

Abstract
BACKGROUND
The angiotensin-converting enzyme (ACE) deletion allele, ACE D, is associated with increased ACE activity and adverse outcomes in cardiovascular disease. Although activation of the renin-angiotensin aldosterone system (RAAS) now appears to play a role in the pathophysiology of atrial fibrillation (AF), it remains to be determined if ACE genotype impacts response to conventional AAD therapy in patients with AF.
OBJECTIVES
To investigate whether response to antiarrhythmic drug (AAD) therapy in patients with AF is modulated by the ACE I/D polymorphism.
METHODS
We studied 213 patients (147 men, 66 women; age 52±15 years) prospectively enrolled in the Vanderbilt AF Registry. AAD therapy outcome was defined prospectively as response if ≥ 75% reduction in symptomatic AF burden or non-response if AF burden was unchanged, necessitating a change in drugs or therapy.
RESULTS
Lone AF (age <65 years, no identifiable cause) was present in 72 (34%) patients, whereas hypertension was the commonest underlying disease in the remaining 141 (41%). AF was paroxysmal in 170 (80%) and persistent in 43 (20%). The frequencies of the DD, ID and II genotypes were in Hardy-Weinberg equilibrium. Lone AF and DD/ID genotypes were highly significant predictors of failure of drug therapy (P<0.005). In patients with lone AF, failure of drug response was 5%, 41% and 47% in patients with II, ID and DD genotypes respectively, (P<0.005, II vs ID/DD).
CONCLUSIONS
These results provide further evidence for a role of RAAS activation in the pathophysiology of AF, and point to a potential role for stratification of therapeutic approaches by ACE genotype.
Keywords: atrial fibrillation, antiarrhythmic drugs, angiotensin-converting-enzyme, genetics
INTRODUCTION
Progress in understanding molecular mechanisms in atrial fibrillation (AF) support the idea that variability in response to drug therapy may reflect differences in disease mechanisms;1 that is, it is entirely possible that response to AF is highly heterogeneous because the arrhythmia itself is not a single pathophysiologic entity, but rather represents a final common arrhythmia response to a variety of disease pathways that culminate in AF. Indeed, work in our own group and others increasingly support the idea that AF risk includes a prominent familial component.2–7 Further, the last several years have also seen increasing evidence that activation of pathways not traditionally linked to arrhythmias may be intimately involved in the development of AF. 8–11 One “non-traditional” AF risk factor is renin-angiotensin-aldosterone system (RAAS) activation. Retrospective analyses suggest that angiotensin-converting-enzyme (ACE) inhibitor therapy is associated with a lower incidence of AF and a placebo-controlled trial found a similar beneficial effect of adding the angiotensin receptor blocker (ARB) irbesartan to amiodarone.12 Additionally, a case control study of 250 Taiwanese subjects with AF and 250 controls identified polymorphisms in this pathway as risk factors for AF although predictors of drug response were not evaluated.13
Antiarrhythmic drugs (AADs) continue to remain a cornerstone of treatment for patients with symptomatic atrial fibrillation (AF), although even the most effective of drugs has a recurrence rate of 30–50% over a year. Further, predicting response to AAD therapy in individual patients also remains highly problematic. The role of genetic heterogeneity in modulating efficacy of antiarrhythmics in patients with AF however, has thus far not been explored.
The ACE gene contains a polymorphism based on the presence (insertion [I]) or absence (deletion [D]) of a 287-base-pair (bp) intronic DNA segment, resulting in three genotypes (DD and II homozygotes, and ID heterozygotes).14, 15 The frequency of ACE DD genotype has been reported to be increased in patients with myocardial infarction,16 dilated cardiomyopathy 17 and sudden death.18 In addition, a pharmacogenetic interaction of the ACE I/D polymorphism with beta-blocker and ACE inhibitor therapy has also recently been demonstrated.19, 20 Subjects with the ACE DD genotype have higher plasma concentration of ACE, 14 higher cardiac ACE concentration 21 and increased renal ACE mRNA expression.22 Thus subjects with the D-allele may be exposed to higher angiotensin II levels than those with the I-allele. Myocardial fibrosis is strongly correlated with RAAS activation, especially angiotensin II and aldosterone23 and chronic exposure to high levels of circulating and/or tissue angiotensin may predispose to both left ventricular hypertrophy and myocardial fibrosis. Since circulating and tissue ACE levels are higher in patients with the D allele,14, 21 we hypothesized that patients with the DD or ID genotype may have reduced response to conventional AADs than patients with the II genotype. Thus the aim of this study was to investigate whether response to AAD therapy in patients with AF is modulated by the ACE I/D polymorphism.
METHODS
Study Population
The study was performed in patients prospectively enrolled in the Vanderbilt AF Registry, which comprises a clinical and genetic registry. Inclusion criteria include age >18 years, a documented history of AF or atrial flutter and attempted maintenance of sinus rhythm with at least 1 conventional AAD. Patients with a history of AF only associated with cardiac surgery were excluded from this study. At enrollment into the Registry, a detailed medical and drug history is obtained in all patients and patients are also asked to complete a symptom questionnaire. This is done in order to assess symptomatic AF burden, which is measured by using an algorithm for scoring the frequency, duration and severity of symptoms (Table 1).24 The algorithm is a modification of the validated University of Toronto AF Severity Scale.25, 26 The symptomatic AF burden scale combines the three measures of severity, (each of which contributes equally to the AF burden score [each measure ranging from 1–10]) to yield scores ranging from 3 to 30. The symptom questionnaire is repeated at 3, 6, and 12 months after starting an AAD or whenever, there was a change to an alternative AAD agent or change in therapy. It is our usual practice that patients experiencing symptoms after commencing an AAD therapy are given an event recorder to document the cause of symptoms. An echocardiogram is obtained on all patients at time of enrollment into the Registry. Written informed consent was obtained from all patients under a protocol approved by the Vanderbilt University Institutional Review Board.
Table 1.
