

Abstract
Emerging evidence indicates that neuroinflammatory responses in astroglia, including chemokine expression, are altered by opioids. Astroglial chemokines, such as CXCL10, are instrumental in response to many neuropathological insults. Opioid mediated disruption of astroglial CXCL10 expression may be detrimental in opioid abusers or patients receiving acute opioid therapy. We have characterized the in vitro effects of opioids on CXCL10 protein expression in human astroglial (A172) cells. The proinflammatory cytokine, tumor necrosis factor (TNF)α induced CXCL10 expression in A172 cells. Using MG-132, helenalin and SN50 [inhibitors of the transcription factor, nuclear factor (NF)-κB], we determined that NF-κB activation is instrumental in TNFα induced CXCL10 expression in A172 astroglia. Morphine exposure during the 24 h TNFα stimulation period did not alter CXCL10 expression. However, fentanyl, a more potent mu opioid receptor (MOR) agonist, inhibited TNFα induced CXCL10 expression. Interestingly, neither the nonselective opioid receptor antagonist, naltrexone nor β-funaltrexamine (β-FNA), a highly selective MOR antagonist, blocked fentanyl mediated inhibition of TNFα induced CXCL10 expression. Rather, β-FNA dose dependently inhibited TNFα induced CXCL10 expression with a greater potency than that observed for fentanyl. Immunoblot analysis indicated that morphine, fentanyl and β-FNA each reduced TNFα induced nuclear translocation of NF-κB p65. These data show that β-FNA and fentanyl inhibit TNFα induced CXCL10 expression via a MOR independent mechanism. Data also suggest that inhibition of TNFα induced CXCL10 expression by fentanyl and β-FNA is not directly related to a reduction in NF-κB p65 nuclear translocation. Further investigation is necessary in order to fully elucidate the mechanism through which these two opioid compounds inhibit CXCL10 expression. Understanding the mechanism by which chemokine expression is suppressed, particularly by the opioid antagonist, β-FNA, may provide insights into the development of safe and effective treatments for neuroinflammation.
Keywords: astrocyte, brain injury, mu opioid receptor, neuroinflammation, tumor necrosis factor α
1. Introduction
Increasing evidence indicates that inflammatory mediators are involved in the neuropathogenesis associated with neurodegenerative diseases (Kadiu et al., 2005), viral infections (Poluektova et al., 2005; Toborek et al., 2005), ischemic stroke (Wang et al., 2004), trauma (Vlodavsky et al., 2006) and neuropathic pain (Myers et al., 2006). Importantly, chemokines have emerged as key molecules involved in neuropathological events and depending on the cellular context can either be neurotoxic or neuroprotective. In particular, CNS levels of the chemokine CXCL10 (formerly referred to as interferon-γ inducible protein or IP-10) are elevated in Alzheimer’s disease (Xia et al., 2000), HIV dementia (Cinque et al., 2005; Kolb et al., 1999), ischemic stroke (Wang et al., 1998; Wang et al., 2000) and following spinal cord injury (Gonzalez et al., 2003). We are primarily interested in CXCL10, which is a member of the CXC or α-chemokine family, all of which have four highly conserved cysteine residues with the first two cysteines separated by a single amino acid (Bajetto et al., 2002; Luster et al., 1985). CXCL10 can also be subclassified as ELR-negative given that it does not contain a conserved tripeptide motif, glutamate-leucine-arginine (ELR) at the N-terminus, before the CXC domain (Belperio et al., 2000). Importantly, chemokines are small secreted proteins that function in both physiological and pathological conditions. CXCL10 is well characterized as a chemoattractant for activated T cells (Taub et al., 1993), monocytes/macrophages (Taub et al., 1993), and microglia (Flynn et al., 2003). CXCL10 is also a potent angiostatic factor (Belperio et al., 2000) and induces astroglial proliferation (Flynn et al., 2003).
Astroglia appear to be a major source of CXCL10 in many neuropathologies. For instance, compared to controls, CXCL10 protein expression was markedly increased in a subpopulation of astrocytes from Alzheimer’s disease brains (Xia et al., 2000). Astroglial expression of CXCL10 has also been observed in ischemic stroke. For example, after occlusion of the middle cerebral artery in rat, CXCL10 mRNA expression in cortical tissues peaked 6 h after occlusion, and a second induction of CXCL10 was noted from 10–15 d post-occlusion (Wang et al., 1998). Immunohistochemical analysis of the ischemic cortex indicated CXCL10 protein predominated in the astrocytes of the cortical, striatal and white matter regions surrounding the lesions, as indicated by co-localization of CXCL10 and glial fibrillary acidic protein (Wang et al., 1998).
