

Abstract
The transcriptional mechanisms governing the development and plasticity of somatopic sensory maps in the cerebral cortex have not been extensively studied. In particular, no studies have addressed the role of epigenetic mechanisms in the development of sensory maps. DNA methylation is one the main epigenetic mechanisms available to mammalian cells to regulate gene transcription. As demethylation results in embryonic lethality, it has been very difficult to study the role of DNA methylation in brain development. We have used cre-lox technology to generate forebrain-specific deletion of DNA methyltransferase 1 (Dnmt1), the enzyme required for the maintenance of DNA methylation. We find that demethylation of neurons in the cerebral cortex results in the failure of development of somatosensory barrel cortex. We also find that in spite of functional thalamocortical neurotransmission, thalamocortical long-term potentiation cannot be induced in slices from Dnmt1 conditional mutants. These studies emphasize the importance of DNA methylation for the development of sensory maps and suggest epigenetic mechanisms may play a role in the development of synaptic plasticity.
Introduction
The mechanisms underlying the development and plasticity of sensory maps in the cerebral cortex is not well understood and represents a major challenge for developmental neuroscience (Goodman and Shatz, 1993; O’Leary et al., 1994; Katz and Shatz, 1996). In the rodent trigeminal-medial lemniscus system, periphery-related patterns representing the whiskers develop in the brainstem and thalamus before developing in the cerebral cortex (Woolsey and Van der Loos, 1970; Van de Loos and Woolsey, 1973; Van der Loos, 1976; Belford and Killackey, 1979). During a critical period in development, these patterns are susceptible to manipulation from the periphery and therefore present an excellent model system for studying the development and plasticity of cortical maps (Van der Loos and Woolsey, 1973; Belford and Killackey 1980; Killackey and Belford, 1980). A number of studies have demonstrated the importance of NMDA receptors (Fox et al., 1996; Iwasato et al., 1997, 2000; Lee et al., 2005) metabotropic glutamate receptors (Hannan et al., 2001), monoamine oxidase-A (MAO-A) (Cases et al., 1996), adenylyl cyclase I (Abdelmajid et al., 1998) protein kinase A R2β (Inan et al., 2006; Watson et al., 2006), PLC-β1 (Hannan et al., 2001) and the signaling molecule FGF8 (Fukuchi-Shimogori and Grove, 2001). However, only a few studies have attempted to delineate the complex transcriptional networks that mediate the generation of cortical maps (Ince-Dunn et al., 2006). Furthermore, there has been no work on whether epigenetic changes underlie or modulate these transcriptional networks.
Epigenetic changes are in part mediated by DNA methylation, one of the main mechanisms available to mammalian cells to regulate gene transcription. DNA methylation in mammals occurs on cytosine, primarily at CpG dinucleotides, in a spatially and temporally regulated manner. DNA methylation is mediated by a family of DNA methyltransferases (Dnmts) that include de novo (Dnmt3a and Dnmt3b) and maintenance methyltransferases (Dnmt1) (Bestor, 2000; Robertson and Wolffe, 2000). Transcriptional downregulation occurs through binding of a number of proteins to methylated DNA including methyl CpG binding protein 2 (MeCP2,) and methyl CpG binding domain proteins 1–4 (MBD1–4). MeCP2 and MBD2 have been shown to recruit histone deacetylases (HDACs) to methylated CpG sites, which in turn causes deacetylation of histones. These changes promote the formation of a silent chromatin state (Jaenisch and Bird, 2003). Mutations in MeCP2 are the main cause of the neurodevelopmental disorder, Rett Syndrome, underscoring the importance of DNA methylation for brain development (Amir et al., 1999). All three DNA methyltransferases are expressed in the central nervous system (CNS) and are dynamically regulated during development and differentiation (Goto et al., 1994; Feng et al., 2005), suggesting that DNA methylation may control transcriptional networks that orchestrate neuronal differentiation, migration, and formation of specific neuronal connections. The DNA methyltransferase Dnmt1 is responsible for maintenance DNA methylation. Without the presence of the maintenance DNA methyltransferase Dnmt1, cells undergoing mitosis become progressively demethylated leading to embryonic lethality (Li et al., 1992).
In addition to the essential role of DNA methylation in embryonic development, recent studies show that it can also modulate activity-dependent changes in gene expression. For example, the activity-dependent expression of BDNF relies on phosphorylation-dependent detachment of a transcriptional repression complex consisting of MeCP2, HDAC1, and mSin3a from the BDNF promoter. Furthermore, depolarization appears to lead to relative demethylation of CpGs near calcium responsive elements in the BDNF promoter, further re-enforcing transcriptional activation (Chen et al., 2003; Martinowich et al., 2003).
To directly examine the role of DNA methylation in the CNS, we have previously induced DNA hypomethylation throughout the entire CNS by conditional knockout of Dnmt1 in neural precursor cells (Fan et al., 2001). We demonstrated that DNA hypomethylation in the CNS disrupts neural control of breathing at birth, leading to neonatal lethality of mutant mice (Fan et al., 2001). However, the neonatal lethality of these mice prevented us from studying the role of DNA methylation in postnatal neural development.
To circumvent this obstacle, we crossed mice bearing the Emx1-cre transgene (Iwasato et al., 2000) to induce deletion of Dnmt1 solely in excitatory neurons of the dorsal forebrain. We have shown that although these mice are viable into adulthood, they develop progressive apoptotic degeneration of dorsal forebrain. Furthermore excitatory cortical neurons in these mice show defects in dendritic branching and action potential repolarization (Hutnick et al., submitted).