Algorithm for scoring symptomatic atrial fibrillation burden*
| Severity parameter | Scoring | |
|---|---|---|
| AF frequency (higher scores denote more frequent AF) | Less than once a year | 1 |
| About once a year | 2 | |
| About 2–4 times a year | 3 | |
| About once a month | 4 | |
| About twice a month | 5 | |
| About once a week | 6 | |
| About 2–3 times a week | 7 | |
| About 4–5 times a week | 8 | |
| Daily or almost daily | 9 | |
| More than once a day | 10 | |
| AF duration (higher scores denote AF of longer duration | A few minutes | 1 |
| <30 minutes | 2 | |
| 30–45 minutes | 3 | |
| About <1 hour | 4 | |
| About 1–4 hours | 5 | |
| About 5–12 hours | 6 | |
| About 13–24 hours | 7 | |
| About 1–3 days | 8 | |
| About 4–7 days | 9 | |
| >7 days | 10 | |
| AF severity (of most recent episode) | 1 = not all severe | 1 – 10 |
| 10 = extremely severe |
AF = atrial fibrillation.
Modified from University of Toronto Atrial Fibrillation Severity Scale.26
Definitions
Lone AF was defined as AF occurring in patients <65 years of age without hypertension or overt structural heart disease by clinical examination, ECG and echocardiography.27 Arterial hypertension was defined by a history of hypertension and/or the presence of antihypertensive therapy. Criteria for coronary artery disease (CAD) included a history of myocardial infarction (MI) or typical angina, previous bypass surgery or angioplasty and drug treatment. Congestive heart failure (CHF) was defined by a history of CHF and/or drug treatment for heart failure. Left atrial and left ventricular measurements from the M mode echocardiograms were made by an experienced physician blinded to the genotype status of the patient. The echocardiograms were evaluated according to the recommendations of the American society of Echocardiography. Diastolic measurements of left ventricular (LV) interval dimensions, ventricular septal thickness, and posterior wall were obtained. LV mass was calculated according to the formula: LV mass = 1.04 [(LVID + PWT + IVST)3 − (LVID)3] − 13.6g,28, 29 where LVID is left ventricular internal dimension, PWT is posterior wall thickness, and IVST is interventricular septal dimension. LV mass index (LVMI) was determined by dividing LV mass by body surface area. A LVMI >131 g/m2 defined LVH.30, 31
Response to AAD therapy
This was defined prospectively and blind to genotype as response when there was ≥ 75% reduction in symptomatic AF burden (based on the composite score for frequency, duration and severity of symptoms) on AAD therapy.24 Non-response was defined as <75% reduction in symptomatic AF burden score necessitating a change to another AAD or to non-pharmacologic therapies such as atrio-ventricular node ablation and pacemaker implantation.
Determination of ACE Genotype
ACE genotyping was performed by laboratory personnel who had no knowledge of the drug response data. ACE genotypes were determined by use of polymerase chain reaction, according to previously published protocols.15 In brief, a set of primers was designed to encompass the polymorphic region in intron 16 of the ACE gene (sense primer 5′ CTGGAGACCACTCCCATCCTTTCT 3′ and antisense primer 5′ GATGTGGCCATCACATTCGTCAGAT 3′). DNA was amplified for 35 cycles, each cycle composed of denaturation at 94°C for 30 seconds, annealing at 58°C for 30 seconds, and extension at 72°C for 30 seconds. The products were separated by electrophoresis on 2% agarose gel and identified by ethidium bromide staining. Each sample found to be DD was verified by reamplification with primers hace5a and hace5c, which recognize the inserted sequence, as previously described.32 The amplification detected the presence of the I allele and produced no product from the DNA with DD genotype.
Statistical Analysis
Data are expressed as mean ± SD. To test for Hardy-Weinberg equilibrium, the expected genotype numbers were calculated from the allele frequencies, and deviation from the observed genotype numbers was determined by chi-square test. To analyze differences between D allele carriers and noncarriers, the ID and DD genotypes were pooled into one group and differences were analyzed by Fisher’s exact test. Statistical significance was defined as P<0.05.
RESULTS
Clinical characteristics
A total of 213 (51% of the entire AF Registry population) enrolled over a 30 month period, were eligible for this study. The reasons for exclusion from this analysis included no trial of AAD for maintenance of sinus rhythm i.e., rate control strategy for management of AF, inadequate or incomplete assessment of symptomatic AF burden prior to initiation of therapy and patients in whom AAD therapy had to be discontinued due to adverse effects. The demographic and baseline clinical characteristics of the study population by genotype are listed in Table 2. The mean age of the cohort was 52 ± 15 years. Genotyping classified 30% (n = 63) of patients as homozygotes for I allele, 45% (n = 98) as heterozygotes, and 25% (n = 52) as homozygotes with D allele. The distribution of clinical characteristics was similar between the 3 groups of patients except for slightly more AAD trials and greater prevalence of echocardiographically-defined LVH in ID/DD than II genotype patients. Importantly, the use of beta-blockers or calcium channel blockers was similar across the three genotypes at baseline. The mean symptomatic AF burden score at baseline was 19 ± 4 indicating a high AF burden.
Table 2.