In recent years, numerous investigators have focused on neutralization of CXCL10 as a therapeutic strategy for decreasing inflammatory-mediated neuropathogenesis (Glaser et al., 2004; Sorensen, 2004). For instance, in a murine model of spinal cord injury (SCI), anti-CXCL10 antibody increased angiogenesis and reduced SCI-induced tissue damage (Glaser et al., 2004). Others have utilized a broad spectrum chemokine inhibitor (NR58-3.14.3) to provide neuroprotection in a rat model of cerebral ischemia-reperfusion injury (Beech et al., 2001). While not specifically targeted to CXCL10, another intriguing strategy that has been employed to attenuate inflammation-mediated neuropathogenesis is treatment with naloxone (Liao et al., 2003; Liu et al., 2000; Liu and Hong, 2003; Liu et al., 2002). Naloxone is well characterized as a non-selective opioid receptor antagonist; however, it has been demonstrated that naloxone reduces neuroinflammation via mechanisms that do not require binding to opioid receptors, including prevention of bacterial lipopolysaccharide-binding to microglia (Liu et al., 2000) and reduced microglial superoxide production (Liu et al., 2002).
The mu-opioid receptor (MOR) agonist, morphine, has both neuroprotective (Peart et al., 2005; Rambhia et al., 2005) and neurotoxic (El-Hage et al., 2005; Khurdayan et al., 2004) properties. While the mechanism for the neuroprotective effects of morphine are not completely understood, there is evidence to suggest that under certain conditions, morphine attenuates oxidative and proinflammatory stress (Lee et al., 2004; Rambhia et al., 2005). In contrast, opioids exacerbate HIV neuropathogenesis, in part by enhancing astroglial release of proinflammatory mediators including chemokines (El-Hage et al., 2005). That is, morphine potentiates HIV-1 Tat-induced chemokine expression in murine astrocytes and attenuation of the morphine effects with the MOR specific antagonist β-funaltrexamine (β-FNA) indicates a MOR dependent mechanism. More recently, these same investigators reported that morphine enhances MOR expression and potentiates HIV-1 Tat-induced chemokine production (El-Hage et al., 2006).
While the above studies demonstrate the role of opioid agents in neuroinflammation, herein, we provide the first characterization of opioid effects on CXCL10 expression in human astroglial cells. More specifically, we have demonstrated that TNFα induced CXCL10 protein expression in human astroglial cells is dose dependently inhibited by two different opioid compounds, fentanyl and β-FNA. Of particular interest is that while typically described as a MOR agonist and MOR antagonist, respectively, fentanyl and β-FNA each inhibit TNFα induced CXCL10 expression through a MOR-independent mechanism.
2. Materials and methods
2.1. Cell culture
The human astrocytoma cell line (A172, ATCC #CRL-1620; American Type Culture Collection, Manassas, VA) was utilized in various pharmacology- and neurochemistry-based studies (Guthikonda et al., 1998; Kubota et al., 2001; Zaheer et al., 1995). Additionally, we have previously characterized proinflammatory pathways in A172 cells (Davis et al., 2002; Davis and Syapin, 2004a; Davis and Syapin, 2004b). A172 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 2 mM L-glutamine, 10% fetal bovine serum, 1% nonessential amino acids, 50 U/ml penicillin, 0.05 mg/ml streptomycin and 2 μg/ml amphotericin B. Cultures were maintained in a humidified incubator at 37°C, 5% CO2 and 95% air and culture medium changed every 48–72 h. Experimental cultures were seeded at a cell density (1 × 104/cm2) in order to provide 80–90% confluence at the time of treatment.
2.2. Proinflammatory stimulation
In order to induce CXCL10 expression and nuclear translocation of NF-κB p65, growth medium in astroglial cultures was replaced with serum free medium containing proinflammatory cytokines as previously described (Davis and Syapin, 2004b; Davis and Syapin, 2004c). The proinflammatory cytokines used were human recombinant TNFα, IL-1β, and IFN-γ (5 ng/ml; Peprotech, Rocky Hill, NJ). The duration of cytokine exposure was 24 h for CXCL10 expression and 0.5 h for NF-κB activation.
