

Abstract
The yeast Mre11-Rad50-Xrs2 (MRX) and Ku complexes regulate single-strand resection at DSB double-strand breaks (DSB), a key early step in homologous recombination (HR). A prior plasmid gap repair study showed that mre11 mutations, which slow single-strand resection, reduce gene conversion tract lengths and the frequency of associated crossovers. Here we tested whether mre11Δ or nuclease-defective mre11 mutations reduced gene conversion tract lengths during HR between homologous chromosomes in diploid yeast. We found that mre11 mutations reduced the efficiency of HR but did not reduce tract lengths or crossovers, despite substantially reduced end-resection at the test (ura3) locus. End-resection is increased in yku70Δ, but this change also had no effect on tract lengths. Thus, heteroduplex formation and tract lengths are not regulated by the extent of end-resection during DSB repair in a chromosomal context. In a plasmid-chromosome DSB repair assay, tract lengths were again similar in wild-type and mre11Δ, but they were reduced in mre11Δ in a gap repair assay. These results indicate that tract lengths are not affected by the extent of end processing when broken ends can invade nearby sites, perhaps because MRX coordination of the two broken ends is dispensable when ends invade nearby sites. Although HR outcome was largely unaffected in mre11 mutants, break-induced replication (BIR) and chromosome loss increased, suggesting that Mre11 function in mitotic HR is limited to early HR stages. Interestingly, yku70Δ suppressed BIR in mre11 mutants. BIR is also elevated in rad51 mutants, but yku70Δ did not suppress BIR in a rad51 background. These results indicate that Mre11 functions in Rad51-independent BIR, and that Ku functions in Rad51-dependent BIR.
1. Introduction
Homologous recombination (HR) and non-homologous end joining (NHEJ) are the two major pathways by which DSB double-strand breaks (DSB) are repaired. DSB can be lethal if unrepaired, and misrepair can lead to genome rearrangements including deletions, inversions, amplifications, and translocations. Both DSB repair pathways operate in eukaryotic cells and play key roles in maintaining genome stability, although their relative efficiencies vary among organisms, by ploidy, and during the cell cycle [1,2]. In the yeast Saccharomyces cerevisiae, most DSB are repaired by HR, particularly in diploids and in S/G2-phase cells where homologous repair templates are readily available [3]. In yeast, HR is mediated by Rad51, the Rad51 paralogs Rad55 and Rad57, Rad54, and RPA. NHEJ involves yKu70, yKu80, Lig4, Lif1, and Lif2. Through Rad50 hook domains, the Mre11-Rad50-Xrs2 (MRX) complex promotes NHEJ by tethering DSB ends [4]. Mre11 has 3′-5′ exonuclease and endonuclease activities that modulate NHEJ outcome [5,6]. Mre11 also functions in HR, at least in part by modulating DSB end processing, as well as in meiotic DSB induction, DSB damage checkpoint signaling, and telomere stability [7,8]. All of these DSB repair proteins are highly conserved from yeast to man. HR is often accurate and conservative. NHEJ can be conservative when precise re-ligation is possible, i.e., when broken ends have complementary overhangs like those produced by nucleases. NHEJ can be mutagenic if imprecise and this outcome is rare with nuclease-induced DSBs in yeast. Imprecise NHEJ may be the only NHEJ outcome when ends are not complementary or require significant processing, such as those produced by ionizing radiation. In yeast, at least 25% of nuclease-induced DSB are repaired by precise NHEJ, but only ~0.1% are repaired by imprecise NHEJ [9,10].
In the early stages of HR, DSB are resected to long 3′ single-stranded tails (ssDNA) by a poorly understood process promoted by MRX and inhibited by the yKu70/yKu80 heterodimer. ssDNA is first bound by RPA, which is replaced by Rad51, producing a nucleoprotein filament that can search for and invade a homologous duplex DNA elsewhere in the genome. Strand invasion and branch migration produces heteroduplex DNA (hDNA), and mismatches in hDNA are repaired by the mismatch repair machinery. This transfers information from an unbroken donor locus to the broken, recipient locus, usually in continuous blocks termed gene conversion tracts that can extend from just a few bp to more than 12 kbp [11,12]. Thus, gene conversion tracts reflect both the extent of hDNA and mismatch repair activity, and represent localized regions of loss of heterozygosity.
Gene conversion is the most common DSB repair outcome in yeast. A fraction of conversions are associated with crossovers, and 50% of crossovers in G2 cells produce large-scale loss of heterozygosity, extending from the crossover point to the telomere [13]. Gene conversion is thought to occur by two related pathways. Synthesis-dependent strand annealing is a non-crossover pathway in which one end invades a donor, primes repair synthesis past the DSB site, then detaches and anneals to the (resected) non-invading strand. Crossovers may arise through two-ended invasions that produce double-Holliday junctions. In either case, failure to engage the second end can result in break induced replication (BIR) in which an invading strand is extended to the end of the chromosome >100 kbp distant. BIR was first identified in rad51Δ mutant cells and is rare or absent in wild-type cells [14–16]. Collapsed replication forks yield a single broken end, and BIR may be an important mechanism for restarting these forks [17]. However, BIR is not an optimum DSB repair pathway because it results in large-scale loss of heterozygosity or non-reciprocal translocations. BIR is thought to occur by an efficient Rad51-dependent strand invasion mechanism that also requires Rad52, Rad55, Rad57, and Rad54, and by an inefficient Rad51-independent mechanism that requires Rad59 (a Rad52 homolog), Tid1 (a Rad54 homolog), and Rad50; the requirement for Rad50 suggests requirements for the other MRX components Mre11 and Xrs2 [15,17,18]. On chromosome III, Rad51-independent BIR is also promoted by a 200 bp “FBI” sequence element located 34 kbp from MAT [19], but its mechanism of action is not clear, nor is it known whether FBI sequences exist on other chromosomes. Gene conversion can occur within 2 hours of DSB induction, but Rad51-dependent BIR requires 4–6 hours and is associated with G2 arrest [18].
Null mutations in any MRX subunit confers hypersensitivity to IR and methyl methanesulfonate (MMS) [20,21]. In mating-type switching, an intrachromosomal gene conversion event initiated by DSB induced in the MAT locus by HO nuclease, MRX defects reduce end-resection and delay, but do not eliminate DSB repair by HR [20,22]. Mre11 has 3′-5′ exonuclease activity on ssDNA and double-stranded DSB (dsDNA), and endonuclease activity on ssDNA with a preference for ssDNA/dsDNA junctions [20,23–27]. Mre11 has 4 conserved nuclease motifs and two classes of nuclease-defective mutations have been described that either prevent or allow MRX complex formation. Mre11 defects that prevent complex formation are similar or identical to mre11Δ, whereas those that form MRX complexes have a milder phenotype [26–28]. Together, the results suggest that Mre11 has nuclease-dependent roles, particularly in meiosis, whereas its ability to complex with Rad50 and Xrs2 is critical for several of its roles in mitotic DSB repair. In these latter roles MRX may be maintaining connections between broken ends (“end coordination”) to facilitate NHEJ and HR [4,29,30].
Symington et al. [31] used a plasmid gap repair assay to show that gene conversion tracts are shorter in mre11Δ, consistent with the idea that reduced end-resection in mre11Δ limits hDNA formation. However, although end-resection is enhanced in yku70Δ mutants [32], conversion tract lengths were similar to wild-type during DSB-induced interchromosomal (allelic) HR [10]. These conflicting results could be explained by differences in ploidy, length of shared homology (i.e., ectopic vs. allelic HR), or interacting partners (plasmid-chromosome vs. interchromosomal). Here we show that end-processing is reduced in mre11Δ, and increased in yku70Δ, yet these changes do not alter allelic tract lengths, suggesting that end-processing does not regulate gene conversion tract lengths during interchromosomal events. We further show that mre11 mutations do not reduce tract lengths when HR is stimulated by DSBs in chromosomal direct repeats, or during plasmid-chromosome HR. However, tract lengths were reduced in mre11Δ when HR was stimulated by a double-strand gap, suggesting that Mre11 regulation of tract lengths depends on whether broken ends invade sites located close or far from each other. DSB-induced allelic HR was reduced in mre11 mutants, and both BIR and chromosome loss were increased, indicating a role for Mre11 in HR initiation. Interestingly, a yku70Δ mutation suppressed BIR, but not chromosome loss, in mre11Δ and nuclease-defective mre11-H125N mutants. Suppression of BIR by yku70Δ is specific to the mre11 mutant background because yku70Δ did not suppress BIR in a rad51 mutant. These results indicate that Mre11 functions in Rad51-dependent BIR, and that yKu70 functions in Rad51-independent BIR.