Patient characteristics by genotype.
| Variables | All (n = 213) | II (n = 63) | ID (n = 98) | DD (n = 52) |
|---|---|---|---|---|
| Age (yrs) | 51 ± 15 | 50 ± 15 | 51 ± 14 | 50 ± 14 |
| Duration of AF (yrs) | 3.1 ± 1.3 | 3.0 ± 1.3 | 2.9 ± 1.0 | 3.0 ± 1.2 |
| Male | 147 (69%) | 44(68%) | 67 (65%) | 36 (69%) |
| Lone AF | 72 (34%) | 19 (30%) | 34 (35%) | 19 (36%) |
| AF type (parox/persis) | 170/43 | 52/11 | 76/22 | 42/10 |
| Trial of AADs | 1.47 ± 0.7 | 1.3 ± 0.6* | 1.51 ± 0.7 | 1.55 ± 0.7 |
| LA size (cm) | 4.5 ± 1.2 | 4.6 ± 1.0 | 4.5 ± 1.0 | 4.5 ± 1.0 |
| HTN | 87 (41%) | 30 (48%) | 38 (39%) | 19 (37%) |
| CAD | 44 (21%) | 13 (21%) | 20 (21%) | 11 (21%) |
| Type II diabetes | 18 (8%) | 6 (10%) | 7 (7%) | 5 (10%) |
| CHF | 42 (20%) | 9 (14%) | 23 (23%) | 10 (19) |
| EF (%) | 51 ± 16 | 51 ± 14 | 50 ± 14 | 52 ± 15 |
| Β-blockers/CCB | 104 (49%) | 30 (48%) | 48 (49%) | 26 (50%) |
| ACE inhibitor/ARB treated (%) | 38 (18%) | 9 (14%) | 21 (21%) | 8 (15%) |
| Echo LVH (%)(LVMI > 131 g/m2) | 36 (17%) | 5 (8%)* | 16 (16%) | 15 (29%) |
ACE, angiotensin-converting enzyme; AF, atrial fibrillation; ARB, angiotensin receptor blocker; CAD, coronary artery disease; CCB, calcium-channel blockers; CHF, congestive heart failure; HTN, hypertension; LA, left atrial; LVH, left ventricular hypertrophy; LVMI, left ventricular mass index; paroxy, paroxysmal; persis, persistent.
P<0.05 vs ID/DD.
ACE Genotype
The frequencies of the II, ID and DD genotypes (30%, 45% and 25%) respectively did not deviate significantly from those predicted by Hardy-Weinberg criteria (27%, 50% and 23% respectively; χ2 = 2.63, P>0.1).
Response to AAD therapy
At least one conventional AAD was ineffective in 61 (29%) patients. When drug response was classified according to ACE genotype, not only was drug failure rate greater in patients with the D allele but also there was graded gene dose effect with ID genotype patients demonstrating an intermediate response to AADs (Figure 1). This association was even more marked in patients with lone AF suggesting that response to conventional AADs may be modulated by the ACE I/D polymorphism (Figure 2). Furthermore, the number of AAD drug trials in the II, ID and DD genotype patients was similar (62, 68 and 68 respectively).
Figure 1.
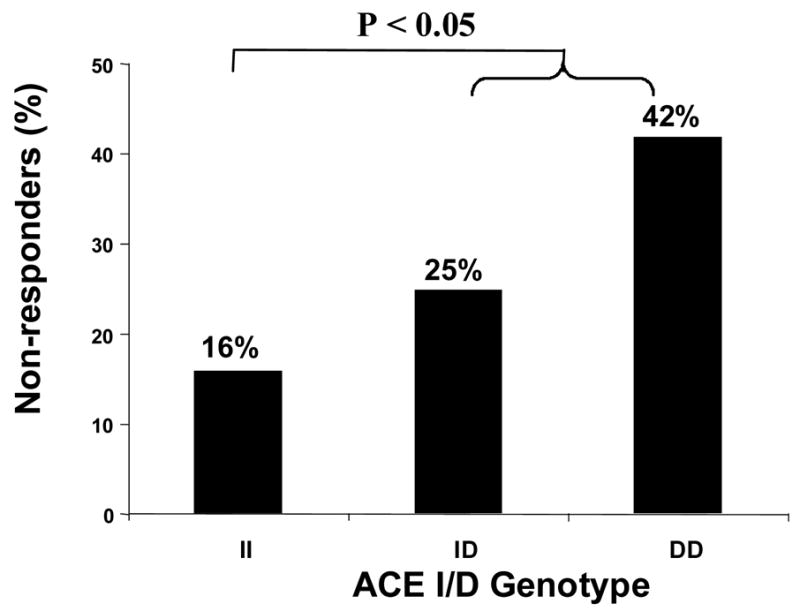
Response to conventional antiarrhythmic drugs (AADs) in patients with atrial fibrillation (AF) according to angiotensin-converting enzyme (ACE) I/D genotype.
Figure 2.
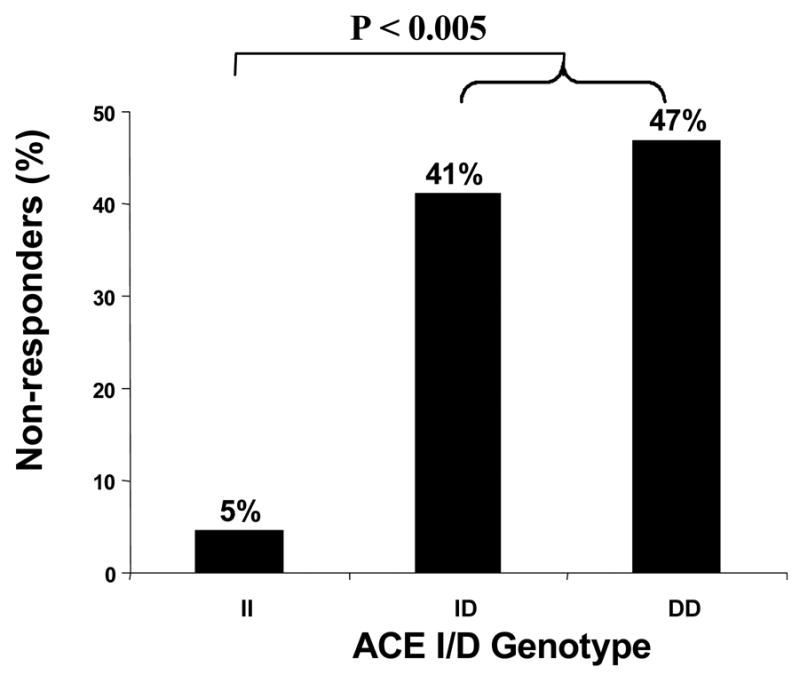
Response to conventional AADs in patients with lone AF according to ACE I/D genotype.