Objectives
While the above studies highlight the importance of DNA methylation in neuronal survival, morphogenesis and ion channel expression, no study has yet addressed what role DNA methylation plays in development of somatopic sensory maps as well as in thalamocortical synaptic transmission and plasticity. Here we show that DNA methylation is essential for development of periphery-related anatomical patterns in somatosensory barrel cortex. We also demonstrate that while thalamocortical axons functionally innervate the cortex in Dnmt1 mutant mice, thalamocortical long-term potentiation is defective. These results highlight the importance of DNA methylation in activity-dependent remodeling of cortical circuits and synaptic plasticity.
Methods
Generation of Emx1-cre;Dnmt1 conditional mutants
All experiments were carried out in accordance to protocols approved by the UCLA Institutional Animal Research committee. Using the cre-loxP binary gene deletion strategy, we crossed female mice homozygous for the Dnmt1 conditional allele (Dnmt12lox) with male mice carrying the emx1-cre insertion (Emx1-cre;Dnmt12lox/+), generating both control/heterozygous(Dnmt12lox/+ or Dnmt12lox/2lox), and conditional mutant (Emx1-cre; Dnmt12lox/2lox) offspring in expected Mendelian ratios. Because of the progressive cortical degeneration in mutants (Hutnick et al., submitted), the brains of mutant animals can be readily discerned from the brains of control animals.
Histology
Postnatal day 8 Dnmt1 emx-cre mutant mice and control littermates were perfused with 4% paraformaldehyde. Brains were removed and cryoprotected in 30% sucrose overnight. For cytochrome oxidase staining, the cortex was then removed, flattened with a microscope slide on dry ice and sectioned tangentially at 50 microns with a cryostat. Cytochrome oxidase histochemistry was performed as follows: Sections were incubated in 4 g of sucrose, 50 mg of cytochrome C, and 50 mg of diaminobenzidine in 100 ml of phosphate buffer at 37°C in a shaker incubator for 3–4 hrs. Sections were then rinsed in phosphate buffer, mounted on subbed slides, and coverslipped with an aqueous medium containing gelatin and glycerol. A second set of control and mutant brains were sectioned in the coronal angle at 10 microns with a cryostat and Nissl stained.
Electrophysiology
Emx1-cre;Dnmt1 mutant mice and control littermates (postnatal day 3–6 (P3–P6)) were anesthetized with halothane and sacrificed by decapitation. The brain was quickly removed and put in chilled artificial cerebrospinal fluid (ACSF) containing in mM 119 NaCl, 26 mM NaHCO3, 11 mM glucose, 2.5 mM KCl, 2.5 mM CaCl2, 1.3 mM MgCl2, 1 mM NaH2PO4 and was bubbled with 95% O2/5% CO2. Slices (300–400-μm thick) containing the somatosensory cortex, RTN, and VP were cut with a vibratome (Leica VT-1000) at an angle that preserves corticothalamic and thalamocortical connectivity (Agmon and Connors, 1991). After incubation at 35°C for 30 min, slices were stored at room temperature (22–24°C) before being transported to a recording chamber. Slices were allowed to recover for at least 1 hour before recording. Electrodes for whole-cell recordings were pulled from borosilicate glass capillaries to a final resistance of 3–6 Mega Ohms. Internal solution for current clamp experiments contained (in mM): 140 K-gluconate, 1.1 EGTA, 0.1 CaCl2, 10 HEPES, 2 Mg-ATP, 1 MgCl2, and 0.3 Na-GTP (pH 7.2). For voltage clamp experiments, the internal solution contained (in mM) 117 Cs-methanesulfonate, 20 HEPES, 0.2 EGTA, 5 TEA-Cl, 3.7 NaCl, 4 Mg-ATP, and 0.3 Na-GTP. Biocytin (3mg/ml) was also added to the internal solution during some recordings. Whole cell recordings were performed at room temperature (22–24 °C) with an Optopatch patch-clamp amplifier (Cairn Research, Kent, UK) in current clamp or voltage clamp configuration from layer IV neurons in control slices. In thalamocortical slices obtained from control mice, layer IV was readily identified by transillumination of the cortex. As cortical lamination was disrupted in mutant cortex, an attempt was made to record from neurons at similar proportional distances from the pial surface in mutant as in control cortical neurons. Thalamocortical synaptic responses were evoked by stimulating the thalamic ventrobasal complex or rarely the immediately adjacent internal capsule with a bipolar tungsten electrode. At times a bipolar stimulating electrode made from theta capillary glass and filled with ACSF was used. Stimuli (100 μs, typically 0.1–1 mA) elicited thalamocortical EPSPs. Current and voltage clamp traces were analogue filtered at 5 kHz, digitized at 10 kHz and digitally filtered at 2–5 kHz, and analyzed using custom made software using Lab View (National Instruments, Austin, TX) and Igor (Wavemetrics, Lake Oswego, OR). For LTP experiments, neurons with accommodating regular spiking action potential firing properties were current clamped using whole-cell patch clamp. Baseline responses were elicited every 30 seconds for 10 minutes. Thalamocortical LTP was induced by delivering 100 stimuli at 1 Hz while depolarizing the neuron to −10 mV (Crair and Malenka, 1995). Thalamocortical responses were elicited and monitored every 30 seconds for at least 40 minutes after LTP induction paradigm. The magnitude of LTP was calculated by dividing average amplitude of 20 responses during the baseline period by the average amplitude of 20 responses obtained at 30 to 40 minutes after pairing protocol. During baseline and after the LTP induction, holding current was injected to set the resting membrane potential to −70 mV. A 10 mV junction potential was subtracted post-hoc from the recordings when a potassium gluconate based internal solution was used.