2.3. Inhibitors of NF-κB activation
To assess the role of NF-κB in TNFα-induced CXCL10 expression, A172 cells were stimulated in the presence of MG-132 (0.14 nM-2.0 μM; carbobenzoxy-L-leucyl-L-leucyl-L-leucinal; Biomol, Plymouth Meeting, PA) or the sesquiterpene lactone, helenalin (2.7 nM-2.0 μM; Biomol). MG-132 inhibits NF-κB activation by blocking the activity of the 26S proteasome (Lee and Goldberg, 1996), and thereby prevents nuclear translocation of NF-κB. The effects of helenalin are known to be further downstream where it prevents NF-κB-DNA binding by specifically alkylating the p65 subunit (Lyss et al., 1998). MG-132 and helenalin were reconstituted in dimethyl sulfoxide at a stock concentration of 10 mM and stored at −80°C and −20°C, respectively (Sanchez et al., 2003). These inhibitors were added to cultures at the time of stimulation with TNFα. A third inhibitor of NF-κB activation, SN50 (Biomol) was also used. SN50, a cell permeable inhibitory peptide containg the nuclear localization sequence of NF-κB p50, prevents nuclear translocation of the active NF-κB complex. Similar to others (Pahan et al., 2001; Yang et al., 2005), cells were preincubated for 1 h prior to stimulation with 50 μM SN50 or 50 μM of the inactive mutant peptide SN50M, used as a control (Biomol).
2.4. Opioid compounds
Opioids [morphine (0.05–150 μM), fentanyl (1.2–150 μM), β-FNA (0.14–100 μM), naltrexone (33–150 μM); all from Sigma, St. Louis, MO] were added to cell cultures at the time of TNFα stimulation, except in the pre-treatment experiments in which opioids were added 5–60 minutes prior to TNFα addition. In the pre-treatment experiments, residual opioids were rinsed from the cells prior to TNFα stimulation.
2.5. ELISA
A standard dual-antibody solid phase immunoassay (ELISA Development Kit, Peprotech) was used for quantitation of secreted CXCL10 in cell culture supernatants, according to the manufacturer’s instructions. Briefly, 100 μl of standards (0–2000 pg/ml) and cell culture supernatants were added to appropriate antibody-coated wells of a 96 well plate. Next, 100 μl of antigen-specific biotinylated detection antibody was added to each well followed by a 2 h incubation at room temperature. Liquid contents were then aspirated and wells washed 3× with wash buffer. Avidin-peroxidase conjugate (100 μl) was then added to each well and incubated for 30 min at room temperature. Wells were then aspirated and washed 3× as before, and 100 μl of ABTS liquid substrate solution (Sigma cat. # A3219) was added to each well followed by 25 min incubation at room temperature. Absorbance was read at 450 nm (with wavelength correction set at 650 nm) on a BIO-TEK HT spectrophotometer.
2.6. Nuclear translocation of NF-κB p65
Nuclear fractions were collected as described previously (Davis and Syapin, 2004c). For immunoblot analysis of nuclear levels of p65, 10 μg protein was electrophoretically separated through a 8% polyacrylamide gel (Bio-Rad, Hercules, CA) in running buffer (25 mM Tris base, 190 mM L-glycine, 0.1% SDS) with the Mini-Protean® 3 Cell (Bio-Rad) for 25 min at 200 V. Proteins were electro-transferred to polyvinylidene difluoride membranes (Bio-Rad) in transfer buffer (25 mM Tris base, 150 mM L-glycine, 20% methanol) with the Mini Trans-Blot® Electrophoretic Transfer Cell (Bio-Rad) for 2 h at 87 V. Membranes were blocked for 1 h at room temperature in Tris sodium Tween 20 (TST) buffer (10 mM Tris base, 100 mM NaCl, 0.1% (v/v) Tween 20, pH 7.5) containing 5% non-fat powdered milk then incubated overnight at 4°C with anti-p65 antibody (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA). Membranes were washed with TST buffer five times for 5 min each, incubated for 1 h at room temperature with horseradish peroxidase-conjugated anti-rabbit IgG antibody (1:5,000; Sigma) and then washed with PBS (5 times for 5 min each). Bands were then visualized using SuperSignal™ West Pico Chemiluminescent Substrate (Pierce, Rockford, IL) and autoradiography (Fuji Medical X-ray film).
2.7. Determination of total cellular protein content
Total cell protein/well was determined using the bicinchoninic acid (BCA) protein assay as previously described (Davis et al., 2002) in order to normalize data when appropriate (i.e., pg chemokine protein produced/mg total cellular protein).
