones typically became smaller in diameter as the focal plane approached the dermal–epidermal junction. About two-thirds of the smaller clones appeared conical, with the apex at the dermal–epidermal junction. Large clones were more cylindrical, with circular bases smaller than the surface patch, suggesting significant lateral expansion at all levels. Many of the clones were oriented at an angle from the vertical. To more clearly visualize the three-dimensional structure of the small, putatively early-stage p53-mutated clones, several whole-mount preparations incubated with primary and secondary antibody were visualized with fluorescent avidin. Confocal microscopy confirmed a conical structure originating at the dermal–epidermal junction and having a broad apex (Fig. 3). The dermal–epidermal junction and hair follicles are the locations of the presumed stem cells in skin (23) and appear to be the source of tumors in experimental animals (24).
Figure 3.
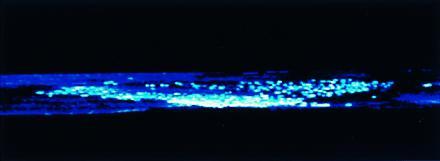
Conical clone of p53-mutated keratinocytes. Shown is the side view of a three-dimensionally reconstructed immunofluorescent cone from an epidermal whole mount. Opacity of the surrounding background epidermis has been adjusted to allow complete false-color visualization of all immunostaining cells in the z-scan. The cone’s apex lies at the bottom of the view, along the plane of the basal surface. Confocal images were captured using a Bio-Rad MRC 600 laser scanning confocal microscope with ×20 objective.
To estimate the number of cells in these clones, we viewed them from the basal surface and counted ≈6400 mutated cells per mm2. Assuming that three of the approximately six cell layers can be counted unobstructed, the clones ranged in size from ≈60 to 3000 cells. Although the geometry of epidermal stem cell compartments differs between body sites (23), a sense of scale facilitates comparison. In murine dorsal skin, an epidermal proliferating unit consists of a column of ≈15 cells: a basal layer of a single stem cell surrounded by ≈10 transit-amplifying cells, plus overlying keratinocytes undergoing squamous differentiation (25). Since the p53-mutated clones we observe are much larger, they appear to have escaped the boundaries of their stem cell compartment. In human epidermis, putative stem cells plus transit-amplifying cells are identified by elevated β1 integrin expression; however, these constitute ≈40% of the basal layer so the arrangement of epidermal proliferating units in human skin is not clear (26).
Tumor Promotion by Sunlight.
Tumor promoters increase tumor incidence only when they follow a tumor initiation step. In addition to being a mutagen, UVB radiation is known to be a tumor promoter in mouse skin (27, 28). If sunlight, which contains UVB and UVA radiation, acts as a tumor promoter in humans, and if promotion involves cells already containing p53 mutations, then the p53-mutant clones should be largest in sun-exposed skin. We therefore used a reticule to quantitate patch area, which is indicative of the volume of the entire clone but is independent of variations in skin thickness between body sites. Ten patches were quantitated from each of a number of sun-exposed skin samples and however many could be found from samples in sun-shielded areas.
Patch size often varied 20-fold in the same individual and was independent of age. The area of each measured patch is plotted in the histogram of Fig. 4. Each exposure category—sun-shielded, intermittently exposed, and chronically exposed—contained a distribution of patch sizes. The average patch size was larger in chronically sun-exposed skin than in sun-shielded skin (P = 0.02). More biologically relevant than the average behavior may be the fact that, though small patches were present in all categories, the upper range extended to larger sizes with increasing exposure. For example, the frequency of patches exceeding 0.05 mm2 was 5-fold greater in chronically exposed skin (25%) than in sun shielded skin (5%) (P = 0.04 by Fisher’s exact test). Therefore sunlight appears to promote the clonal expansion of p53-mutant cells.
Figure 4.