Results
Periphery-related patterns fail to develop in the somatosensory barrel cortex of Emx1-cre;Dnmt1 conditional mutant mice
Nissl stained coronal sections of control and mutant brains at P7 demonstrated indistinct lamination of the cerebral cortex in mutant animals (Figure 1A). To determine whether transcriptional regulation of gene expression through DNA methylation is essential for development of cortical sensory maps, we chose to study the development of whisker-related barrels in the somatosensory cortex of Emx1-cre;Dnmt1 mutant mice. We chose to study the development of barrels at P8 because at later ages neurodegeneration of the cortex in Emx1-cre;Dnmt1 mutants confounds analysis of cortical columnar structure. As expected, cytochrome oxidase staining of flattened sections through the cortex of control littermates at P8 showed normal development of forepaw and whisker related patterns (Figure 1B, left). In conditonal mutant mice, however, no whisker or forepaw related pattern could be discerned (Figure 1B, right). This indicated that DNA methylation is essential for development of periphery related patterns in the cerebral cortex.
Figure 1.
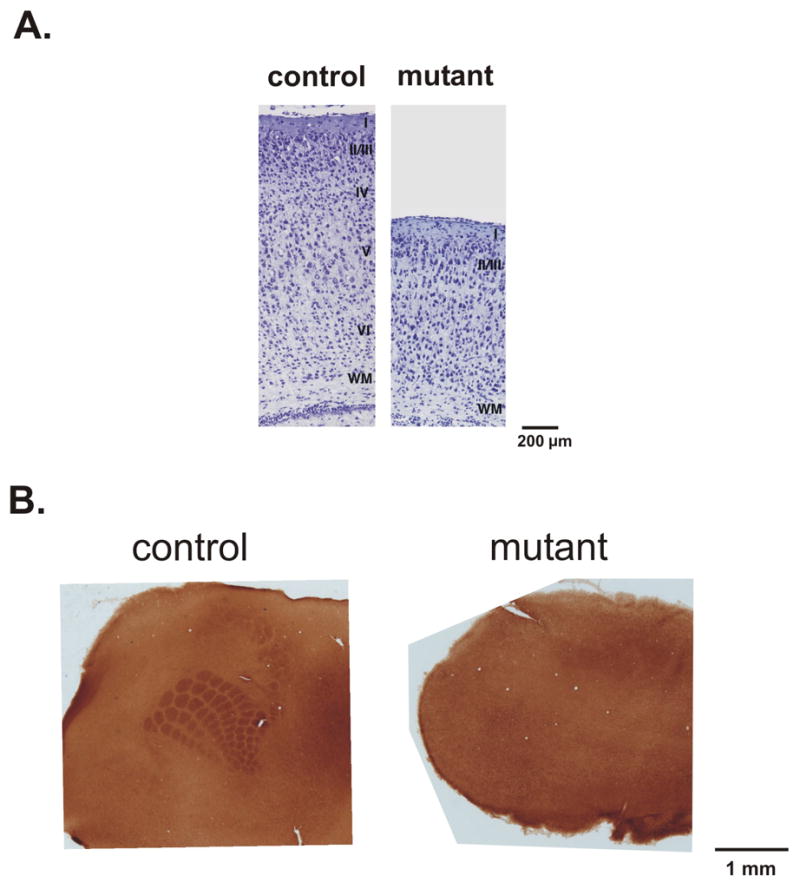
A. Photomicrographs of Nissl stained coronal sections through the dorsal forebrain of control and Emx1-cre;Dnmt1 mutant mice at P7. Note indistinct development of cortical layers in the mutant cortex. B. Cytochrome oxidase stained flattened tangential sections of somatosensory cortex from control and Emx1-cre;Dnmt1 mutant animals at P8. Note the lack of development of periphery related patterns in the section obtained from Emx1-cre;Dnmt1 mutant mouse.
Thalamocortical neurotransmission is intact despite the failure of development of periphery related patterns
To determine whether the failure of development of periphery-related patterns in barrel cortex results from the failure of in-growth of functional thalamocortical afferents or the failure of thalamocortical synaptogenesis, we performed whole-cell recordings from cortical neurons in thalamocortical slices of control and Emx1-cre;Dnmt1 mutants at P3-P6. In slices obtained from control cortices layer IV could be readily identified by transillumination and in these slices all recordings were performed in layer IV. As neocortical layers were indistinct in Emx1-cre;Dnmt1 conditional mutants, recordings were performed at similar proportional distances from the pial surface as in control slices. Stimulation of thalamocortical axons elicited excitatory postsynaptic potentials/currents (EPSP/Cs) in cortical neurons of both control and Emx1-cre;Dnmt1 mutants (Figure 2A). This clearly shows that even in the cortex of Emx1-cre;Dnmt1 mutants, functional thalamocortical connections develop. Thalamocortical EPSCs recorded from neurons in mutants at −70 mV had slightly more prolonged onset latencies than EPSCs recorded from neurons in control mice (8.7 +/− 0.8 ms in controls, n=5; 11.9 +/− 3.0 ms in mutants, n=9; p<0.05) (Figure 2B). EPSCs recorded in mutant neurons also had more prolonged 20–80% rise times compared to control (0.9 +/− 0.2 ms in controls, n=5; 1.4 +/− 0.5 ms in mutants, n=9; p<0.05), but the time constant of decay was not statistically different between the two groups (5.6 +/− 0.7 ms in controls, n=5; 5.2 +/− 1.1 ms in mutants, n=9; p>0.05) (Figure 2B). This demonstrates that while the thalamocortical synapses may be located at electrotonically more distant location in mutants compared to control, the failure of development of somatosensory barrels is not the result of the failure of ingrowth of functional thalamocortical fibers or failure of the generation of thalamocortical synapses in mutant mice.