There also appeared to be a differential response to the class of AADs i.e., patients with the D allele had a poorer response to class IA, IC and III drugs than patients with I allele (Table 3). Response to amiodarone therapy did not appear to be modulated by ACE genotype. The effect of ACE inhibitor/ARB therapy on response to AADs could not be evaluated due to limited number of patients in each of the I/D genotypes.
Table 3.
Drug failure according to class of antiarrhythmic drugs.
| II (n = 11) | ID (n = 26) | DD (n = 24) | |
|---|---|---|---|
| Class IA | 2/8 (25%) | 5/13 (38%) | 4/9 (44%) |
| Class IC | 4/24 (17%)* | 4/9 (44%) | 11/21 (52%) |
| Class III | 4/23 (17%)* | 8/17 (47%) | 4/8 (50%) |
| Amiodarone | 1/7 (14%) | 9/29 (31%) | 5/30 (17%) |
Note: Denominators are greater than # of patients who failed each class of drug since many patients were tried on more than one AAD agent.
P<0.05 vs ID/DD genotype patients.
DISCUSSION
This study demonstrates a pharmacogenetic interaction between the ACE I/D polymorphism and efficacy of AAD therapy in patients with lone AF. Further, it provides additional evidence for the increasingly important role of RAAS activation in the pathophysiology of AF. Of the 3–5 million Americans with AF,33 it is estimated that ~750,000 (10–15%) have lone AF.34 This means that approximately 450,000 patients with lone AF will have the ACE DD genotype.
These findings suggest that this genetic subset not only have poor response to conventional AADs but raises the possibility that these subjects may also especially benefit from ACE inhibition to increase antiarrhythmic drug response. Thus, determination of ACE genotype may help target therapy for patients with lone AF.
Subjects with one or two D alleles of the ACE I/D polymorphism have approximately 25% and 50% higher ACE levels than subjects with the II genotype. These higher ACE levels are observed both in plasma14 and tissue sites.21 Although, evidence for increased conversion of angiotensin I to angiotensin II is limited, no data are available as far as tissue angiotensin II levels are concerned.35 It is possible that at tissue sites such as the atria, enhanced formation of angiotensin II may have the strongest functional implications and patients with the DD genotype may have more structural alterations than patients with ID or II genotypes. Although in our cohort, there appeared to be no significance difference in left atrial size between the genotypes, it is possible that more comprehensive evaluation of atrial size and function (such as diastolic function and left atrial volumes [not available in our subjects]), may provide evidence of structural abnormalities in patients with the DD genotype.
In prior population-based studies, lone AF was present in approximately 2% to 11% of the population.27, 34 Referral bias is one likely reason why a disproportionate number of patients with lone AF were evaluated in our center (34%). As a genetic registry, there is a bias towards enrollment of younger subjects with AF rather than elderly subjects with limited life expectancy. This may have also contributed to a greater proportion of patients having lone AF in our cohort than in other population based studies.
There is increasing evidence suggesting a role for the renin-angiotensin system in the pathophysiology of AF. Angiotensin II has been shown to increase atrial pressure and stretch, and through atrial enlargement and electrophysiologic remodeling, promotes AF.36, 37
Angiotensin II is also a potent promoter of fibrosis, leading to cardiac myoblast proliferation and reduced collagenase activity.38, 39 On biopsy, atria from patients with refractory lone AF exhibit inflammatory infiltrates, myocytes necrosis and fibrosis.40 The presence of atrial fibrosis may explain not only intraatrial conduction disturbances and the persistent susceptibility for AF in these patients but also, reduced response to conventional AADs in patients with the D allele.41 Increased ACE expression and alterations in the angiotensin II receptor expression have also been observed in the atria of AF patients. Furthermore, an ACE inhibition-dependent increase in levels of activated extracellular signal-regulated kinases Erk1/Erk2 and Erk-activating kinases MEK1/2 was found in patients with AF by examining atrial tissue samples obtained during open-heart surgery.42, 43 Additional support for the role of increased RAAS activation in the pathophysiology of AF comes from data which show that the ACE inhibitor enalapril has been shown to decrease mitogen-activated protein kinase activation, atrial fibrosis, and AF promotion in a dog model of heart failure.44 Enalapril was also shown to decrease atrial structural and functional remodeling in the same model.45 In addition, the angiotensin II receptor antagonist candesartan was reported to prevent the promotion of AF by suppressing the development of structural remodeling (interstitial fibrosis) in a dog rapid pacing model.46
Data from our study suggests that there is a differential response to the class of AAD therapy. Subjects with ID or DD genotypes had a poorer response to class IA, IC and III drugs than patients with II genotype. Response to amiodarone, whose pharmacologic actions not only block cardiac potassium channels but also sodium channels and a multitude of other targets, was not modulated by the ACE genotype. However, the number of patients in each class of AAD was limited. Consequently, these results are only hypothesis generating and will require confirmation in a larger cohort of patients with symptomatic AF.
In our study, we found that patients with II genotype had at least as much hypertension as seen with the other genotypes, yet had markedly less evidence of LVH. Previous studies relating ACE genotype to LV mass have been conflicting, possibly due to small size47, 48 and to the confounding of cross-sectional population studies by deaths attributable to ACE genotype associated disease, including LVH itself.49, 50 Importantly however, there is increasing evidence that activation of the RAAS influences myocardial growth independently of hypertension51, 52 and a recent study showed an association between the ACE I/D polymorphism and LVH independent of hypertension.53 Thus, our observation that LVH was less common in the II genotype patients, is consistent with the known association of ACE I/D with LVH. Furthermore, it may also provide some insight into why ACE I/D genotype predicted response to AAD therapy in our patients.
Study limitations
Although this was a retrospective analysis, patients were enrolled prospectively and more importantly, response to AAD therapy was defined a priori without knowledge of the ACE genotype. Another limitation relates to the fact that the decision to switch from one AAD to another was left to the discretion of the attending physician and was not determined by the symptomatic AF burden score. Nevertheless, change in drug therapy did correlate with either unchanged or <25% reduction in the symptomatic AF burden score when compared to baseline. However, this does highlight the importance of not only validating our findings in an independent cohort of patients but doing so in an appropriately designed randomized pharmacogenetic trial with valid endpoints.