2. Materials and Methods
2.1. Plasmid DNA and yeast strains
Plasmids were manipulated and prepared as described [33,34]. Yeast culture and chromosome modification were described earlier [16,35,36]. Structures of all modified chromosomes were confirmed by Southern and PCR analyses. Plasmid pFH800 carries a GAL1 promoter-driven HO nuclease gene (GALHO) on a TRP1/ARS1/CEN4 (TAC) plasmid [37]. A pUC19 derivative carrying ura3 inactivated by an EcoRI linker at NcoI, and HIS3/ARS1/CEN4 (pUraRHAC-R), is identical to pUHAC described previously [38] except that ura3 included 9 silent RFLP mutations. MRE11 was deleted in appropriate strains by two-step transformation using pSK-Scmre11Δ [21]. The mre11Δ mutation was complemented with wild-type MRE11 using pSK-ScMRE11, with derivatives carrying nuclease-defective alleles mre11-2, mre11-3, mre11-4, and mre11-11 [21], and with a derivative of plasmid pScM11-314 [39] carrying an mre11-H125N mutation created with primer 5′-ATATCAGGTAATAATGATGATGCGTCGGGG [40]. Yeast strain genotypes are shown in Table 1. yku70Δ strains described previously [41] were mated with mre11Δ mutants, and diploids were sporulated to create mre11Δ yku70Δ double mutants. RAD51 in pUC19 was mutated to rad51-K191R (hereafter rad51KR) using primer 5′-GAATTCAGGACCGGCCGTTCCCAGCTATGTC, URA3 was inserted, and the plasmid was cleaved with BstEII and transformed into appropriate strains. The resulting Ura+ transformants were isolated, grown nonselectively, and Ura− derivatives were selected on medium with 5-fluoro-orotic acid [42]. rad51KR was identified by mapping PCR products with EagI, and confirmed by DNA sequencing and by Southern hybridization analysis. rad51KR yku70Δ double mutants were created by mating/sporulation as above.
Table 1.
Yeast strains.
| Name | Genotype | Source |
|---|---|---|
| JW3082 | MATa-inc ade2-101 his3-200 lys2-801::pHSSGALHO::LYS2 trp1-Δ1 leu2-Δ1 ura3-X764- LEU2-ura3R-HO432 | [36] |
| BW3566 | JW3082 mre11Δ | This study |
| JC3441 | MATa-inc ade2-101 his3-200:HIS3:telV lys2-801::pHSSGALHO::LYS2 trp1-Δ1 leu2-Δ1 RscRI-ura3R-HO432-LEU2 | [16] |
| DY3427 | MATα ade2-101 his3-200 lys2-801 trp1-Δ1 leu2-Δ1 RscBam-ura3-X764-LEU2 | [16] |
| JC3517-13 | Diploid product of DY3427 × JC3441 | [16] |
| BW3575 | JC3441 mre11Δ | This study |
| BW3575 | DY3427 mre11Δ | This study |
| BW3592 | JC3517-13 mre11Δ | This study |
| JC3548 | JC3441 yku70Δ | [41] |
| JC3549 | DY3427 yku70Δ | [41] |
| JC3550 | JC3517-13 yku70Δ | [41] |
| BW3591 | JC3441 mre11Δ yku70Δ | This study |
| SK3799 | DY3427 mre11Δ yku70Δ | This study |
| SK3800 | JC3517-13 mre11Δ yku70Δ | This study |
| JC3654 | JC3441 rad51KR | This study |
| JC3655 | DY3427 rad51KR | This study |
| JC3656 | JC3517-13 rad51KR | This study |
| SK3801 | JC3441 rad51KR yku70Δ | This study |
| SK3802 | DY3427 rad51KR yku70Δ | This study |
| SK3803 | JC3517-13 rad51KR yku70Δ | This study |
2.2. Chromosomal DSB repair assays
Allelic HR was analyzed as described [16] with the following modifications. Two-day old colonies from synthetic complete medium lacking tryptophan (-Trp plates) were transferred to 1.5 ml of YPGly (1.0% yeast extract, 2.0% peptone, 2.0% glycerol) and incubated for 24 h, split into two tubes per culture and the medium was replaced with 1.5 ml of YPD (2.0% dextrose; represses GALHO) or YPGal (2% galactose; GALHO-induced), incubated for 6 h before seeding to medium lacking tryptophan. The broken chromosome has a telomere-proximal HIS3 (HIS3:telV) used to monitor crossovers, chromosome loss, and BIR. Ura+ His+, Ura+ His−, and Ura− His− products were identified with appropriate selective medium. Ura− His+ recombinants were distinguished from parental colonies by re-induction of GALHO on YPGal and transfer to uracil omission medium as Ura+ papillae arise only in parental colonies [36,43]. A variety of sectored colonies arise (e.g., Ura+/− His+ and Ura+ His+/−), most likely resulting from independent G2 events, and each sector was scored as a single colony. Some colonies have parental sectors and were scored as one parental and one recombinant colony. HR frequencies were calculated as the number of recombinants per YPD colony scored. For each determination, 3–4 independent populations were tested and 1000–1500 colonies were scored per population. The fraction of His− products arising by G2 crossover is equivalent to the frequency of His+ recombinants with two copies of HIS3:telV (termed His++) identified by PCR among 60–100 His+ products per strain. Total G2 crossover frequencies were calculated as double the fraction of His− gene conversions among all HR products (doubling accounts for the fact that only half of crossovers result in HIS3:telV loss). Crossovers were also estimated from just Ura+ products (i.e., the ratio of Ura+ His− : Ura+ expressed as a percentage); this approach avoids uncertainties associated with loss of HIS3:telV by BIR or chromosome loss. Chromosome loss was determined directly as described [44], and BIR comprises non-loss, non-crossover His− products [16]. Gene conversion tract lengths were estimated as the fraction of Ura− recombinants among all (Ura+ + Ura−) recombinants (excluding BIR and chromosome loss products). In rare cases we identified Ura− His+ products that had lost the donor chromosome and retained a parental recipient chromosome, presumably as a result of spontaneous loss of the unbroken (donor) chromosome.
DSB-dependent cell killing was assayed with continuous GALHO induction by seeding equivalent numbers of cells to tryptophan omission medium with 2% glucose or 2% galactose and incubating for 3 days. DSB-dependent cell killing was calculated by dividing the number of galactose-grown colonies by the number of glucose-grown colonies and expressing the ratio as a percentage [41]. All statistical analyses were performed by using t tests unless noted otherwise.
2.3. Plasmid-chromosome HR assays
HR was analyzed between EcoRI digested pUraRHAC-R transformed by using lithium acetate [45] into wild-type and mre11Δ strains (DY3427 and BW3575, respectively), each with ura3-X764 on chromosome V. Pools of >100 His+ transformants were collected and genomic DNA was isolated and cleaved with EcoRI prior to transformation into E. coli DH10B cells by electroporation [46]. Cleavage by EcoRI linearizes plasmids that circularized in yeast by precise NHEJ, and because linear DNA is ineffective at transforming E. coli, this eliminates NHEJ products from the analysis. For each transformation, 10% the E. coli transformants were seeded to LB-agar with ampicillin (to estimate the number of transformants), and the remainder were grown overnight as pools in 5 ml of liquid LB medium with ampicillin from which plasmid DNA was isolated and analyzed by agarose gel electrophoresis. Each pool comprised >100 transformants. The relative fractions of products with long and short gene conversion tracts (co-conversion of X764 or not, respectively) were determined by densitometric analysis of specific gel bands using a NucleoTech gel analysis system. Gap repair was analyzed by cleaving pUHAC-R with PpuMI and ApaI prior to selection of His+ transformants as above. Tract lengths were estimated by determining fractions of Ura+ and Ura− colonies, and fractions of parental molecules were determined by EcoRI digestion of plasmid DNA pools from pools of His+ transformants as above.
2.4. Analysis of single-strand DNA resection
Cells were cultured in YPGly as above, and harvested at various times after transfer to YPGal. Genomic DNA was prepared and DNA concentrations were measured by quantitative real-time PCR of NDC1 as described [44]. Equal quantities of non-denatured DNA were spotted on a nylon membrane using a BioRad dot blot apparatus locus and hybridized with a denatured 32P-labeled 660 bp URA3 fragment amplified with primers 5′-CGCATATGTGGT GTTGAAGAA and 5′-TCTTTGTCGCTCTTCGCAAT.
3. Results
3.1. Mre11 defects reduce DSB-induced allelic HR efficiency, but do not alter HR outcome
Mating-type switching is delayed but not eliminated in mre11Δ cells [20]. Although mating-type switching is a well-studied form of DSB-induced HR, it measures only one outcome, namely gene conversion without an associated crossover. To determine whether Mre11 influences DSB-induced HR outcomes in a chromosomal context, we introduced mre11Δ mutations into diploid cells carrying one ura3 allele inactivated by an HO site insertion at position 432, and a second allele inactivated by a +1 frameshift mutation 332 bp downstream (X764) (Fig. 1A). DSBs were created at the HO site following induction of GALHO, yielding Ura+ products when gene conversion tracts do not reach X764, and Ura− products when tracts extend beyond X764. Thus, changes in tract length alter the fractions of Ura+ and Ura− products. Additional phenotypically silent markers within and flanking ura3 have been used to monitor tract lengths [16], but these are analyzed only if the Ura+ : Ura− ratio is altered. Crossovers, BIR, and chromosome loss are monitored with a telomere-proximal HIS3 gene. This non-selective assay provides simultaneous measures of HR efficiency, and HR outcomes including gene conversion tract lengths and crossover frequencies, BIR, and chromosome loss. We also determined the effect of mre11Δ on chromosomal direct repeat HR using these same ura3 alleles flanking LEU2 (Fig. 1B), which provides measures of long and short-tract gene conversions, and deletions that result from crossovers or single-strand annealing.