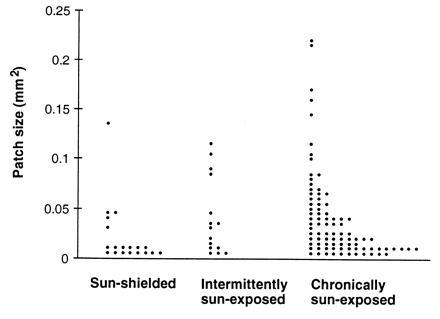
Increase of clone size with sun exposure. Dot histogram shows the area in mm2 of individual p53-mutated patches from sun-shielded, intermittently exposed, and chronically sun-exposed skin. Each point represents an individual clone.
The p53 Mutation Frequency in Normal Skin.
The total area of p53-immunopositive clones in a skin sample constitutes a measure of p53 mutation frequency. Any base substitutions leading to overexpression of p53 protein would be represented. This frequency was calculated by averaging the clone areas measured in each sample and then multiplying by the number of clones per cm2 in the same sample. The p53 mutation frequency ranged from 10−4 to 10−3 in sun-shielded skin and from 10−3 to 4 × 10−2 in sun-exposed skin. Thus as much as 4% of sun-exposed normal epidermis contains p53-mutated keratinocytes. It should be kept in mind that even sun-shielded skin receives sunlight exposure.
DISCUSSION
Tumorigenic Mutations.
The multiple genetic hit model of carcinogenesis (29, 30) requires that ordinary individuals have many cancer-prone mutant cells awaiting further genetic hits. The very high frequency of mutant keratinocyte clones we observe in skin from individuals with typical levels of sun exposure indicates that this situation is not a theoretical possibility but a reality. These cells were often arranged spatially as conical clones arising from putative stem-cell compartments, supporting the idea that stem cells are the cell of origin. A stable population of p53-mutant stem cells offers an explanation of the ability of childhood sun exposure to influence adult cancer frequency (1, 2). The age independence of clone frequency is consistent with substantial mutagenesis during childhood as well as with the fact that most human skin precancers (31) and UV-induced clusters of p53-overexpressing cells in mouse skin (7) regress in the absence of continued exposure.
Tumor Promotion.
The mechanism by which sunlight mutates the p53 gene of a single cell is understood in some detail (22). In contrast, the means by which this single mutated cell expands beyond its stem cell compartment has not been demonstrated. The clone size data found here confirms the existence of a selection pressure from sunlight that favors p53-mutant cells, even when the clones have no cancer- or precancer-like phenotype. Small clone sizes can be attained even with the minimal exposure experienced by sun-shielded skin, but chronic sun exposure appears to be necessary for clone size to exceed ≈0.05 mm2. Since average clone size is age-independent, the picture of normal skin that emerges is one of p53-mutant clones in various stages of waxing and waning as sun exposure is offset by regression. Sunlight’s promotion activity actually appears to contribute a greater number of mutant cells to the skin than does its initial mutagenic effect because, in chronically exposed skin, the aggregate size of clones >0.05 mm2 (sizes that are clearly exposure-dependent) exceeds that of the more numerous smaller clones.
Clonal expansion of a p53-mutated cell would be favored if a p53 mutation conferred resistance to: (i) squamous differentiation, including apoptosis and shedding (32); (ii) transforming growth factor β, a growth inhibitor for keratinocytes (33); (iii) apoptosis resulting from loss of cell–matrix interactions (34); or (iv) apoptosis resulting from UV radiation (5). We feel that the latter model is most likely to be correct because UV-induced apoptosis has a clear relation to sunlight and is p53-dependent (5), though one p53 point mutation retains apoptosis competence (35). In contrast, the other effects are modest or p53-independent (5, 36).
Apoptosis-resistance of p53-mutated cells would mean that sunlight can act as a tumor promoter by killing normal cells and sparing the mutants (5). Normal keratinocytes that happen to be in the S phase of the cell cycle when UVB-irradiated subsequently develop pycnotic nuclei and eosinophilic cytoplasm. Double-labeling experiments (not shown) demonstrate that these cells have the DNA double-strand breaks typical of cells dying by apoptosis. This tissue-protective mechanism has been termed “cellular proofreading” (22). However, about half of damaged p53+/− cells and 90% of damaged p53−/− cells are resistant to UV-induced apoptotic death. After surviving irradiation, these mutant cells could then clonally expand into vacated compartments. These events are a type of tumor promotion because apoptotic selection can act only after the initial p53 mutation. The observation of larger clones of p53-mutated cells in sun-exposed skin confirms a key prediction of this model in humans. Models for tumor promotion developed in other systems are viable, but do not require that cells defective in a p53 pathway be clonally expanded in preference to their neighbors. Tumor promotion may be a result, rather than a single mechanism.