A conventional AF burden score usually assesses the frequency and duration of AF. However, we incorporated severity of symptoms as an equally weighted measure of symptomatic AF burden because studies from the Canadian Trials of Atrial Fibrillation (CTAF) have shown that measures of subjective well-being, in addition, to conventional measures of severity of AF are important when assessing the therapeutic efficacy of treatments for AF.26
Measurement of symptomatic AF burden requires that patients keep records of when symptoms start and end and symptom severity, and thereby is prone to sampling error. However, symptomatic AF burden is not only easy to measure but a sub-study of the CTAF showed that it correlated with different rates of arrhythmia recurrence in AF trials.54 Furthermore, we and others have used symptomatic AF burden as an endpoint to assess response to AAD therapy after catheter ablation of AF.55, 56 The ACC/AHA Clinical Data Standards for AF published in 2004 have also advocated evaluating symptomatic AF burden using a similar scoring system based on frequency, duration and severity of symptoms during AF.57
Although asymptomatic episodes of AF are not measured when evaluating symptomatic burden, AADs may decrease the occurrence of asymptomatic AF.58 Furthermore, it is possible that negative dromotropic agents such as beta-blockers and calcium-channel blockers, may influence the subjective experience of AF symptoms and thereby could favorably affect the symptomatic AF burden score. However, at baseline the proportion of patients receiving these agents was similar across the three genotypes arguing against this bias.
In conclusion, our data do support the hypothesis that the ACE I/D polymorphism modulates response to AAD therapy in patients with lone AF in a manner consistent with the known effect of the D allele on ACE activity. However, this hypothesis can only be truly evaluated in the context of a clinical trial, and these findings should be confirmed in randomized studies before tailoring AAD therapy for AF based on ACE genotype. The results of this study strongly support the need for randomized pharmacogenetic trials and suggest that genetic assessment of therapeutic efficacy should be incorporated into all future trials of AAD therapy for AF. If the current findings are confirmed, one-third of all lone AF patients with the DD genotype could be selected for aggressive neurohormonal therapy. Further investigations will hopefully clarify the role of ACE genotyping in the clinical management of patient with lone AF.
Acknowledgments
This work was supported by NIH awards HL075266 to DD and HL65962 to DMR.
Abbreviation List
- AAD
antiarrhythmic drug
- ACE
angiotensin-converting enzyme
- AF
atrial fibrillation
- LV
left ventricular
- RAAS
renin-angiotensin aldosterone system
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nattel S. New ideas about atrial fibrillation 50 years on. Nature. 2002;415(6868):219–226. doi: 10.1038/415219a. [DOI] [PubMed] [Google Scholar]
- 2.Brugada R, Tapscott T, Czernuszewicz GZ, Marian AJ, Iglesias A, Mont L, Brugada J, Girona J, Domingo A, Bachinski LL, Roberts R. Identification of a genetic locus for familial atrial fibrillation. N Engl J Med. 1997;336(13):905–911. doi: 10.1056/NEJM199703273361302. [DOI] [PubMed] [Google Scholar]
- 3.Chen YH, Xu SJ, Bendahhou S, Wang XL, Wang Y, Xu WY, Jin HW, Sun H, Su XY, Zhuang QN, Yang YQ, Li YB, Liu Y, Xu HJ, Li XF, Ma N, Mou CP, Chen Z, Barhanin J, Huang W. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. 2003;299(5604):251–254. doi: 10.1126/science.1077771. [DOI] [PubMed] [Google Scholar]
- 4.Ellinor PT, Shin JT, Moore RK, Yoerger DM, MacRae CA. Locus for atrial fibrillation maps to chromosome 6q14–16. Circulation. 2003;107(23):2880–2883. doi: 10.1161/01.CIR.0000077910.80718.49. [DOI] [PubMed] [Google Scholar]
- 5.Darbar D, Herron KJ, Ballew JD, Jahangir A, Gersh BJ, Shen WK, Hammill SC, Packer DL, Olson TM. Familial atrial fibrillation is a genetically heterogeneous disorder. J Am Coll Cardiol. 2003;41(12):2185–2192. doi: 10.1016/s0735-1097(03)00465-0. [DOI] [PubMed] [Google Scholar]
- 6.Fox CS, Parise H, D’Agostino RB, Sr, Lloyd-Jones DM, Vasan RS, Wang TJ, Levy D, Wolf PA, Benjamin EJ. Parental atrial fibrillation as a risk factor for atrial fibrillation in offspring. JAMA. 2004;291(23):2851–2855. doi: 10.1001/jama.291.23.2851. [DOI] [PubMed] [Google Scholar]
- 7.Yang Y, Xia M, Jin Q, Bendahhou S, Shi J, Chen Y, Liang B, Lin J, Liu Y, Liu B, Zhou Q, Zhang D, Wang R, Ma N, Su X, Niu K, Pei Y, Xu W, Chen Z, Wan H, Cui J, Barhanin J. Identification of a KCNE2 gain-of-function mutation in patients with familial atrial fibrillation. Am J Hum Genet. 2004;75(5):899–905. doi: 10.1086/425342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carnes CA, Chung MK, Nakayama T, Nakayama H, Baliga RS, Piao S, Kanderian A, Pavia S, Hamlin RL, McCarthy PM, Bauer JA, Van Wagoner DR. Ascorbate attenuates atrial pacing-induced peroxynitrite formation and electrical remodeling and decreases the incidence of postoperative atrial fibrillation. Circ Res. 2001;89(6):E32–38. doi: 10.1161/hh1801.097644. [DOI] [PubMed] [Google Scholar]