Fig. 1.
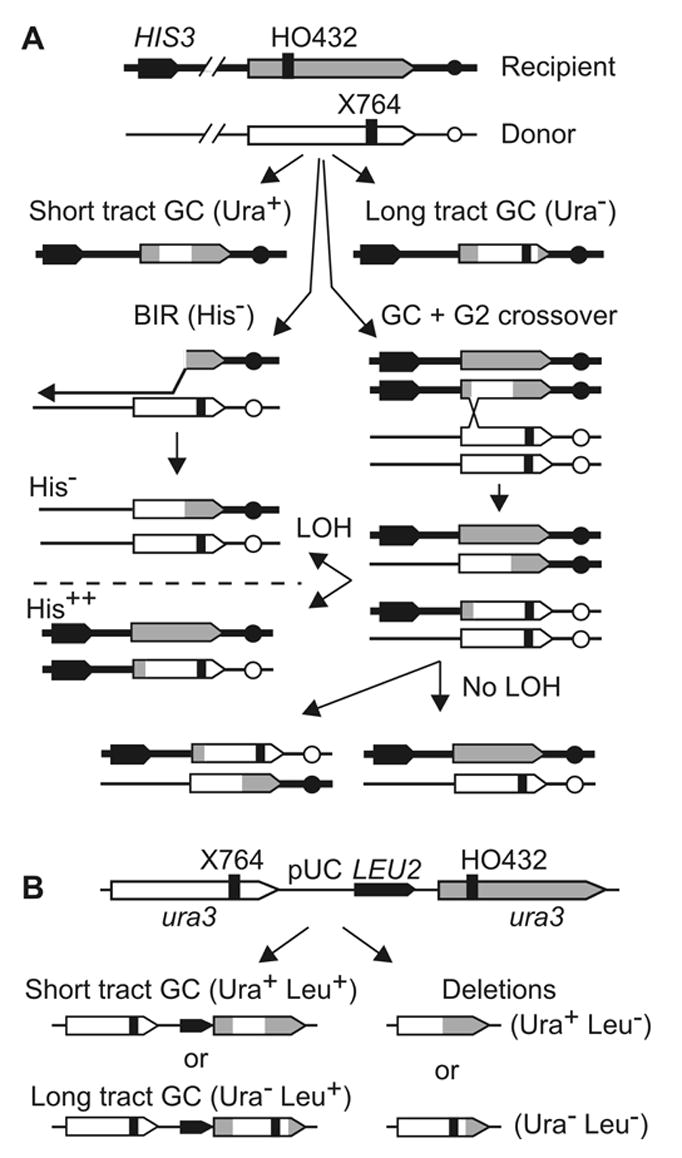
Allelic and direct repeat HR substrates and outcomes. (A) Allelic HR substrate has ura3 alleles inactivated by frameshift mutations comprising an HO site (HO432) and X764. The chromosome carrying the HO site is marked with HIS3:telV. HR produces short- and long-tract gene conversions (GC); only the converted (recipient) chromosome is shown. Some conversions have associated crossovers and in G2 cells, half of crossovers result in LOH at HIS3:telV (His− or His++ products) and half remain heterozygous. Some G2 cells experience only a single DSB (as shown) due to limited access of HO to its site in ura3. BIR produces only His− products (shown above the dotted line). His− products also arise by chromosome loss (not shown) that are also Ura−. (B) Direct repeat substrate has the same ura3 alleles flanking pUC19 and LEU2. The four principal HR product types (with associated phenotype) are shown below.
We found that DSB-induced allelic HR product distributions were similar in wild-type, mre11Δ, and mre11-H125N (Fig. 2A), but total HR frequencies were reduced by ~1.5-fold in mre11Δ and mre11-H125N (Fig. 2B). These modest defects in DSB-induced allelic HR are consistent with the mild effects of MRX defects on mating-type switching [15,20,22], but contrast with the strong defects of mre11Δ, mre11-2, mre11-4 mutants in the repair of IR-induced DSBs [21], and the strong defect of mre11Δ in IR-induced HR [39]. The modest reduction in allelic HR in mre11Δ contrasts with the nearly complete elimination of HR in rad51 and rad52 mutants [47] (unpublished results).
Fig. 2.
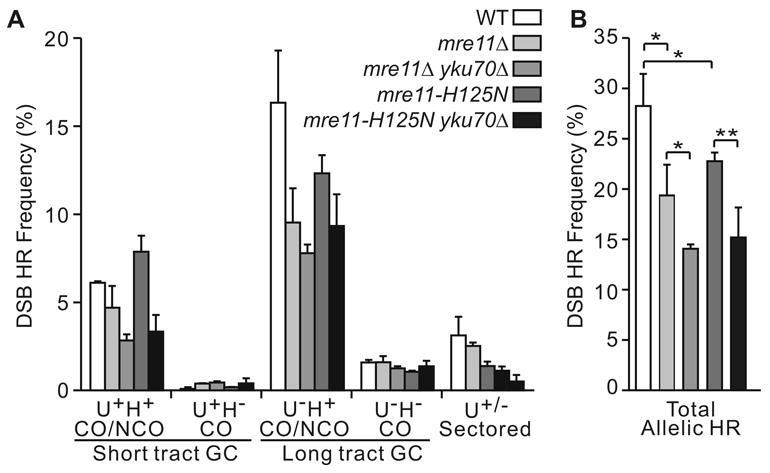
DSB-induced allelic HR product spectra. (A) Gene conversion (GC) frequencies are shown for wild-type (WT), mre11Δ, and mre11-H125N with or without yku70Δ. Values for each product class are averages ±SD for 4 determinations. His− products are crossovers (CO) but His+ may be crossover or non-crossover (NCO) products. These values do not include BIR or chromosome loss events. (B) Total allelic HR frequencies from panel A. * indicates P < 0.05; ** indicates P < 0.01.
End resection is reduced in mre11Δ cells [32]. Symington et al. [31] examined HR between a transformed, gapped plasmid and a chromosomal donor locus and observed shorter gene conversion tracts in mre11Δ. These results led to the proposal that shorter ssDNA tails limit hDNA formation that is reflected in shorter tract lengths. In that study, mre11Δ also displayed reduced crossovers, consistent with the observation that crossovers are more frequent among products with long gene conversion tracts [48,49]. The allelic and direct repeat HR systems employed in this study gave a different result; allelic conversion tract lengths and crossovers were not reduced in mre11Δ or mre11-H125N (Fig. 3A, B). In fact, total crossovers, and crossovers among Ura+ products, increased in mre11Δ. In agreement with prior measurements of end-processing at MAT, ssDNA resection at ura3 was significantly reduced in mre11Δ, and increased in yku70Δ (Fig. 3C), excluding the possibility of differential regulation of resection by Mre11 at MAT and ura3, or in the different genetic backgrounds. Allelic gene conversion tract lengths were also unaffected in yku70Δ and mre11Δ yku70Δ mutants (Fig. 3A) [10].
Fig. 3.
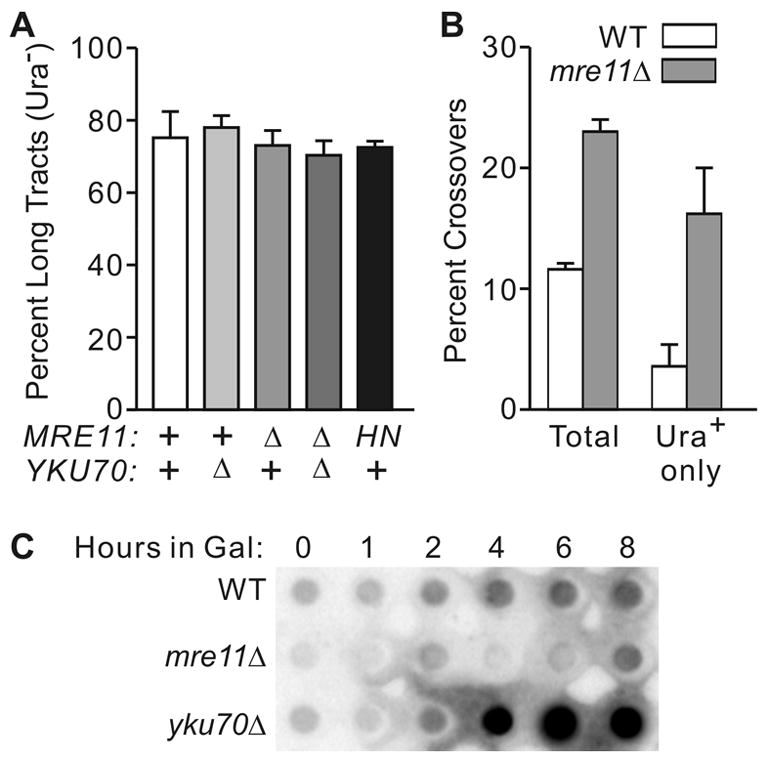
Allelic gene conversion tract lengths do not correlate with extent of end-processing. (A) Tract lengths were estimated as the percentage of long-tract products (Ura−) in wild-type (+), deletion mutants of mre11 or yku70 (Δ), or mre11-H125N (HN). Values are averages ±SD for 3–8 determinations. (B) Percent crossovers estimated as His− fractions among all products, or just Ura+ classes. (C) End-processing measured by dot-blot of native genomic DNA from wild-type, mre11Δ, and yku70Δ diploid cells isolated before and at indicated times after GALHO induction.