Figure 2.
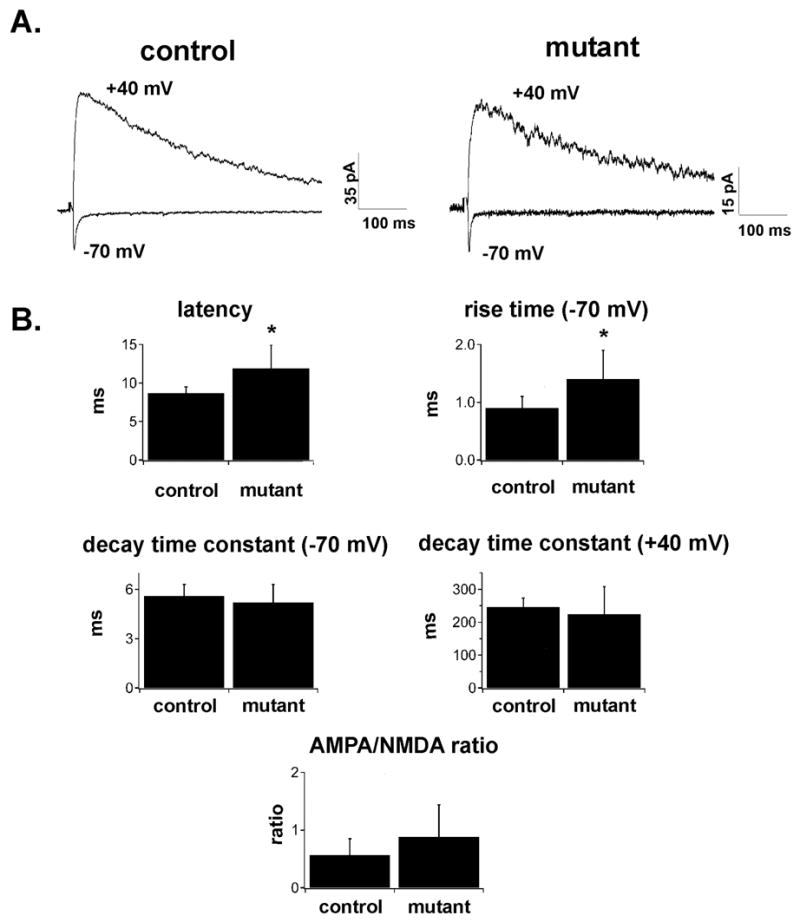
A. Thalamocortical EPSCs evoked at −70 and +40 mV in slices obtained from control (left) and mutant (right) animals at P4–P5. Stimulation artifacts have been blanked. Note that both slowly decaying NMDA-receptor mediated currents and rapidly decaying non-NMDA mediated currents can be evoked in slices obtained from both control and mutant animals. B. Bar graphs of latency, rise time, decay time constant (at −70 and +40 mV), and AMPA/NMDA ratio of thalamocortical EPSCs evoked in slices obtained from control or Emx1-cre;Dnmt1 mutant animals at P4–P6. Asterisks denote statistical significance at p<0.05 level. Note that thalamocortical EPSCs in mutants have slightly more prolonged latencies and rise times, but the decay time constant at −70 and +40 mV, as well and AMPA/NMDA ratio is not statistically different between the two groups.
As NMDA receptor mediated responses have been shown to be essential for development of periphery-related patterns in barrel cortex (Iwasato et al., 2000), we assessed the ratio of NMDA to AMPA mediated EPSCs. Thalamocortical EPSCs were elicited first at −70 mV, where the current is largely carried by AMPA-type glutamate receptors. The voltage was then stepped to +40 mV where in addition to the fast AMPA component, a slowly-decaying, NMDA-receptor mediated current can be revealed. As described above, AMPA-mediated currents were observed in both mutant and control animals. In addition, at +40 mV, large slowly decaying currents typical of NMDA receptor mediated currents could be elicited in slices obtained from both control and mutant animals. The time constant of decay of the EPSC at +40 mV was not different in the two groups (246.7 +/− 26.7 ms in controls, n=5; 224.1 +/− 84.1 ms in mutants; p>0.05) (Figure 2). The peak amplitudes of the EPSC at −70 mV (−24.8 +/− 15.3 pA in control (n=5); −27.3 +/− 24.2 pA in mutant (n=9)) and +40 mV (53.0 +/− 35.4 pA in control (n=5); 31.2 +/− 21.0 in mutant (n=9)) were not significantly different in the two groups (p>0.05). We also calculated the ratio of AMPA/NMDA currents by dividing the peak amplitude of the EPSC at −70mV by the amplitude of the EPSC at +40 mV at 50 ms after the stimulus. This ratio was not significantly different in control and mutant neurons (Figure 2; 0.57 +/− 0.28 for control neurons (n=5); 0.88 +/− 0.56 for mutant neurons (n=9); p=0.27). These experiments were performed without blockade of GABA-A receptor and thus inhibitory currents could have a confounding effect. However at this age GABA-A currents are commonly not observed after stimulation of thalamocortical fibers (Agmon and O’Dowd, 1992). Furthermore, recordings performed at the glutamate receptor reversal potential failed to show evidence of polysynaptic GABA-A currents (data not shown). Taken together, our data indicate that the failure of the development of periphery-related patterns likely does not result from a lack of NMDA receptor activity.