2.8. Determination of cell viability
Cell viability was assessed using the MTT assay according to a modified version of the procedure described by Carmichael et al., (1987). Briefly, cells were incubated for 45 min in culture medium containing 0.55 mg/ml 3-[4,5-Dimethylthiazol-2-yl]-2,5,-diphenyltetrazolium bromide (MTT). Media was then removed and cells were dissolved in 1 ml DMSO and absorbance measured at 492 nm using a BIO-TEK HT spectrophotometer.
2.9. Statistical analysis
Prism™ version 4.0 software (GraphPad Inc., San Diego, CA) was used for figure presentation, curve fitting, percent transformations and statistical analysis. Analyses included one-way analysis of variance (ANOVA) with Neuman-Kuels multiple comparison post hoc test as well as nonlinear regression in which the Hill slope was not constrained. Data are presented as mean + S.E.M. or mean IC50 values with their 95% confidence interval (CI). A probability (p) of < 0.05 was accepted as demonstrating statistically significant differences between groups; data points with any common letters above them are not statistically different from each other. The number of replicate measures and independent experiments from which the data were obtained are provided in the individual figure legends.
3. Results
3.1. Cytokine-induced CXCL10 expression
In pilot studies, we determined that CXCL10 protein expression is induced by a cytokine mixture containing IL-1β (5 ng/ml) + TNFα (30 ng/ml) + IFN-γ (100 ng/ml). In the present study, we showed that TNFα and to a lesser degree, IL-1β, are each sufficient to induce CXCL10 in A172 cells (Fig. 1). In fact, a relatively low concentration of TNFα (5 ng/ml) is stimulatory. Conversely, IFN-γ at 100 ng/ml does not induce CXCL10 in A172 cells.
Fig. 1.
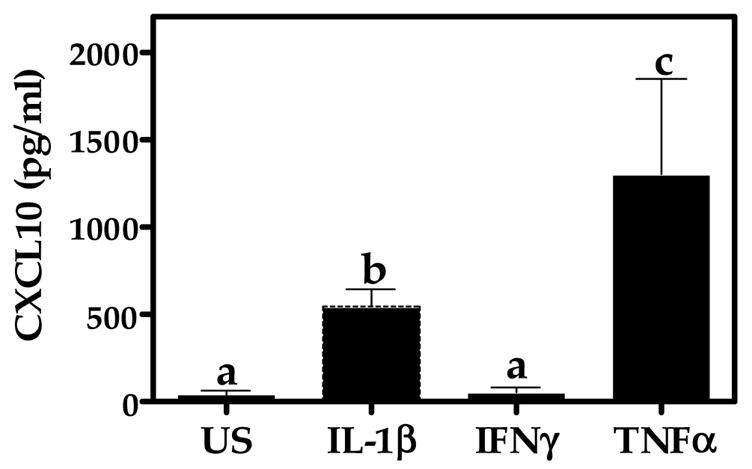
Cytokine-induced CXCL10 protein expression in A172 cells. Astroglial cells were unstimulated (US) or exposed for 24 h to human recombinant TNFα, IL-1β, or IFN-γ (5 ng/ml). A standard dual-antibody solid phase immunoassay (ELISA) was used for quantitation of secreted CXCL10 in cell culture supernatants. Data represent mean + S.E.M. of duplicate measures from 3 independent experiments. Data points with any common letters above them are not statistically different from each other as determined by one-way analysis of variance (ANOVA) with Neuman-Kuels’ multiple comparisons.
3.2 Role of NF-κB in TNF-α-induced CXCL10 expression
The role of NF-κB in CXCL10 expression in human astroglial cells is not well defined. Therefore, we assessed the ability of mechanistically distinct NF-κB inhibitors, MG-132, helenalin and SN50 to modulate CXCL10 expression. We determined that MG-132 and helenalin dose-dependently inhibit CXCL10 protein expression in human astroglial cells with IC50 values of 26 and 969 nM, respectively (Fig. 2). Additionally, we have determined that blockade of the NF-κB p50 nuclear recognition sequence with the peptide inhibitor SN50 prevents TNFα induced CXCL10 expression in A172 cells (Fig. 8).
Fig. 2.