3.2. Mre11 defects do not alter DSB-induced direct repeat HR efficiency or outcome
IR-induced direct repeat HR is strongly reduced in mre11Δ [39]. In contrast, there was no difference in HO-induced direct repeat HR product spectra or total HR frequencies between wild-type and mre11Δ, or nuclease-defective mre11-2, mre11-3, mre11-4, and mre11-11 mutants (Fig. 4A, B). Direct repeat conversion tract lengths were also unaffected in mre11 mutants (Fig. 4C). These results indicate that gene conversion tract lengths (and by inference, hDNA formation) are not affected by the extent of single-strand resection in a chromosomal context.
Fig. 4.
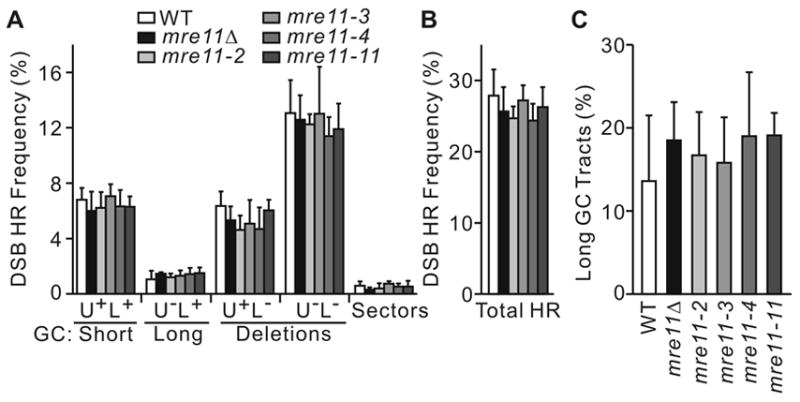
Mre11 has little or no role in DSB-induced direct repeat HR. (A) Direct repeat HR product spectra in mre11Δ cells with wild-type complementing vector (mre11Δ/MRE11), empty vector control (mre11Δ), or vectors expressing nuclease-defective mre11 alleles. Values are averages ±SD for 4 determinations. There were no significant differences in frequencies of individual HR product classes, or total HR.
3.3. mre11Δ reduces gene conversion tract lengths during double-strand gap repair but not DSB repair
To further investigate why Mre11 influences tract lengths in a transformed/gapped plasmid-chromosome assay [31] but not chromosomal HR, we tested whether tract lengths were reduced in mre11Δ in two plasmid-chromosome HR assays. In the first assay, HR was initiated by a DSB in the plasmid-borne copy of ura3 prior to transformation into haploid wild-type or mre11Δ cells. As in the chromosomal assays above, the plasmid-chromosome assay was non-selective for long or short tract gene conversions; selection was for plasmid transformation using a HIS3 gene, and against NHEJ products (see Fig. 5A and Materials and Methods). As shown in Fig. 5B and C, mre11Δ did not reduce the fraction of long tract products, but instead there was a slight increase. Thus, reduced end-processing in mre11Δ does not decrease tract lengths during chromosomal or plasmid-chromosome gene conversion induced by a DSB. Because the plasmid included a CEN element, crossovers produce lethal dicentric chromosomes and are not detected. However, this limitation does not account for the lack of an effect of mre11Δ on tract lengths (see Discussion). In the second assay, we introduced a 222 bp double-strand gap into the plasmid-borne ura3 prior to transformation (Fig. 5D), similar to the 238 bp gap tested by Symington et al. [31]. As above, selection was for His+ transformants; we did not select against NHEJ because the two NHEJ events required to recreate parental molecules were expected to be rare. Parental molecules were recovered and may have arisen from incompletely digested plasmid DNA prior to transformation. However this did not confound the interpretation because the fractions of parental molecules were similar in wild-type and mre11Δ (~10%; data not shown). The remaining 90% were recombinants distributed among short- and long-tract gene conversions. We scored 199 and 202 colonies from wild-type and mre11Δ, respectively, and consistent with Symington et al. [31], long-tracts were significantly reduced in mre11Δ (Fig. 5E; P = 0.005, Fisher exact test). Together, the results indicate that Mre11 influences tract lengths during gap repair, but not DSB repair.
Fig. 5.
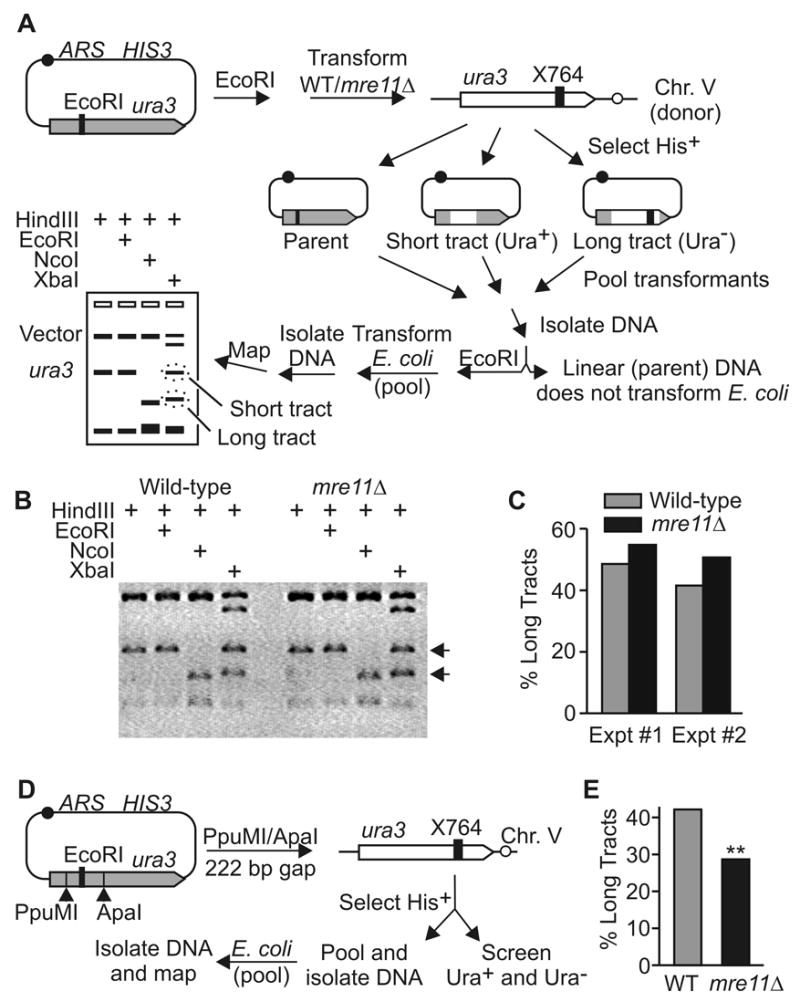
Plasmid-chromosome HR. (A) HR between chromosome V and a linear, transformed plasmid carrying ura3 inactivated by an EcoRI linker in NcoI (position 432). Selected His+ transformants were pooled, and isolated genomic DNA was digested with EcoRI. This linearizes parental molecules, and only the circular gene conversion products efficiently transform E. coli. Ampicillin-resistant E. coli transformants were pooled and mapped to determine the ratio of long and short tract HR products. The first three lanes are controls: HindIII excises a 1.2 kbp ura3 fragment from the vector; smaller fragments result from HindIII sites in HIS3. The HindIII/EcoRI digest confirms that no parental molecules are present. The HindIII/NcoI digest confirms that all molecules arose by gene conversion (ura3 cleaved by NcoI). The HindIII/XbaI digest reveals the relative fractions of molecules with long or short tracts (ura3 cleaved by XbaI or not cleaved, respectively). (B) mre11Δ does not reduce plasmid-chromosome tract lengths. The fourth lane of each set (HindIII/XbaI) reveals similar fractions of long and short tract HR products. (C) Scanning densitometric analysis of agarose gels from two independent experiments (Expt #1 data are from gel shown in panel B). (D) Gap repair assay; symbols are described in panel A. (E) Percentage of long tracts among gap repair products, calculated as the ratio of Ura− recombinants per total recombinants (Ura+ + Ura−).