Acknowledgments
We thank Dr. D. Ward for the gift of Cy-3 avidin and Drs. S. Rockwell and H. Sharma for scientific advice. This work was supported by American Cancer Society Grant CN-102 and National Institutes of Health Grant CA55737 to D.E.B. and a Munson Foundation gift to D.J.L.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
References
- 1.Marks R, Jolley D, Lectsas S, Foley P. Med J Aust. 1990;152:62–66. doi: 10.5694/j.1326-5377.1990.tb124456.x. [DOI] [PubMed] [Google Scholar]
- 2.Kricker A, Armstrong B K, English D R, Heenan P J. Int J Cancer. 1991;48:650–662. doi: 10.1002/ijc.2910480504. [DOI] [PubMed] [Google Scholar]
- 3.Nelson M A, Einspahr J G, Alberts D S, Balfour C A, Wymer J A, Welch K L, Salasche S J, Bangert J L, Grogan T M, Bozzo P O. Cancer Lett (Shannon, Irel) 1994;85:23–29. doi: 10.1016/0304-3835(94)90234-8. [DOI] [PubMed] [Google Scholar]
- 4.Taguchi M, Watanabe S, Yashima K, Murakami Y, Sekiya T, Ikeda S. J Invest Dermatol. 1994;103:500–503. doi: 10.1111/1523-1747.ep12395643. [DOI] [PubMed] [Google Scholar]
- 5.Ziegler A, Jonason A S, Leffell D J, Simon J A, Sharma H W, Kimmelman J, Remington L, Jacks T, Brash D E. Nature (London) 1994;372:773–776. doi: 10.1038/372773a0. [DOI] [PubMed] [Google Scholar]
- 6.Urano Y, Asano T, Yoshimoto K, Iwahana H, Kubo Y, Kato S, Sasaki S, Takeuchi N, Uchida N, Nakanishi H, Arase S, Itakura M. J Invest Dermatol. 1995;104:928–932. doi: 10.1111/1523-1747.ep12606204. [DOI] [PubMed] [Google Scholar]
- 7.Berg R J W, van Kranen H J, Rebel H G, de Vries A, van Vloten W A, van Kreijl C F, van der Leun J C, de Gruijl F R. Proc Natl Acad Sci USA. 1996;93:274–278. doi: 10.1073/pnas.93.1.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ren Z P, Pontén F, Nister M, Pontén J. Int J Cancer. 1996;69:174–179. doi: 10.1002/(SICI)1097-0215(19960621)69:3<174::AID-IJC4>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 9.Nakazawa H, English D, Randell P L, Nakazawa K, Martel N, Armstrong B K, Yamasaki H. Proc Natl Acad Sci USA. 1994;91:360–364. doi: 10.1073/pnas.91.1.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aguilar F, Harris C C, Sun T, Hollstein M, Cerutti P. Science. 1994;264:1317–1319. doi: 10.1126/science.8191284. [DOI] [PubMed] [Google Scholar]
- 11.Ahomadegbe J C, Barrois M, Fogel S, Le Bihan M L, Douc-Rasy S, Duvillard P, Armand J P, Riou G. Oncogene. 1995;10:1217–1227. [PubMed] [Google Scholar]
- 12.Greenblatt M S, Bennett W P, Hollstein M, Harris C C. Cancer Res. 1994;54:4855–4878. [PubMed] [Google Scholar]
- 13.Marrs J M, Voorhees J J. J Invest Dermatol. 1971;56:174–181. doi: 10.1111/1523-1747.ep12260779. [DOI] [PubMed] [Google Scholar]
- 14.Narisawa Y, Hashimoto K, Kohda H. Br J Dermatol. 1993;129:541–546. doi: 10.1111/j.1365-2133.1993.tb00481.x. [DOI] [PubMed] [Google Scholar]