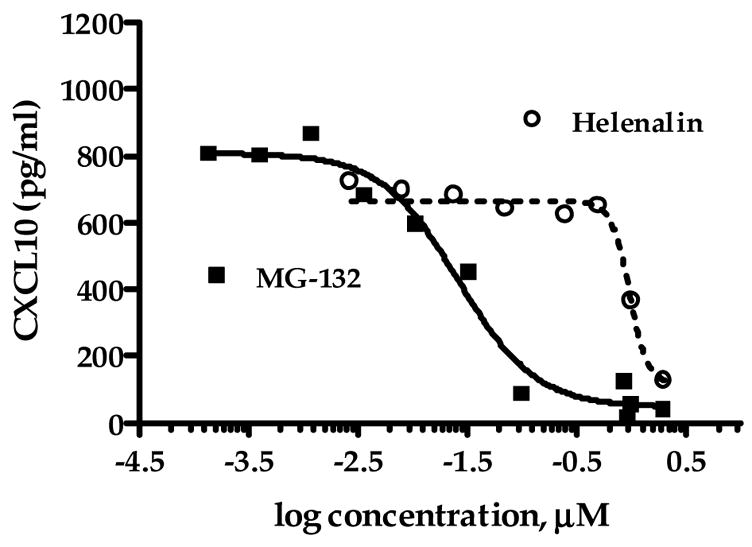
Inhibitors of NF-κB activation reduce TNFα-stimulated CXCL10 protein expression in A172 cells. Astroglial cells were exposed for 24 h to human recombinant TNFα (5 ng/ml) in the presence or absence of MG-132 (0.14 nM-2.0 μM; carbobenzoxy-L-leucyl-L-leucyl-L-leucinal) or the sesquiterpene lactone, helenalin (2.7 nM-2.0 μM). A standard dual-antibody solid phase immunoassay (ELISA) was used for quantitation of secreted CXCL10 in cell culture supernatants. Data points represent the mean + S.E.M. of duplicate measures from 2–7 independent experiments. Curves are from nonlinear regression in which the Hill slope was not constrained. The IC50 value and 95% confidence interval (CI) for MG-132 are 26 nM (95% CI: 17–41 nM) and 969 nM (95% CI: 754 nM-1.25 μM) for helenalin.
Fig. 8.
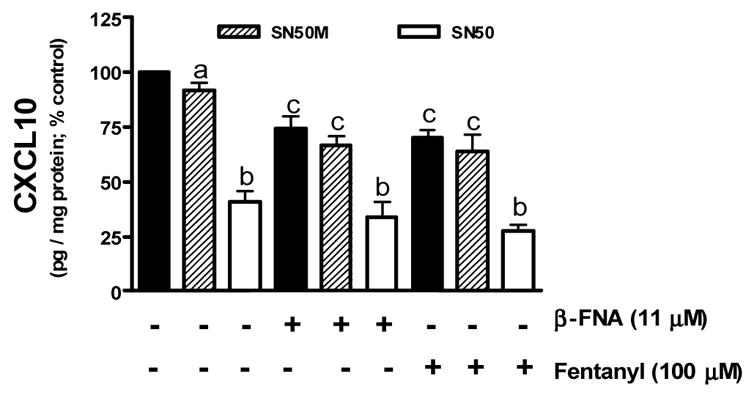
Inhibition of TNFα stimulated CXCL10 expression by the NF-κB inhibitor SN50 and selected opioids β-FNA and fentanyl. Astroglial cells were preincubated for 1 h with SN50 (50 μM) or the mutant peptide SN50M (50 μM). Cells were then stimulated with TNFα (5 ng/ml) in the presence or absence of β-FNA (11 μM) or fentanyl (100 μM) for 24 h. A standard dual-antibody solid phase immunoassay (ELISA) was used for quantitation of secreted CXCL10 in cell culture supernatants. Data points represent the mean + S.E.M. of duplicate measures from 3 independent experiments. Data points with any common letters above them are not statistically different from each other as determined by one-way analysis of variance (ANOVA) with Neuman-Kuels’ multiple comparisons.
3.3. Inhibition of CXCL10 protein expression by opioids
Exposure to morphine for 24 h did not significantly alter TNFα-induced CXCL10 expression in A172 cells (Fig. 3A). The more potent MOR agonist, fentanyl, dose-dependently inhibited TNFα-induced CXCL10 expression, albeit at relatively high concentrations (IC50 = 328 μM; Fig. 3B). The highly selective MOR antagonist, β-FNA, failed to prevent the inhibitory effects of fentanyl on CXCL10 expression. Interestingly, β-FNA was actually a relatively potent inhibitor of TNFα-induced CXCL10 expression (IC50 = 7.6 μM; Fig. 3C). The inhibitory effects of β-FNA and fentanyl were not a consequence of cytotoxicity as all four opioids were determined to be non-toxic under the experimental condition utilized (Fig. 4).