3.4. Mre11 defects increase BIR, chromosome loss, and DSB-dependent cell killing
HR is the major DSB repair pathway in yeast, particularly in diploid cells, and is critical for cell survival of DSB damage [3]. In mre11Δ, allelic HR is reduced (Fig. 2B) and mating-type switching is delayed [20,50], suggesting that the limited end-processing in mre11Δ inhibits an initial stage of HR. In agreement with this view, BIR and chromosome loss were increased in diploid mre11Δ and mre11-H125N cells (Fig. 6A, B). BIR and chromosome loss were higher in mre11Δ than mre11-H125N, consistent with prior studies showing that this nuclease mutant, which is capable of forming of an MRX complex, has a weaker phenotype than mre11Δ [26–28]. It is likely that any mutation that prevents MRX complex formation, such as rad50Δ, would also show high levels of BIR and chromosome loss. We observed about 25% more DSB-dependent cell killing in mre11Δ than wild-type, but this was fully suppressed by the nuclease-defective mre11-H125N allele, indicating that this function requires the MRX complex but not the Mre11 nuclease (Fig. 6C). The enhanced BIR, chromosome loss, and cell killing in mre11 mutants, together with the lack of change in HR outcome, indicates that Mre11 promotes DSB-induced HR at an early stage (see Discussion).
Fig. 6.
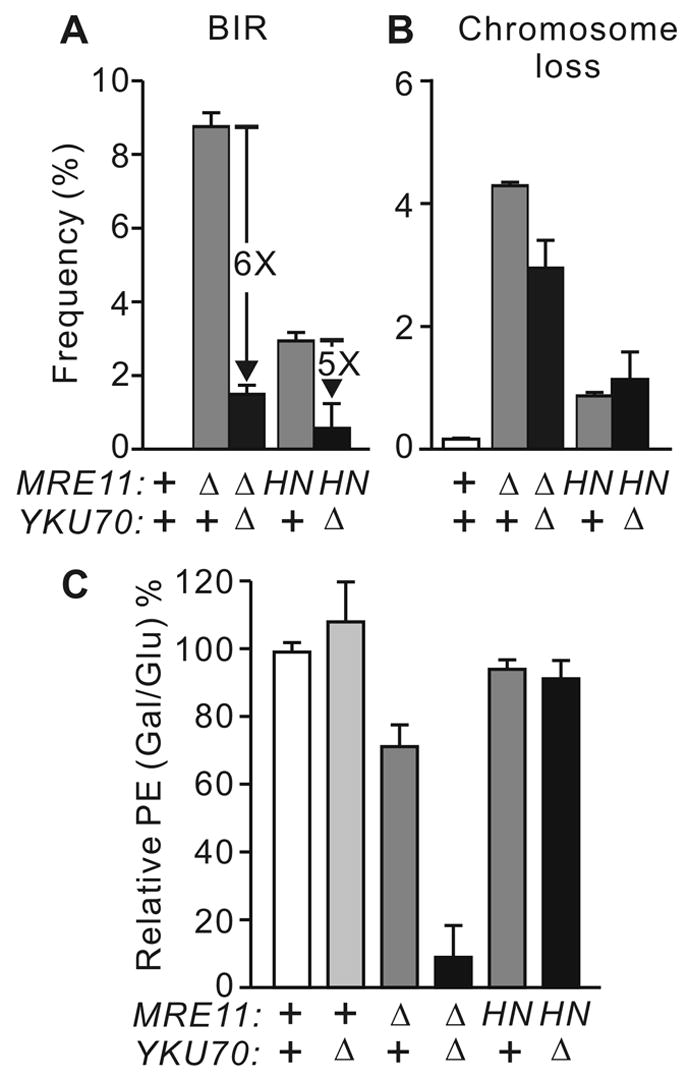
mre11 mutations increase BIR, chromosome loss, and DSB-induced cell killing. All values are averages ±SD for 3–4 determinations. (A) BIR in mre11 mutants is suppressed 5- to 6-fold by yku70Δ. (B) yku70Δ does not suppress chromosome loss in mre11 mutants. (C) DSB-dependent cell killing is enhanced in mre11Δ and further enhanced in this background by yku70Δ.
3.5. yku70Δ suppresses BIR, but not chromosome loss in mre11 mutants
Ku and Mre11 have opposing roles in end-processing, and yku70Δ partially suppresses the radiosensitivity of mre11Δ cells [39]. The latter result suggests that Ku may interfere with the repair of IR-induced DSBs when Mre11 is absent. In addition, yku70Δ mutation in otherwise wild-type cells increases the frequency of DSB-induced HR, most likely because those DSBs normally repaired by precise NHEJ are shunted to HR [10]. We therefore tested whether Ku interferes with DSB repair by HR in mre11 mutant backgrounds by assaying HR in diploid mre11Δ yku70Δ and mre11-H125N yku70Δ cells. Surprisingly, DSB-induced HR was reduced to a small, but significant degree by yku70Δ mutation in both mre11 backgrounds (Fig. 2B), and yku70Δ enhanced DSB-dependent cell killing in mre11Δ, but not in mre11-H125N (Fig. 6C). Thus, in mre11 mutants, yku70Δ enhances cell survival after IR exposure, but reduces HR and cell survival after nuclease-induced DSBs. The reduced repair/survival in yku70Δ depends on the formation of MRX but not Mre11 nuclease activity. Gene conversion tract lengths and crossover frequencies were unaffected by yku70Δ alone, or in combination with mre11Δ or mre11-H125N [10] (Fig. 3A and data not shown).
Interestingly, BIR in the mre11Δ background was strongly suppressed by yku70Δ, and a similar suppression was observed in mre11-H125N (Fig. 6A), but yku70Δ had little or no effect on chromosome loss in either mre11 mutant (Fig. 6B). To determine whether yku70Δ is a general BIR suppressor, we examined the effect of yku70Δ in a rad51KR (ATPase-defective) background (Fig. 7). rad51KR is similar to mre11Δ in showing a modest reduction in DSB-induced HR, and increased levels of BIR and chromosome loss. However, unlike mre11 mutants, yku70Δ did not suppress BIR in rad51KR. Thus, yku70Δ suppression of BIR is specific to the mre11 mutant background. Prior studies suggest that BIR in mre11 mutants is Rad51-dependent [15], and our results indicate that yKu70 also functions in the Rad51-dependent BIR pathway. Note that in wild-type cells, Ku suppresses HR, presumably by shunting DSBs to NHEJ [10]. However, in rad51KR mutant cells, yku70Δ reduces HR (Fig. 7), similar to its effect in mre11Δ (Fig. 2B). Thus, Ku appears to promote HR in cells with modest HR defects.
Fig. 7.
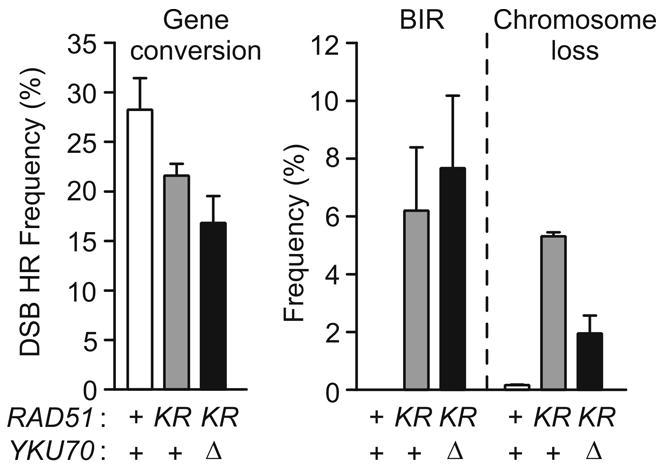
yku70Δ does not suppress BIR in rad51KR. Data are presented as shown in Figs. 2B, 6A, and 6B.
4. Discussion
4.1. End-processing regulates gene conversion tract lengths during double-strand gap repair but not DSB repair
The MRX complex is involved in several aspects of the cellular DNA damage response including DSB repair by NHEJ and HR, meiotic DSB formation, telomere maintenance, checkpoint activation, and checkpoint adaptation [8,32]. In this study we focused on Mre11 regulation of HR efficiency and outcome. mre11 mutations include those that eliminate MRX complex formation, like mre11Δ and mre11-D16A, and less severe mutations like mre11-H125N that permit complex formation but lack nuclease activity [27]. Although mre11 mutants show profound sensitivity to IR and severe reductions in IR-induced HR [21,39], HR-mediated MAT switching is delayed but not eliminated [20], and allelic and direct repeat HR stimulated by HO nuclease is mildly reduced or unaffected (Figs. 2 and 4). These results indicate that the strong defect in IR-induced HR [39] reflects a requirement for Mre11 to process “ragged” ends at DSBs produced by IR, rather than an HR defect per se. We found that both mre11Δ and mre11-H125N reduced HO-induced allelic HR, and increased BIR and chromosome loss, but the phenotype was stronger in mre11Δ. In addition, mre11 mutations did not affect HR outcome. These results indicate that MRX function in DSB repair by allelic HR is limited to early HR stages, and that this depends on MRX complex formation, and to a lesser extent, on Mre11 nuclease activity. The Mre11 nuclease is critical for meiotic HR, presumably reflecting its role in removing covalently bound Spo11 from broken ends [20,51].