Thalamocortical long-term potentiation (LTP) is deficient in cortical neurons of Emx1-cre;Dnmt1 conditional mutants
In a number of mouse strains where periphery-related patterns do not develop in barrel cortex, thalamocortical LTP has been shown to be deficient (Iwasato et al., 2003; Lu et al., 2003). This has prompted investigators to conclude that strengthening of thalamocortical synapses through LTP-like phenomena during early development may be essential for development of periphery–related patterns in barrel cortex (Lu et al., 2003; Inan et al., 2006). This enticed us to determine whether thalamocortical long-term potentiation can be elicited in Emx1-cre;Dnmt1 mutant cortex.
We elicited thalamocortical EPSPs in both control and mutants at P3–P6, during the critical developmental window when thalamocortical LTP has been shown to be elicited (Crair and Malenka, 1995). After eliciting a stable baseline for 10 minutes at −70 mV, cortical neurons were depolarized to −10 mV and 100 presynaptic stimuli were delivered at 1 Hz to elicit LTP (Crair and Malenka, 1995). Neurons were then hyperpolarized back to −70 mV and thalamocortical responses were monitored for at least 40 minutes after the pairing protocol. Layer IV cortical neurons from control slices showed robust LTP 30–40 minutes after the pairing protocol, (195.4 +/− 95.4 %; n=5) (Figure 3A,C). As has been demonstrated before (Crair and Malenka, 1995), the increase in the amplitude of responses developed slowly over 5 to 15 minutes before stabilizing for the duration of the recording (Figure 3B). In contrast, the same pairing protocol in mutant neurons did not elicit significant LTP (97.3 % +/− 45.6 %; n=6) at the same time point (Figure 3A,C). The magnitude of potentiation was significantly different in control and mutant neurons (p<0.05) (Figure 3C). This indicates that while thalamocortical transmission is intact in Dnmt1-emx-cre mutant cortex, thalamocortical synapses in mutant slices cannot be readily strengthened by pairing synaptic stimulation with neuronal depolarization.
Figure 3.
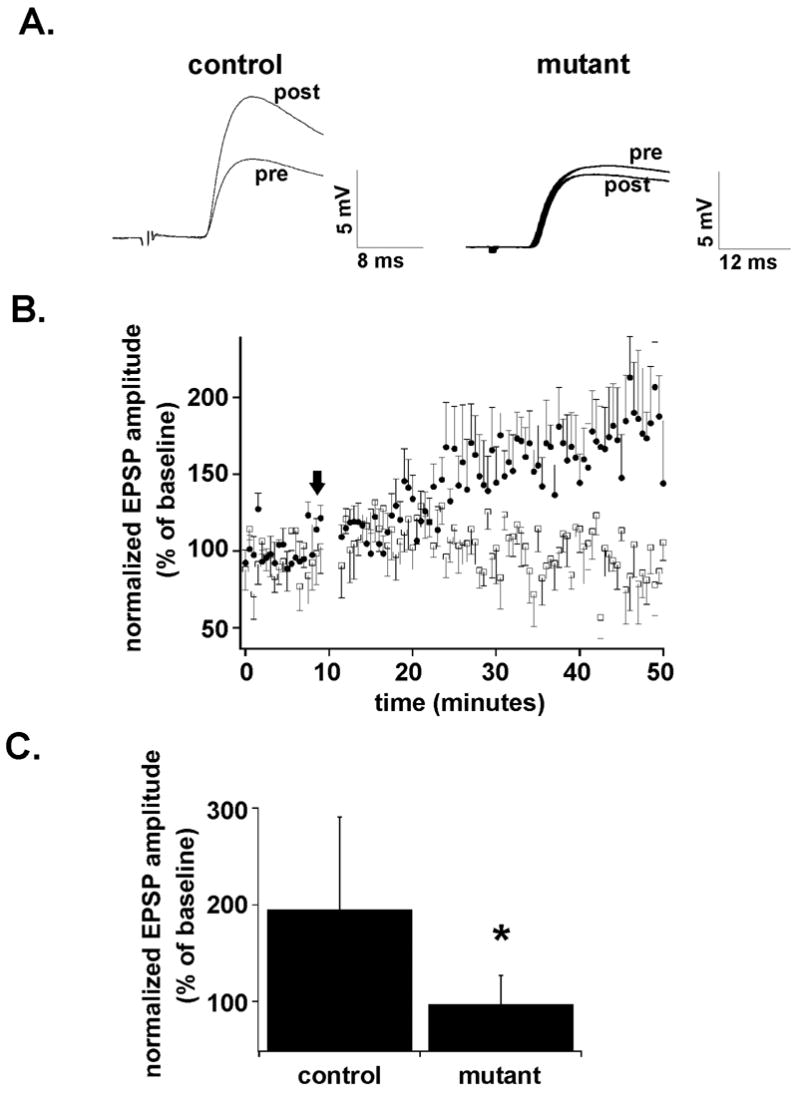
A. Thalamocortical EPSPs evoked in slices from control and Emx1-cre;Dnmt1 mutant animals 0–10 minutes before (pre) and 30–40 minutes after (post) pairing 100 stimuli at 1 Hz with depolarization of the postsynaptic cell. Note robust long-term potentiation in control but not in the mutant cell. B. Graph demonstrating the amplitude of the EPSP normalized to the average baseline value in slices obtained from control (solid circles) and mutant (open squares) animals before and after the pairing protocol. Error bars represent standard of error values. The dark arrow represents the onset of pairing protocol. C. Bar graph demonstrating the mean normalized EPSP amplitude 30–40 minutes after the pairing protocol in control and mutant neurons. Error bars represent standard of deviation values. Note that there is a significant difference between the amount of potentiation induced in control versus mutant animals (asterisk).