- 9.Chung MK, Martin DO, Sprecher D, Wazni O, Kanderian A, Carnes CA, Bauer JA, Tchou PJ, Niebauer MJ, Natale A, Van Wagoner DR. C-reactive protein elevation in patients with atrial arrhythmias: inflammatory mechanisms and persistence of atrial fibrillation. Circulation. 2001;104(24):2886–2891. doi: 10.1161/hc4901.101760. [DOI] [PubMed] [Google Scholar]
- 10.Dernellis J, Panaretou M. C-reactive protein and paroxysmal atrial fibrillation: evidence of the implication of an inflammatory process in paroxysmal atrial fibrillation. Acta Cardiol. 2001;56(6):375–380. doi: 10.2143/AC.56.6.2005701. [DOI] [PubMed] [Google Scholar]
- 11.Dernellis J, Panaretou M. Relationship between C-reactive protein concentrations during glucocorticoid therapy and recurrent atrial fibrillation. Eur Heart J. 2004;25(13):1100–1107. doi: 10.1016/j.ehj.2004.04.025. [DOI] [PubMed] [Google Scholar]
- 12.Madrid AH, Bueno MG, Rebollo JM, Marin I, Pena G, Bernal E, Rodriguez A, Cano L, Cano JM, Cabeza P, Moro C. Use of irbesartan to maintain sinus rhythm in patients with long-lasting persistent atrial fibrillation: a prospective and randomized study. Circulation. 2002;106(3):331–336. doi: 10.1161/01.cir.0000022665.18619.83. [DOI] [PubMed] [Google Scholar]
- 13.Tsai CT, Lai LP, Lin JL, Chiang FT, Hwang JJ, Ritchie MD, Moore JH, Hsu KL, Tseng CD, Liau CS, Tseng YZ. Renin-angiotensin system gene polymorphisms and atrial fibrillation. Circulation. 2004;109(13):1640–1646. doi: 10.1161/01.CIR.0000124487.36586.26. [DOI] [PubMed] [Google Scholar]
- 14.Rigat B, Hubert C, Alhenc-Gelas F, Cambien F, Corvol P, Soubrier F. An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J Clin Invest. 1990;86(4):1343–1346. doi: 10.1172/JCI114844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rigat B, Hubert C, Corvol P, Soubrier F. PCR detection of the insertion/deletion polymorphism of the human angiotensin converting enzyme gene (DCP1) (dipeptidyl carboxypeptidase 1) Nucleic Acids Res. 1992;20(6):1433. doi: 10.1093/nar/20.6.1433-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Samani NJ, Thompson JR, O’Toole L, Channer K, Woods KL. A meta-analysis of the association of the deletion allele of the angiotensin-converting enzyme gene with myocardial infarction. Circulation. 1996;94(4):708–712. doi: 10.1161/01.cir.94.4.708. [DOI] [PubMed] [Google Scholar]
- 17.Raynolds MV, Bristow MR, Bush EW, Abraham WT, Lowes BD, Zisman LS, Taft CS, Perryman MB. Angiotensin-converting enzyme DD genotype in patients with ischaemic or idiopathic dilated cardiomyopathy. Lancet. 1993;342(8879):1073–1075. doi: 10.1016/0140-6736(93)92061-w. [DOI] [PubMed] [Google Scholar]
- 18.Marian AJ, Yu QT, Workman R, Greve G, Roberts R. Angiotensin-converting enzyme polymorphism in hypertrophic cardiomyopathy and sudden cardiac death. Lancet. 1993;342(8879):1085–1086. doi: 10.1016/0140-6736(93)92064-z. [DOI] [PubMed] [Google Scholar]
- 19.McNamara DM, Holubkov R, Janosko K, Palmer A, Wang JJ, MacGowan GA, Murali S, Rosenblum WD, London B, Feldman AM. Pharmacogenetic interactions between beta-blocker therapy and the angiotensin-converting enzyme deletion polymorphism in patients with congestive heart failure. Circulation. 2001;103(12):1644–1648. doi: 10.1161/01.cir.103.12.1644. [DOI] [PubMed] [Google Scholar]
- 20.McNamara DM, Holubkov R, Postava L, Janosko K, MacGowan GA, Mathier M, Murali S, Feldman AM, London B. Pharmacogenetic interactions between angiotensin-converting enzyme inhibitor therapy and the angiotensin-converting enzyme deletion polymorphism in patients with congestive heart failure. J Am Coll Cardiol. 2004;44(10):2019–2026. doi: 10.1016/j.jacc.2004.08.048. [DOI] [PubMed] [Google Scholar]
- 21.Danser AH, Schalekamp MA, Bax WA, van den Brink AM, Saxena PR, Riegger GA, Schunkert H. Angiotensin-converting enzyme in the human heart. Effect of the deletion/insertion polymorphism. Circulation. 1995;92(6):1387–1388. doi: 10.1161/01.cir.92.6.1387. [DOI] [PubMed] [Google Scholar]
- 22.Mizuiri S, Hemmi H, Kumanomidou H, Iwamoto M, Miyagi M, Sakai K, Aikawa A, Ohara T, Yamada K, Shimatake H, Hasegawa A. Angiotensin-converting enzyme (ACE) I/D genotype and renal ACE gene expression. Kidney Int. 2001;60(3):1124–1130. doi: 10.1046/j.1523-1755.2001.0600031124.x. [DOI] [PubMed] [Google Scholar]
- 23.Sun Y, Weber KT. Angiotensin II and aldosterone receptor binding in rat heart and kidney: response to chronic angiotensin II or aldosterone administration. J Lab Clin Med. 1993;122(4):404–411. [PubMed] [Google Scholar]
- 24.Darbar D, Roden DM. Symptomatic burden as an endpoint to evaluate interventions in patients with atrial fibrillation. Heart Rhythm. 2005;2:544–549. doi: 10.1016/j.hrthm.2005.01.028. [DOI] [PubMed] [Google Scholar]