Although 5′-3′ end-processing is reduced in mre11Δ [20,32,52] (Fig. 3C), the Mre11 exonuclease is unlikely to be involved in end-processing because it has the opposite (3′-5′) polarity. Instead, the Mre11 ssDNA endonuclease might act on DNA unwound by MRX or a helicase, or MRX might promote other exonuclease activities, such as Exo1 [50]. Such regulatory roles for MRX are consistent with Mre11 being among the first proteins recruited to DSBs [53–55]. We recently showed that Rad51 recruitment to DSBs is delayed for at least two hours after DSB induction in mre11Δ [56]. Here we shown that this results in only a modest (1.5-fold) reduction in HR when assayed over a period of days (Fig. 2), suggesting that in the absence of MRX, sufficient Rad51 is eventually recruited to broken ends to catalyze HR. The modest reduction in HR and increases in BIR and chromosome loss may reflect HR failure at early times after DSB induction. Defects in other proteins that act early in HR, such as Rad51, also reduce HR and increase BIR and chromosome loss [14,15] (see below).
Crossovers are more frequently associated with long gene conversion tracts [31,48,49]. Thus, tract length regulation is important because it affects both the extent of localized loss of heterozygosity, and the frequency of large-scale loss of heterozygosity associated with crossovers. Yeast proteins known to regulate tract lengths and/or crossovers during DSB repair include Sgs1, Top3, Srs2, and Rad9 [49,57] (unpublished results). Gene conversion tract lengths reflect both the extent of hDNA formation, and mismatch repair within hDNA. hDNA can form directly during strand invasion and by branch migration of joint molecules. mre11Δ displayed shorter gene conversion tracts and reduced crossovers in a plasmid-chromosome gap repair assay, suggesting that hDNA formation is proportional to the extent of end resection [31]. However, tracts were not shorter in mre11Δ when gene conversion was stimulated by DSBs in allelic, direct repeat, or plasmid-chromosome systems, nor did mre11Δ decrease allelic crossovers (Figs. 3 and 5). Our plasmid-chromosome HR assay does not detect crossovers, but this limitation cannot explain why tract lengths were not reduced in mre11Δ because crossovers occur less often among products with short tracts [31,48,49]. DSB-induced gene conversion tract lengths are increased in sgs1Δ [49] and it was recently shown that Sgs1 and Mre11 are in a large complex in unstressed cells, and in a smaller complex when the DNA damage checkpoint is activated [58]. Thus, a tract length reduction due to decreased end processing in mre11 might be balanced by an increase if Sgs1 function is compromised in mre11. However, this model does not explain why mre11 reduces tract lengths during gap repair but not DSB repair [31] (Fig. 5).
Our data indicate that Mre11 does not regulate tract lengths when ends can invade nearby sequences in a donor duplex. In addition to regulating end-processing, MRX (through its Rad50 subunit) is thought to play a role in end-coordination during DSB repair. MRX may have a lesser role in coordinating ends when ends invade nearby sites. When ends are forced to invade distant sites (during gap repair), hDNA formation and tract length may be controlled to a greater degree by the extent of end-processing. The lack of correlation between tract lengths and end-processing for DSB-initiated events suggests that in this situation, hDNA forms primarily by branch migration after strand invasion occurs in a limited region, rather than by strand invasion over the full length of a single-stranded tail. In this view, hDNA formation by branch migration will be similar whether both a donor duplex and an invading duplex must be unwound, or if only the donor duplex must be unwound (because the invading strand has been resected to ssDNA), which is consistent with branch migration between two duplexes being an energy-neutral process that proceeds by random walk [59,60]. Although branch migration might be expected to be impeded in a chromatin environment, nucleosome eviction by the INO80 chromatin remodeling complex is sharply reduced in mre11Δ [56], yet hDNA formation in mre11Δ (reflected in tract lengths) appears unaffected. This suggests that nucleosomes do not inhibit branch migration.
Symington et al. [31] suggested that shorter gene conversion tracts in mre11Δ could explain its spontaneous hyperrecombination phenotype [61] as co-conversion of heteroallelic markers is less likely when tracts are short. There are three other ways to explain spontaneous hyperrecombination in mre11Δ that are independent of tract length changes. First, spontaneous DSBs may be shunted from NHEJ to HR, although this is inconsistent with the fact that Ku defects do not increase spontaneous HR [62], and the HR deficiency of mre11Δ (Fig. 2B). Second, the Rad50 subunit of MRX is a member of the SMC protein family that functions in a manner analogous to cohesin to promote repair between sister chromatids, rather than between homologs. Thus, spontaneous hyperrecombination in mre11Δ could reflect a shift in repair from sister chromatid to homolog interactions [8]. Finally, MRX is important for S-phase checkpoint arrest [63,64], and hyperrecombination is a common phenotype of checkpoint defective cells [65].
4.2. Mre11 and Ku function in distinct BIR pathways
Gene conversion requires strand invasion and coordination of the two broken ends. If strand invasion is inefficient, or if the two ends are not coordinated, BIR or chromosome loss can result. Through its Rad50 subunit, MRX might play an important role in coordinating ends both for NHEJ and gene conversion. However, this cannot explain the fact that both BIR and chromosome loss are elevated in mre11-H125N (Fig. 6A, B), yet the MRX complex forms normally in this mutant [27]. It is more likely that the reduced recruitment of Rad51 to DSBs in mre11Δ [56] reduces the efficiency of strand invasion, with the failure of one or both ends to invade the donor resulting in BIR or chromosome loss.
Two BIR pathways have been defined, including an efficient Rad51-dependent pathway that also requires Rad52, Rad55, Rad57, and Rad54, and an inefficient Rad51-independent pathway that requires Rad50, Rad59, and Tid1 [15,17]. [Note that gene conversion is much more efficient than either BIR pathway [18]]. The requirement for Rad50 in Rad51-independent BIR implicates Mre11 and Xrs2 in this pathway. Thus, the BIR observed in mre11 mutants is probably Rad51-dependent, and the suppression of BIR in mre11Δ by yku70Δ suggests that Ku has an important role in Rad51-dependent BIR. This conclusion gains support from the observation that yku70Δ does not affect BIR in rad51KR. Thus, Ku and Mre11 function in distinct BIR pathways; these proteins also have opposing roles in checkpoint adaptation to a persistent DSB [32].
In wild-type cells, DSB repair by HR is increased in yku70Δ because Ku promotes DSB repair by NHEJ [10]. However, NHEJ is defective in mre11Δ [8] and in mre11Δ and mre11-H125N, Ku promotes both HR and BIR (Fig. 2B and 6A). How might Ku promote these strand invasion-dependent processes? At MAT, BIR in rad51 mutants preferentially initiates in or near a “facilitator of BIR” (FBI) site and Ku regulation of end-processing might promote BIR in mre11 mutants by controlling access to a similar site near ura3. However, this is unlikely because BIR in mre11 mutants is Rad51-dependent which does not require the FBI site [18], and because mre11Δ and mre11Δ yku70Δ show similar rates of end-processing [32] but different levels of BIR. Ku can tether DNA ends [66] and potentially promote HR in mre11Δ by coordinating ends, but this does not explain its positive role in BIR because BIR does not require end-coordination, and because Ku lacks affinity for ssDNA and intact duplex DNA [67]. Although Ku does not bind ssDNA, it somehow regulates end-processing even after long ssDNA tails have been produced [32], perhaps because Ku migrates inward after initial binding to dsDNA ends [68]. Ku may therefore remain associated with ssDNA-dsDNA junctions, and this could affect Rad51 loading because the Escherichia coli Rad51 homolog, RecA, is known to load at ssDNA-dsDNA junctions [69]. Finally, the end-binding/protection properties of Rad52 are reminiscent of Ku [67,70], end-resection is slowed by yeast Rad52 [71], and human Rad52 binds to 3′ termini of resected DNA [70]. Ku might promote BIR and HR in mre11Δ by protecting a fraction of ends from degradation and “handing off” these ends to Rad52, thereby promoting BIR and HR. Such a role might be evident only when Ku is unable to fulfill its normal role in NHEJ, as in NHEJ-defective mre11 mutants. yku70Δ suppresses BIR but not chromosome loss in mre11Δ and in mre11-H125N, perhaps because both strand invasion (HR/BIR) and NHEJ are compromised in yku70Δ.