Conclusions
Conditional deletion of Dnmt1 in dorsal forebrain disrupts the development of periphery-related anatomical patterns in somatosensory barrel cortex.
While thalamocortical axons functionally innervate the cortex in Emx1-cre;Dnmt1 mutant mice, thalamocortical long-term potentiation is defective.
Discussion
To study the role of DNA methylation on postnatal cortical development we have used cre-lox technology to specifically delete the DNA methyltransferase Dnmt1 in neural precursors of excitatory neurons of the dorsal forebrain. We have previously shown that the cortex is poorly laminated in these mutant mice and undergoes progressive neurodegeneration (Hutnick et al., submitted). Here we show that while thalamocortical fibers functionally innervate excitatory cortical neurons of Emx1-cre;Dnmt1 mice, periphery-related anatomical patterns do not form in the somatosensory barrel cortex of mutant mice. Furthermore, we show that deletion of Dnmt1 in excitatory cortical neurons abolishes thalamocortical LTP, highlighting the importance of DNA methylation for the developmental plasticity at the thalamocortical synapse.
Periphery-related anatomical patterns fail to develop in the somatosensory barrel cortex of Emx1-cre;Dnmt1 mutant mice
The failure of development of periphery-related patterns in somatosensory cortex of Emx1-cre;Dnmt1 mutant mice may be a direct effect of demethylation on the development of cortical modules or may arise as a consequence of early apoptotic cell death and cortical laminar disorganization in these mutant mice. Answering this essential question will depend on examining the development of periphery-related patterns in Dnmt1 mutant mice where cell death and cortical migration phenotypes have been rescued through genetic means. Targeting deletion of Dnmt1 to specific subsets of excitatory cortical neurons (such as layer IV progenitors) may further allow dissection of the complexities of this model. Direct effects of demethylation may include inappropriate expression of transcription factors, axon guidance cell adhesion proteins, or molecules such as BDNF which have been shown to be critical for activity dependent development of thalamocortical circuits (Cabelli et al. 1995; 1997). In fact, as activity-dependent expression of BDNF requires release of methyl-binding proteins from the BDNF promoter, BDNF remains an important candidate for mediating some of the effects of demethylation (Chen et al., 2003; Martinowich et al., 2003). In addition, increasing BDNF expression in Mecp2 mice with a conditional BDNF transgene extends the lifespan, rescues locomotor defects, and reverses an electrophysiological deficit observed in Mecp2 mutants, again linking transcriptional control of BDNF to DNA methylation binding proteins (Chang et al., 2006). Studies in our laboratory are in progress to characterize genome-wide differences in mRNA expression in specific subsets of cortical neurons in Emx1-cre;Dnmt1 mice and control littermates to better delineate the specific molecular mechanisms of cortical disorganization in these mice.
It is also possible that disruption of thalamocortical plasticity in Emx1-cre;Dnmt1 mutants resulted in disruption of the development of somatosensory cortical barrels. Considerable controversy exists on whether thalamocortical long-term potentiation is essential for barrel formation. Barrel formation and thalamocortical plasticity are both disrupted by adenylyl cyclase I deletion (Abdel Majid et al., 1998; Lu et al., 2003). NMDA receptor blockade (Crair and Malenka, 2005), and PKA RIIβ deletion both prevent thalamocortical LTP during early development (Watson et al., 2006) but cortical deletion of the critical NMDA receptor subunit NR1 (Iwasato et al., 2000) and deletion of PKA RIIβ (Watson et al., 2006; Inan et al., 2006), while blurring barrel boundaries, do not completely disrupt formation of somatosensory cortical barrels. Finally, while studies have suggested the importance of GluR1 trafficking for thalamocortical long-term potentiation (Lu et al., 2003), barrels form normally in GluR1 KO mice (Watson et al., 2006), again suggesting that thalamocortical LTP may not be essential for the development of somatosensory cortical barrels. This would suggest that the lack of thalamocortical LTP is unlikely to be the sole direct cause of the lack of development of somatosensory cortical barrels in Emx1-cre;Dnmt1 mutants.
Thalamocortical neurotransmission is intact despite the failure of development of periphery related patterns
While we could elicit both AMPA and NMDA mediated EPSCs in Dnmt1 mutant cortex, the rise time of EPSCs at −70 mV were significantly more prolonged. The small though statistically significant difference in latency may be related to possible changes in thalamocortical axonal length or ramifications in mutant cortex. The increased rise time of thalamocortical EPSCs may be the result of aberrant synapse formation at electrotonically more distant locations in the mutant. This is supported by our previous study that demonstrated increased dendritic complexity in pyramidal neurons from Emx1-cre;Dnmt1 mutants (Hutnick et al., submitted). Our studies do not show a statistically significant difference in the amplitude EPSCs at −70 and +40 mV; however, minimal stimulation or recordings from synaptically connected neurons in thalamus and cortex will be necessary to more accurately quantify changes in unitary EPSC amplitude.