- 15.Zhang L, Cui X, Schmitt K, Hubert R, Navidi W, Arnheim N. Proc Natl Acad Sci USA. 1992;89:5847–5851. doi: 10.1073/pnas.89.13.5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brash D E, Rudolph J A, Simon J A, Lin A, McKenna G J, Baden H P, Halperin A J, Pontén J. Proc Natl Acad Sci USA. 1991;88:10124–10128. doi: 10.1073/pnas.88.22.10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ziegler A, Leffell D J, Kunala S, Sharma H W, Gailani M, Simon J A, Halperin A J, Baden H P, Shapiro P E, Bale A E, Brash D E. Proc Natl Acad Sci USA. 1993;90:4216–4220. doi: 10.1073/pnas.90.9.4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lehmann E L. Nonparametrics: Statistical Methods Based on Ranks. San Francisco: Holden–Day; 1977. [Google Scholar]
- 19.Hall P A, McKee P H, Menage H, Dover R, Lane D P. Oncogene. 1993;8:203–207. [PubMed] [Google Scholar]
- 20.Campbell C, Quinn A G, Angus B, Farr P M, Rees J L. Cancer Res. 1993;53:2697–2699. [PubMed] [Google Scholar]
- 21.Hutchinson F. Mutat Res. 1994;309:11–15. doi: 10.1016/0027-5107(94)90037-x. [DOI] [PubMed] [Google Scholar]
- 22.Brash D E, Ziegler A, Jonason A, Simon J A, Kunala S, Leffell D J. J Invest Dermatol Symp Proc. 1996;1:136–142. [PubMed] [Google Scholar]
- 23.Miller S J, Lavker R M, Sun T T. Semin Dev Biol. 1993;4:217–240. [Google Scholar]
- 24.Miller S J, Sun T T, Lavker R M. J Invest Dermatol. 1993;100:288S–294S. doi: 10.1111/1523-1747.ep12470169. [DOI] [PubMed] [Google Scholar]
- 25.Potten C S, Morris R J. In: Stem Cells. Lord B I, Dexter T M, editors. Vol. 10. Cambridge, U.K.: Company of Biologists; 1988. pp. 45–62. [Google Scholar]
- 26.Jones P H, Harper S, Watt F M. Cell. 1995;80:83–93. doi: 10.1016/0092-8674(95)90453-0. [DOI] [PubMed] [Google Scholar]
- 27.Epstein J H, Epstein W L. J Invest Dermatol. 1962;39:455–460. [Google Scholar]
- 28.Blum H F. In: The Biologic Effects of Ultraviolet Radiation. Urbach F, editor. Oxford: Pergamon; 1969. pp. 543–549. [Google Scholar]
- 29.Nowell P C. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 30.Fearon E R, Vogelstein B. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 31.Marks R, Foley P, Goodman G, Hage B H, Selwood T S. Br J Dermatol. 1986;115:649–655. doi: 10.1111/j.1365-2133.1986.tb06644.x. [DOI] [PubMed] [Google Scholar]
- 32.McCall C A, Cohen J J. J Invest Dermatol. 1991;97:111–114. doi: 10.1111/1523-1747.ep12478519. [DOI] [PubMed] [Google Scholar]
- 33.Reiss M, Vellucci V F, Zhou Z-L. Cancer Res. 1993;53:899–904. [PubMed] [Google Scholar]
- 34.Frisch S M, Francis H. J Cell Biol. 1994;124:619–626. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li G, Mitchell D L, Ho V C, Reed J C, Tron V A. Am J Pathol. 1996;148:1113–1123. [PMC free article] [PubMed] [Google Scholar]
- 36.Missero C, Calautti E, Eckner R, Chin J, Tsai L H, Livingston D M, Dotto G P. Proc Natl Acad Sci USA. 1995;92:5451–5455. doi: 10.1073/pnas.92.12.5451. [DOI] [PMC free article] [PubMed] [Google Scholar]