Acknowledgments
We thank Kim Paffett for technical assistance, and Mary Ann Osley and members of the Nickoloff laboratory for helpful comments. This research was supported by grant CA55302 from the National Cancer Institute to J.A.N., and by grants GM56888 and GM59413 from the National Institute of General Medical Sciences to J.H.J.P.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nickoloff JA, Haber JE. Mating-type control of DNA repair and recombination in Saccharomyces cerevisiae. In: Nickoloff JA, Hoekstra MF, editors. DNA Damage and Repair, Vol. 3: Advances from Phage to Humans. Humana Press; Totowa, NJ: 2001. pp. 107–124. [Google Scholar]
- 2.Critchlow SE, Jackson SP. DNA end-joining: from yeast to man. Trends Biochem Sci. 1998;23:394–398. doi: 10.1016/s0968-0004(98)01284-5. [DOI] [PubMed] [Google Scholar]
- 3.Paques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wiltzius JJ, Hohl M, Fleming JC, Petrini JH. The Rad50 hook domain is a critical determinant of Mre11 complex functions. Nat Struct Mol Biol. 2005;12:403–407. doi: 10.1038/nsmb928. [DOI] [PubMed] [Google Scholar]
- 5.Paull TT, Gellert M. A mechanistic basis for Mre11-directed DNA joining at microhomologies. Proc Natl Acad Sci USA. 2000;97:6409–6414. doi: 10.1073/pnas.110144297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Durant ST, Nickoloff JA. Good timing in the cell cycle for precise DNA repair by BRCA1. Cell Cycle. 2005;4:1216–1222. doi: 10.4161/cc.4.9.2027. [DOI] [PubMed] [Google Scholar]
- 7.Petrini JHJ, Maser RS, Bressan DA. The MRE11-RAD50 complex: diverse functions in the cellular DNA damage response. In: Nickoloff JA, Hoekstra MF, editors. DNA Damage and Repair, Vol. III: Advances from Phage to Humans. Humana Press; Totowa, NJ: 2001. pp. 147–172. [Google Scholar]
- 8.Haber JE. The many interfaces of Mre11. Cell. 1998;95:583–586. doi: 10.1016/s0092-8674(00)81626-8. [DOI] [PubMed] [Google Scholar]
- 9.Moore JK, Haber JE. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:2164–2173. doi: 10.1128/mcb.16.5.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clikeman JA, Khalsa GJ, Barton SL, Nickoloff JA. Homologous recombinational repair of double-strand breaks in yeast is enhanced by MAT heterozygosity through yKu-dependent and -independent mechanisms. Genetics. 2001;157:579–589. doi: 10.1093/genetics/157.2.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palmer S, Schildkraut E, Lazarin R, Nguyen J, Nickoloff JA. Gene conversion tracts in Saccharomyces cerevisiae can be extremely short and highly directional. Nucleic Acids Res. 2002;31:1164–1173. doi: 10.1093/nar/gkg219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Symington LS, Petes T. Expansions and contractions of the genetic map relative to the physical map of yeast chromosome III. Mol Cell Biol. 1988;8:595–604. doi: 10.1128/mcb.8.2.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nickoloff JA. Recombination: mechanisms and roles in tumorigenesis. In: Bertino JR, editor. Encyclopedia of Cancer. 2. Elsevier Science (USA); San Diego: 2002. pp. 49–59. [Google Scholar]
- 14.Malkova A, Ivanov EL, Haber JE. Double-strand break repair in the absence of RAD51 in yeast: a possible role for break-induced DNA replication. Proc Natl Acad Sci USA. 1996;93:7131–7136. doi: 10.1073/pnas.93.14.7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Signon L, Malkova A, Naylor ML, Klein H, Haber JE. Genetic requirements for RAD51- and RAD54-independent break-induced replication repair of a chromosomal double-strand break. Mol Cell Biol. 2001;21:2048–2056. doi: 10.1128/MCB.21.6.2048-2056.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nickoloff JA, Sweetser DB, Clikeman JA, Khalsa GJ, Wheeler SL. Multiple heterologies increase mitotic double-strand break-induced allelic gene conversion tract lengths in yeast. Genetics. 1999;153:665–679. doi: 10.1093/genetics/153.2.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davis AP, Symington LS. RAD51-dependent break-induced replication in yeast. Mol Cell Biol. 2004;24:2344–2351. doi: 10.1128/MCB.24.6.2344-2351.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malkova A, Naylor ML, Yamaguchi M, Ira G, Haber JE. RAD51-dependent break-induced replication differs in kinetics and checkpoint responses from RAD51-mediated gene conversion. Mol Cell Biol. 2005;25:933–944. doi: 10.1128/MCB.25.3.933-944.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malkova A, Signon L, Schaefer CB, Naylor ML, Theis JF, Newlon CS, Haber JE. RAD51-independent break-induced replication to repair a broken chromosome depends on a distant enhancer site. Genes Dev. 2001;15:1055–1060. doi: 10.1101/gad.875901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moreau S, Ferguson JR, Symington LS. The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Mol Cell Biol. 1999;19:556–566. doi: 10.1128/mcb.19.1.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bressan DA, Olivares HA, Nelms BE, Petrini JHJ. Alteration of N-terminal phosphoesterase signature motifs inactivates Saccharomyces cerevisiae Mre11. Genetics. 1998;150:591–600. doi: 10.1093/genetics/150.2.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ivanov EL, Sugawara N, White CI, Fabre F, Haber JE. Mutations in XRS2 and RAD50 delay but do not prevent mating-type switching in Saccharomyces cerevisiae. Mol Cell Biol. 1994;14:3414–3425. doi: 10.1128/mcb.14.5.3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Furuse M, Nagase Y, Tsubouchi H, Murakamimurofushi K, Shibata T, Ohta K. Distinct roles of two separable in vitro activities of yeast mre11 in mitotic and meiotic recombination. EMBO J. 1998;17:6412–6425. doi: 10.1093/emboj/17.21.6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paull TT, Gellert M. The 3′ to 5′-exonuclease activity of Mre11 facilitates repair of DNA double-strand breaks. Mol Cell. 1998;1:969–979. doi: 10.1016/s1097-2765(00)80097-0. [DOI] [PubMed] [Google Scholar]
- 25.Trujillo KM, Sung P. DNA structure-specific nuclease activities in the Saccharomyces cerevisiae Rad50-Mre11 complex. J Biol Chem. 2001;276:35458–35464. doi: 10.1074/jbc.M105482200. [DOI] [PubMed] [Google Scholar]
- 26.Usui T, Ohta T, Oshiumi H, Tomizawa J, Ogawa H, Ogawa T. Complex formation and functional versatility of Mre11 of budding yeast in recombination. Cell. 1998;95:705–716. doi: 10.1016/s0092-8674(00)81640-2. [DOI] [PubMed] [Google Scholar]
- 27.Krogh BO, Llorente B, Lam A, Symington LS. Mutations in Mre11 phosphoesterase motif I that impair Saccharomyces cerevisiae Mre11-Rad50-Xrs2 complex stability in addition to nuclease activity. Genetics. 2005;171:1561–1570. doi: 10.1534/genetics.105.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewis LK, Storici F, Van Komen S, Calero S, Sung P, Resnick MA. Role of the nuclease activity of Saccharomyces cerevisiae Mre11 in repair of DNA double-strand breaks in mitotic cells. Genetics. 2004;166:1701–1713. doi: 10.1534/genetics.166.4.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Jager M, van Noort J, van Gent DC, Dekker C, Kanaar R, Wyman C. Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol Cell. 2001;8:1129–1135. doi: 10.1016/s1097-2765(01)00381-1. [DOI] [PubMed] [Google Scholar]
- 30.Moreno-Herrero F, de Jager M, Dekker NH, Kanaar R, Wyman C, Dekker C. Mesoscale conformational changes in the DNA-repair complex Rad50/Mre11/Nbs1 upon binding DNA. Nature. 2005;437:440–443. doi: 10.1038/nature03927. [DOI] [PubMed] [Google Scholar]
- 31.Symington LS, Kang LE, Moreau S. Alteration of gene conversion tract length and associated crossing over during plasmid gap repair in nuclease-deficient strains of Saccharomyces cerevisiae. Nucleic Acids Res. 2000;28:4649–4656. doi: 10.1093/nar/28.23.4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee SE, Moore JK, Holmes A, Umezu K, Kolodner RD, Haber JE. Saccharomyces Ku70, Mre11/Rad50, and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell. 1998;94:399–409. doi: 10.1016/s0092-8674(00)81482-8. [DOI] [PubMed] [Google Scholar]
- 33.Taghian DG, Nickoloff JA. Subcloning strategies and protocols. In: Harwood A, editor. Basic DNA and RNA Protocols. Humana Press; Totowa, NJ: 1996. pp. 221–235. [DOI] [PubMed] [Google Scholar]