Impaired thalamocortical LTP in Emx1-cre;Dnmt1 mutant mice
While we could elicit both NMDA and non-NMDA mediated responses in cortical neurons in Emx1-cre;Dnmt1 conditional mutants, pairing postsynaptic depolarization with synaptic stimulation did not elicit thalamocortical LTP in these mice. Several observations indicated that thalamocortical LTP may be affected by demethylation. First, the activity-dependent expression of BDNF (which is essential for conversion of silent thalamocortical synapse to functional synapses) (Itami et al., 2003) relies on the release of methylated CpG binding protein-2 (MECP-2) from the BDNF promoter (Chen et al., 2003; Martinowich et al., 2003); second, demethylating agents (Levenson et al., 2006) or loss of MECP-2 (Morretti et al., 2006) modulate LTP in cortex and hippocampus. All of these effects might either be due to an influence of the presynaptic release machinery or the postsynaptic receptor complexes. Since Dnmt1 deletion is restricted to excitatory cortical neurons that are by definition postsynaptic to the thalamocortical inputs, our studies are informative on the locus of the potential plasticity deficits.
As cortical lamination is disorganized in Emx1-cre;Dnmt1 mutant mice, one mechanism underlying the lack of thalamocortical LTP may be that thalamocortical axons formed synapses with displaced cells that were not destined to become layer IV neurons. These neurons may lack the molecular machinery for thalamocortical LTP during the critical period. Alternatively, transcriptional misregulation through demethylation may have impaired the adenylate cyclase/PKA pathway or further downstream processes that are essential for trafficking of AMPA receptors into thalamocortical synapses. Again, definitive answers to these questions will rely on development of techniques to demethylate individual neurons that have already migrated into position and acquired their neuronal identity.
This study highlights the importance of DNA methylation for the development of somatotopic sensory maps and the strengthening of thalamocortical synapses during early development. Future studies will address the mechanisms underlying the developmental defects caused by hypomethylation.
Acknowledgments
This work was supported by NIH RO1 grants NS51411 and NS44405 to GF. LH was supported by NINDS NRSA 5F31NS051 and the ARCS Foundation. PG was supported by NIH KO8 NS56210. FES was supported by NS41317 and DC007678.
References
- Abdel-Majid RM, Leong WL, Schalkwyk LC, Smallman DS, Wong ST, Storm DR, Fine A, Dobson MJ, Guernsey DL, Neumann PE. Loss of adenylyl cyclase I activity disrupts patterning of mouse somatosensory cortex. Nature Genetics. 1998;19:289–291. doi: 10.1038/980. [DOI] [PubMed] [Google Scholar]
- Agmon A, Connors BW. Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience. 1991;41:365–379. doi: 10.1016/0306-4522(91)90333-j. [DOI] [PubMed] [Google Scholar]
- Agmon A, O’Dowd DK. NMDA receptor-mediated currents are prominent in the thalamocortical synaptic response before maturation of inhibition. Journal of Neurophysiology. 1992;68(1):345–9. doi: 10.1152/jn.1992.68.1.345. [DOI] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked Mecp2, encoding methyl-CpG-binding protein 2. Nature Genetics. 1999;23(2):185–8. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Belford GR, Killackey HP. Vibrissae representation in subcortical trigeminal centers of the neonatal rat. Journal of Comparative Neurology. 1979;183:305–322. doi: 10.1002/cne.901830207. [DOI] [PubMed] [Google Scholar]
- Belford GR, Killackey HP. The sensitive period in the development of the trigeminal system of the neonatal rat. Journal of Comparative Neurology. 1980;193(2):335–50. doi: 10.1002/cne.901930203. [DOI] [PubMed] [Google Scholar]
- Bestor TH. The DNA methyltransferases of mammals. Human Molecular Genetics. 2000;9:2395–2402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- Cabelli RJ, Hohn A, Shatz CJ. Inhibition of ocular dominance column formation by infusion of NT-4/5 or Bdnf. Science. 1995;267(5204):1662–6. doi: 10.1126/science.7886458. [DOI] [PubMed] [Google Scholar]
- Cabelli RJ, Shelton DL, Segal RA, Shatz CJ. Blockade of endogenous ligands of trkB inhibits formation of ocular dominance columns. Neuron. 1997;19(1):63–76. doi: 10.1016/s0896-6273(00)80348-7. [DOI] [PubMed] [Google Scholar]
- Cases O, Vitalis T, Seif I, De Maeyer E, Sotelo C, Gaspar P. Lack of barrels in the somatosensory cortex of monoamine oxidase A-deficient mice: role of a serotonin excess during the critical period. Neuron. 1996;16:297–307. doi: 10.1016/s0896-6273(00)80048-3. [DOI] [PubMed] [Google Scholar]
- Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49(3):341–348. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Derepression of Bdnf transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302(5646):885–9. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- Crair MC, Malenka RC. A critical period for long-term potentiation at thalamocortical synapses. Nature. 1995;375:325–328. doi: 10.1038/375325a0. [DOI] [PubMed] [Google Scholar]