- 25.Dorian PJW, Paquette M, Newman D, Wood K, Ayers GM, Camm J, Akhtar M, Luderitz B. The impairment of health-related quality of life in patients with intermittent atrial fibrillation: implications for the assessment of investigational therapy. J Am Coll Cardiol. 2000;36:1303–1309. doi: 10.1016/s0735-1097(00)00886-x. [DOI] [PubMed] [Google Scholar]
- 26.Dorian P, Paquette M, Newman D, Green M, Connolly SJ, Talajic M, Roy D. Quality of life improves with treatment in the Canadian Trial of Atrial Fibrillation. Am Heart J. 2002;143(6):984–990. doi: 10.1067/mhj.2002.122518. [DOI] [PubMed] [Google Scholar]
- 27.Kopecky SL, Gersh BJ, McGoon MD, Whisnant JP, Holmes DR, Jr, Ilstrup DM, Frye RL. The natural history of lone atrial fibrillation. A population-based study over three decades. N Engl J Med. 1987;317(11):669–674. doi: 10.1056/NEJM198709103171104. [DOI] [PubMed] [Google Scholar]
- 28.Devereux RB, Reichek N. Echocardiographic determination of left ventricular mass in man. Anatomic validation of the method. Circulation. 1977;55(4):613–618. doi: 10.1161/01.cir.55.4.613. [DOI] [PubMed] [Google Scholar]
- 29.Devereux RB, Alonso DR, Lutas EM, Gottlieb GJ, Campo E, Sachs I, Reichek N. Echocardiographic assessment of left ventricular hypertrophy: comparison to necropsy findings. Am J Cardiol. 1986;57(6):450–458. doi: 10.1016/0002-9149(86)90771-x. [DOI] [PubMed] [Google Scholar]
- 30.Levy D, Savage DD, Garrison RJ, Anderson KM, Kannel WB, Castelli WP. Echocardiographic criteria for left ventricular hypertrophy: the Framingham Heart Study. Am J Cardiol. 1987;59(9):956–960. doi: 10.1016/0002-9149(87)91133-7. [DOI] [PubMed] [Google Scholar]
- 31.Houghton JL, Frank MJ, Carr AA, von Dohlen TW, Prisant LM. Relations among impaired coronary flow reserve, left ventricular hypertrophy and thallium perfusion defects in hypertensive patients without obstructive coronary artery disease. J Am Coll Cardiol. 1990;15(1):43–51. doi: 10.1016/0735-1097(90)90173-m. [DOI] [PubMed] [Google Scholar]
- 32.Lindpaintner K, Pfeffer MA, Kreutz R, Stampfer MJ, Grodstein F, LaMotte F, Buring J, Hennekens CH. A prospective evaluation of an angiotensin-converting-enzyme gene polymorphism and the risk of ischemic heart disease. N Engl J Med. 1995;332(11):706–711. doi: 10.1056/NEJM199503163321103. [DOI] [PubMed] [Google Scholar]
- 33.Feinberg WM, Blackshear JL, Laupacis A, Kronmal R, Hart RG. Prevalence, age distribution, and gender of patients with atrial fibrillation. Analysis and implications. Arch Intern Med. 1995;155(5):469–473. [PubMed] [Google Scholar]
- 34.Brand FN, Abbott RD, Kannel WB, Wolf PA. Characteristics and prognosis of lone atrial fibrillation. 30-year follow-up in the Framingham Study. JAMA. 1985;254(24):3449–3453. [PubMed] [Google Scholar]
- 35.Ueda S, Meredith PA, Morton JJ, Connell JM, Elliott HL. ACE (I/D) genotype as a predictor of the magnitude and duration of the response to an ACE inhibitor drug (enalaprilat) in humans. Circulation. 1998;98(20):2148–2153. doi: 10.1161/01.cir.98.20.2148. [DOI] [PubMed] [Google Scholar]
- 36.Ravelli F, Allessie M. Effects of atrial dilatation on refractory period and vulnerability to atrial fibrillation in the isolated Langendorff-perfused rabbit heart. Circulation. 1997;96(5):1686–1695. doi: 10.1161/01.cir.96.5.1686. [DOI] [PubMed] [Google Scholar]
- 37.Nakashima H, Kumagai K, Urata H, Gondo N, Ideishi M, Arakawa K. Angiotensin II antagonist prevents electrical remodeling in atrial fibrillation. Circulation. 2000;101(22):2612–2617. doi: 10.1161/01.cir.101.22.2612. [DOI] [PubMed] [Google Scholar]
- 38.Pages G, Lenormand P, L’Allemain G, Chambard JC, Meloche S, Pouyssegur J. Mitogen-activated protein kinases p42mapk and p44mapk are required for fibroblast proliferation. Proc Natl Acad Sci U S A. 1993;90(18):8319–8323. doi: 10.1073/pnas.90.18.8319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brilla CG, Zhou G, Matsubara L, Weber KT. Collagen metabolism in cultured adult rat cardiac fibroblasts: response to angiotensin II and aldosterone. J Mol Cell Cardiol. 1994;26(7):809–820. doi: 10.1006/jmcc.1994.1098. [DOI] [PubMed] [Google Scholar]
- 40.Frustaci A, Chimenti C, Bellocci F, Morgante E, Russo MA, Maseri A. Histological substrate of atrial biopsies in patients with lone atrial fibrillation. Circulation. 1997;96(4):1180–1184. doi: 10.1161/01.cir.96.4.1180. [DOI] [PubMed] [Google Scholar]
- 41.Everett THt, Li H, Mangrum JM, McRury ID, Mitchell MA, Redick JA, Haines DE. Electrical, morphological, and ultrastructural remodeling and reverse remodeling in a canine model of chronic atrial fibrillation. Circulation. 2000;102(12):1454–1460. doi: 10.1161/01.cir.102.12.1454. [DOI] [PubMed] [Google Scholar]
- 42.Goette A, Arndt M, Rocken C, Spiess A, Staack T, Geller JC, Huth C, Ansorge S, Klein HU, Lendeckel U. Regulation of angiotensin II receptor subtypes during atrial fibrillation in humans. Circulation. 2000;101(23):2678–2681. doi: 10.1161/01.cir.101.23.2678. [DOI] [PubMed] [Google Scholar]