- 34.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 35.Sweetser DB, Hough H, Whelden JF, Arbuckle M, Nickoloff JA. Fine-resolution mapping of spontaneous and double-strand break-induced gene conversion tracts in Saccharomyces cerevisiae reveals reversible mitotic conversion polarity. Mol Cell Biol. 1994;14:3863–3875. doi: 10.1128/mcb.14.6.3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cho JW, Khalsa GJ, Nickoloff JA. Gene conversion tract directionality is influenced by the chromosome environment. Curr Genet. 1998;34:269–279. doi: 10.1007/s002940050396. [DOI] [PubMed] [Google Scholar]
- 37.Nickoloff JA, Chen EYC, Heffron F. A 24-base-pair sequence from the MAT locus stimulates intergenic recombination in yeast. Proc Natl Acad Sci USA. 1986;83:7831–7835. doi: 10.1073/pnas.83.20.7831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nickoloff JA, Singer JD, Heffron F. In vivo analysis of the Saccharomyces cerevisiae HO nuclease recognition site by site-directed mutagenesis. Mol Cell Biol. 1990;10:1174–1179. doi: 10.1128/mcb.10.3.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bressan DA, Baxter BK, Petrini JHJ. The Mre11-Rad50-Xrs2 protein complex facilitates homologous recombination-based double-strand break repair in Saccharomyces cerevisae. Mol Cell Biol. 1999;19:7681–7687. doi: 10.1128/mcb.19.11.7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deng WP, Nickoloff JA. Site-directed mutagenesis of virtually any plasmid by eliminating a unique site. Anal Biochem. 1992;200:81–88. doi: 10.1016/0003-2697(92)90280-k. [DOI] [PubMed] [Google Scholar]
- 41.Paffett KS, Clikeman JA, Palmer S, Nickoloff JA. Overexpression of Rad51 inhibits double-strand break-induced homologous recombination but does not affect gene conversion tract lengths. DNA Repair. 2005;4:687–698. doi: 10.1016/j.dnarep.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 42.Boeke JD, Lacroute F, Fink GR. A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol Gen Genet. 1984;197:345–346. doi: 10.1007/BF00330984. [DOI] [PubMed] [Google Scholar]
- 43.Weng Y-s, Whelden J, Gunn L, Nickoloff JA. Double-strand break-induced gene conversion: examination of tract polarity and products of multiple recombinational repair events. Curr Genet. 1996;29:335–343. doi: 10.1007/BF02208614. [DOI] [PubMed] [Google Scholar]
- 44.Lo YC, Kurtz RB, Nickoloff JA. Analysis of chromosome/allele loss in genetically unstable yeast by quantitative real-time PCR. Biotechniques. 2005;38:685–690. doi: 10.2144/05385BM01. [DOI] [PubMed] [Google Scholar]
- 45.Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gunn L, Nickoloff JA. Rapid transfer of low copy number episomal plasmids from Saccharomyces cerevisiae to Escherichia coli by electroporation. Mol Biotech. 1995;3:79–84. doi: 10.1007/BF02789103. [DOI] [PubMed] [Google Scholar]
- 47.Krogh BO, Symington LS. Recombination proteins in yeast. Annu Rev Genet. 2004;38:233–271. doi: 10.1146/annurev.genet.38.072902.091500. [DOI] [PubMed] [Google Scholar]
- 48.Jinks-Robertson S, Michelitch M, Ramcharan S. Substrate length requirements for efficient mitotic recombination in Saccharomyces cerevisiae. Mol Cell Biol. 1993;13:3937–3950. doi: 10.1128/mcb.13.7.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lo YC, Paffett KS, Amit O, Clikeman JA, Sterk R, Brenneman MA, Nickoloff JA. Sgs1 regulates gene conversion tract lengths and crossovers independently of its helicase activity. Mol Cell Biol. 2006;26:4086–4094. doi: 10.1128/MCB.00136-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moreau S, Morgan EA, Symington LS. Overlapping functions of the Saccharomyces cerevisiae Mre11; Exo1 and Rad27 nucleases in DNA metabolism. Genetics. 2001;159:1423–1433. doi: 10.1093/genetics/159.4.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Keeney S, Giroux CN, Kleckner N. Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell. 1997;88:375–384. doi: 10.1016/s0092-8674(00)81876-0. [DOI] [PubMed] [Google Scholar]
- 52.Tsubouchi H, Ogawa H. A novel mre11 mutation impairs processing of double-strand breaks of DNA during both mitosis and meiosis. Mol Cell Biol. 1998;18:260–268. doi: 10.1128/mcb.18.1.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mirzoeva OK, Petrini JH. DNA damage-dependent nuclear dynamics of the Mre11 complex. Mol Cell Biol. 2001;21:281–288. doi: 10.1128/MCB.21.1.281-288.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lisby M, Barlow JH, Burgess RC, Rothstein R. Choreography of the DNA damage response: Spatiotemporal relationships among checkpoint and repair proteins. Cell. 2004;118:699–713. doi: 10.1016/j.cell.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 55.Shroff R, Arbel-Eden A, Pilch D, Ira G, Bonner WM, Petrini JH, Haber JE, Lichten M. Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr Biol. 2004;14:1703–1711. doi: 10.1016/j.cub.2004.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsukuda T, Fleming AB, Nickoloff JA, Osley MA. Chromatin remodeling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature. 2005;438:379–383. doi: 10.1038/nature04148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ira G, Malkova A, Liberi G, Foiani M, Haber JE. Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell. 2003;115:401–411. doi: 10.1016/s0092-8674(03)00886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chiolo I, Carotenuto W, Maffioletti G, Petrini JH, Foiani M, Liberi G. Srs2 and Sgs1 DNA helicases associate with Mre11 in different subcomplexes following checkpoint activation and CDK1-mediated Srs2 phosphorylation. Mol Cell Biol. 2005;25:5738–5751. doi: 10.1128/MCB.25.13.5738-5751.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meselson M. Formation of hybrid DNA by rotary diffusion during genetic recombination. J Mol Biol. 1972;71:795–798. doi: 10.1016/s0022-2836(72)80040-8. [DOI] [PubMed] [Google Scholar]
- 60.Panyutin IG, Hsieh P. The kinetics of spontaneous DNA branch migration. Proc Natl Acad Sci USA. 1994;91:2021–2025. doi: 10.1073/pnas.91.6.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chamankhah M, Xiao W. Molecular cloning and genetic characterization of the Saccharomyces cerevisiae NGS1/MRE11 gene. Curr Genet. 1998;34:368–374. doi: 10.1007/s002940050408. [DOI] [PubMed] [Google Scholar]
- 62.Mages GJ, Feldmann HM, Winnacker EL. Involvement of the Saccharomyces cerevisiae HDF1 gene in DNA double-strand break repair and recombination. J Biol Chem. 1996;271:7910–7915. doi: 10.1074/jbc.271.14.7910. [DOI] [PubMed] [Google Scholar]
- 63.Chahwan C, Nakamura TM, Sivakumar S, Russell P, Rhind N. The fission yeast Rad32 (Mre11)-Rad50-Nbs1 complex is required for the S-phase DNA damage checkpoint. Mol Cell Biol. 2003;23:6564–6573. doi: 10.1128/MCB.23.18.6564-6573.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Andrews CA, Clarke DJ. MRX (Mre11/Rad50/Xrs2) mutants reveal dual intra-S-phase checkpoint systems in budding yeast. Cell Cycle. 2005;4:1073–1077. [PubMed] [Google Scholar]
- 65.Bill CA, Nickoloff JA. Ultraviolet light-induced and spontaneous recombination in eukaryotes: roles of DNA damage and DNA repair proteins. In: Nickoloff JA, Hoekstra MF, editors. DNA Damage and Repair, vol. 3: Advances from Phage to Humans. Humana Press; Totowa, NJ: 2001. pp. 329–357. [Google Scholar]
- 66.Featherstone C, Jackson SP. Ku: a DNA repair protein with multiple cellular functions? Mutat Res. 1999;434:3–15. doi: 10.1016/s0921-8777(99)00006-3. [DOI] [PubMed] [Google Scholar]
- 67.Mimori T, Hardin JA. Mechanism of interaction between Ku protein and DNA. J Biol Chem. 1986;261:10375–10379. [PubMed] [Google Scholar]
- 68.de Vries E, van Driel W, Bergsma WG, Arnberg AC, van der Vliet PC. HeLa nuclear protein recognizing DNA termini and translocating on DNA forming a regular DNA-multimeric protein complex. J Mol Biol. 1989;208:65–78. doi: 10.1016/0022-2836(89)90088-0. [DOI] [PubMed] [Google Scholar]
- 69.Morimatsu K, Kowalczykowski SC. RecFOR proteins load RecA protein onto gapped DNA to accelerate DNA strand exchange: a universal step of recombinational repair. Mol Cell. 2003;11:1337–1347. doi: 10.1016/s1097-2765(03)00188-6. [DOI] [PubMed] [Google Scholar]
- 70.Van Dyck E, Stasiak AZ, Stasiak A, West SC. Binding of double-strand breaks in DNA by human Rad52 protein. Nature. 1999;398:728–731. doi: 10.1038/19560. [DOI] [PubMed] [Google Scholar]
- 71.Sugawara N, Haber JE. Characterization of double-strand break-induced recombination: homology requirements and single-stranded DNA formation. Mol Cell Biol. 1992;12:563–575. doi: 10.1128/mcb.12.2.563. [DOI] [PMC free article] [PubMed] [Google Scholar]