- Feng J, Chang H, Li E, Fan G. Dynamic expression of de novo DNA methyltransferases Dnmt3a and Dnmt3b in the central nervous system. Journal of Neuroscience Research. 2005;79:734–746. doi: 10.1002/jnr.20404. [DOI] [PubMed] [Google Scholar]
- Fox K, Schlaggar BL, Glazewski S, O’Leary DDM. Glutamate receptor blockade at cortical synapses disrupts development of thalamocortical and columnar organization in somatosensory cortex. Proceedings of the National Acadamy of Sciences. 1996;93:5584–5589. doi: 10.1073/pnas.93.11.5584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto K, Numata M, Komura JI, Ono T, Bestor TH, Kondo H. Expression of DNA methyltransferase gene in mature and immature neurons as well as proliferating cells in mice. Differentiation. 1994;56:39–44. doi: 10.1046/j.1432-0436.1994.56120039.x. [DOI] [PubMed] [Google Scholar]
- Inan M, Lu HC, Albright MJ, She WC, Crair MC. Barrel map development relies on PKARIIβ-mediated cAMP signaling. Journal of Neuroscience. 2006;26:4338–4349. doi: 10.1523/JNEUROSCI.3745-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ince-Dunn G, Hall BJ, Hu SC, Ripley B, Huganir RL, Olson JM, Tapscott SJ, Ghosh A. Regulation of thalamocortical patterning and synaptic maturation by NeuroD2. Neuron. 2006;49(5):683–95. doi: 10.1016/j.neuron.2006.01.031. [DOI] [PubMed] [Google Scholar]
- Itami C, Kimura F, Kohno T, Matsuoka M, Ichikawa M, Tsumoto T, Nakamura S. Brain-derived neurotrophic factor-dependent unmasking of “silent” synapses in the developing mouse barrel cortex. Proceedings of the National Academy of Sciences. 2003;100(22):13069–74. doi: 10.1073/pnas.2131948100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasato T, Datwani A, Wolf AM, Nishiyama H, Taguchi Y, Tonegawa S, Knopfel T, Erzurumlu RS, Itohara S. Cortex-restricted disruption of NMDAR1 impairs neuronal patterns in the barrel cortex. Nature. 2000;406:726–731. doi: 10.1038/35021059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nature Genetics. 2003;33(Suppl):245–54. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- Killackey HP, Belford GR. Central correlates of peripheral pattern alterations in the trigeminal system of the rat. Brain Research. 1980;183(1):205–10. doi: 10.1016/0006-8993(80)90131-6. [DOI] [PubMed] [Google Scholar]
- Lee LJ, Iwasato T, Itohara S, Erzurumlu RS. Exuberant thalamocortical axon arborization in cortex-specific NMDAR1 knockout mice. Journal of Comparative Neurology. 2005;485(4):280–92. doi: 10.1002/cne.20481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson JM, Roth TL, Lubin FD, Miller CA, Huang IC, Desai P, Malone LM, Sweatt JD. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. Journal of Biological Chemistry. 2006;281(23):15763–73. doi: 10.1074/jbc.M511767200. [DOI] [PubMed] [Google Scholar]
- Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69(6):915–26. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- Lu HC, She WC, Plas DT, Neumann PE, Janz R, Crair MC. Adenylyl cyclase I regulates AMPA receptor trafficking during mouse cortical ‘barrel’ map development. Nature Neuroscience. 2003;6(9):939–47. doi: 10.1038/nn1106. [DOI] [PubMed] [Google Scholar]
- Ma PK, Woolsey TA. Cytoarchitectonic correlates of the vibrissae in the medullary trigeminal complex of the mouse. Brain Research. 1984;306:374–379. doi: 10.1016/0006-8993(84)90390-1. [DOI] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent Bdnf gene regulation. Science. 2003;302(5646):890–3. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- Moretti P, Levenson JM, Battaglia F, Atkinson R, Teague R, Antalffy B, Armstrong D, Arancio O, Sweatt JD, Zoghbi HY. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. Journal of Neuroscience. 2006;26(1):319–27. doi: 10.1523/JNEUROSCI.2623-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary DDM, Ruff NL, Dyck RH. Development, critical period plasticity, and adult reorganizations of mammalian somatosensory system. Current Opinions in Neurobiology. 1994;4:535–544. doi: 10.1016/0959-4388(94)90054-x. [DOI] [PubMed] [Google Scholar]
- Robertson KD, Wolffe AP. DNA methylation in health and disease. Nature Reviews Genetics. 2000;1:11–19. doi: 10.1038/35049533. [DOI] [PubMed] [Google Scholar]
- Van der Loos H. Barreloids in mouse somatosensory thalamus. Neuroscience Letters. 1976;2:1–6. doi: 10.1016/0304-3940(76)90036-7. [DOI] [PubMed] [Google Scholar]
- Van der Loos H, Woolsey TA. Somatosensory cortex: structural alterations following early injury to sense organs. Science. 1973;179:395–398. doi: 10.1126/science.179.4071.395. [DOI] [PubMed] [Google Scholar]
- Watson RF, Abdel-Majid RM, Barnett MW, Willis BS, Katsnelson A, Gillingwater TH, McKnight GS, Kind PC, Neumann PE. Involvement of protein kinase A in patterning of the mouse somatosensory cortex. Journal of Neuroscience. 2006;26(20):5393–401. doi: 10.1523/JNEUROSCI.0750-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolsey A, Van der Loos H. The structural organization of layer IV in the somatosensory region (SI) of the mouse cerebral cortex. Brain Research. 1970;17:205–242. doi: 10.1016/0006-8993(70)90079-x. [DOI] [PubMed] [Google Scholar]