- 43.Goette A, Staack T, Rocken C, Arndt M, Geller JC, Huth C, Ansorge S, Klein HU, Lendeckel U. Increased expression of extracellular signal-regulated kinase and angiotensin-converting enzyme in human atria during atrial fibrillation. J Am Coll Cardiol. 2000;35(6):1669–1677. doi: 10.1016/s0735-1097(00)00611-2. [DOI] [PubMed] [Google Scholar]
- 44.Li D, Shinagawa K, Pang L, Leung TK, Cardin S, Wang Z, Nattel S. Effects of angiotensin-converting enzyme inhibition on the development of the atrial fibrillation substrate in dogs with ventricular tachypacing-induced congestive heart failure. Circulation. 2001;104(21):2608–2614. doi: 10.1161/hc4601.099402. [DOI] [PubMed] [Google Scholar]
- 45.Shi Y, Li D, Tardif JC, Nattel S. Enalapril effects on atrial remodeling and atrial fibrillation in experimental congestive heart failure. Cardiovasc Res. 2002;54(2):456–461. doi: 10.1016/s0008-6363(02)00243-2. [DOI] [PubMed] [Google Scholar]
- 46.Kumagai K, Nakashima H, Urata H, Gondo N, Arakawa K, Saku K. Effects of angiotensin II type 1 receptor antagonist on electrical and structural remodeling in atrial fibrillation. J Am Coll Cardiol. 2003;41(12):2197–2204. doi: 10.1016/s0735-1097(03)00464-9. [DOI] [PubMed] [Google Scholar]
- 47.Gharavi AG, Lipkowitz MS, Diamond JA, Jhang JS, Phillips RA. Deletion polymorphism of the angiotensin-converting enzyme gene is independently associated with left ventricular mass and geometric remodeling in systemic hypertension. Am J Cardiol. 1996;77(15):1315–1319. doi: 10.1016/s0002-9149(96)00198-1. [DOI] [PubMed] [Google Scholar]
- 48.Kupari M, Perola M, Koskinen P, Virolainen J, Karhunen PJ. Left ventricular size, mass, and function in relation to angiotensin-converting enzyme gene polymorphism in humans. Am J Physiol. 1994;267(3 Pt 2):H1107–1111. doi: 10.1152/ajpheart.1994.267.3.H1107. [DOI] [PubMed] [Google Scholar]
- 49.Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322(22):1561–1566. doi: 10.1056/NEJM199005313222203. [DOI] [PubMed] [Google Scholar]
- 50.Cambien F, Poirier O, Lecerf L, Evans A, Cambou JP, Arveiler D, Luc G, Bard JM, Bara L, Ricard S, et al. Deletion polymorphism in the gene for angiotensin-converting enzyme is a potent risk factor for myocardial infarction. Nature. 1992;359(6396):641–644. doi: 10.1038/359641a0. [DOI] [PubMed] [Google Scholar]
- 51.Iwashima Y, Horio T, Kuroda S, Takishita S, Kawano Y. Influence of plasma aldosterone on left ventricular geometry and diastolic function in treated essential hypertension. Hypertens Res. 2002;25(1):49–56. doi: 10.1291/hypres.25.49. [DOI] [PubMed] [Google Scholar]
- 52.Miller JA, Scholey JW. The impact of renin-angiotensin system polymorphisms on physiological and pathophysiological processes in humans. Curr Opin Nephrol Hypertens. 2004;13(1):101–106. doi: 10.1097/00041552-200401000-00014. [DOI] [PubMed] [Google Scholar]
- 53.Saeed M, Saleheen D, Siddiqui S, Khan A, Butt ZA, Frossard PM. Association of angiotensin converting enzyme gene polymorphisms with left ventricular hypertrophy. Hypertens Res. 2005;28(4):345–349. doi: 10.1291/hypres.28.345. [DOI] [PubMed] [Google Scholar]
- 54.Dorian P, Mangat I. Quality of life variables in the selection of rate versus rhythm control in patients with atrial fibrillation: observations from the Canadian Trial of Atrial Fibrillation. Card Electrophysiol Rev. 2003;7(3):276–279. doi: 10.1023/B:CEPR.0000012395.33292.cd. [DOI] [PubMed] [Google Scholar]
- 55.Darbar D, Munger TM, Hammill SC, Packer DL. Differential response to antiarrhythmic drug therapy in focal-triggered substrate mediated atrial fibrillation following failed ablation. PACE. 2001;24:657–658. [Google Scholar]
- 56.Oral H, Knight BP, Tada H, Ozaydin M, Chugh A, Hassan S, Scharf C, Lai SW, Greenstein R, Pelosi F, Jr, Strickberger SA, Morady F. Pulmonary vein isolation for paroxysmal and persistent atrial fibrillation. Circulation. 2002;105(9):1077–1081. doi: 10.1161/hc0902.104712. [DOI] [PubMed] [Google Scholar]
- 57.McNamara RL, Brass LM, Drozda JP, Jr, Go AS, Halperin JL, Kerr CR, Levy S, Malenka DJ, Mittal S, Pelosi F, Jr, Rosenberg Y, Stryer D, Wyse DG, Radford MJ, Goff DC, Jr, Grover FL, Heidenreich PA, Peterson ED, Redberg RF. ACC/AHA key data elements and definitions for measuring the clinical management and outcomes of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Data Standards (Writing Committee to Develop Data Standards on Atrial Fibrillation) Circulation. 2004;109(25):3223–3243. doi: 10.1161/01.CIR.0000131893.41821.D1. [DOI] [PubMed] [Google Scholar]
- 58.Page RL, Tilsch TW, Connolly SJ, Schnell DJ, Marcello SR, Wilkinson WE, Pritchett EL. Asymptomatic or “silent” atrial fibrillation: frequency in untreated patients and patients receiving azimilide. Circulation. 2003;107(8):1141–1145. doi: 10.1161/01.cir.0000051455.44919.73. [DOI] [PubMed] [Google Scholar]