Fig. 3.
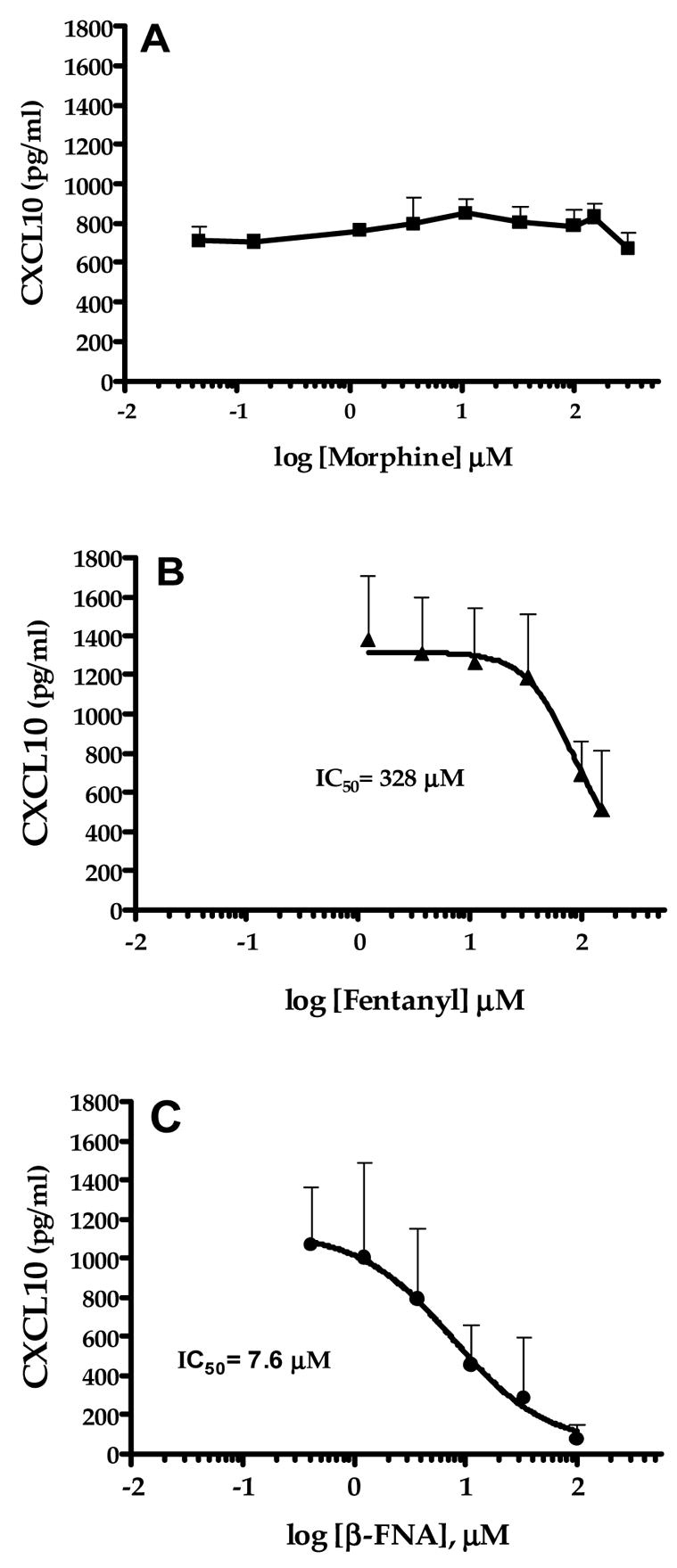
Modulation of TNFα-stimulated CXCL10 protein expression in A172 cells by selected opioids. Astroglial cells were exposed for 24 h to human recombinant TNFα (5 ng/ml) in the presence or absence of morphine (0.05–150 μM), fentanyl (1.2–150 μM) or β-funaltrexamine (β-FNA; 0.14–100 μM). A standard dual-antibody solid phase immunoassay (ELISA) was used for quantitation of secreted CXCL10 in cell culture supernatants. Data points represent the mean + S.E.M. of duplicate measures from 2–5 independent experiments. Curves are from nonlinear regression analysis in which the Hill slope was not constrained. The IC50 values are presented for each curve and the 95% confidence interval (CI) for each opioid are: morphine = not determined, fentanyl = 14.5–522.1 μM and β-FNA = 2.1–27.5 μM.
Fig. 4.
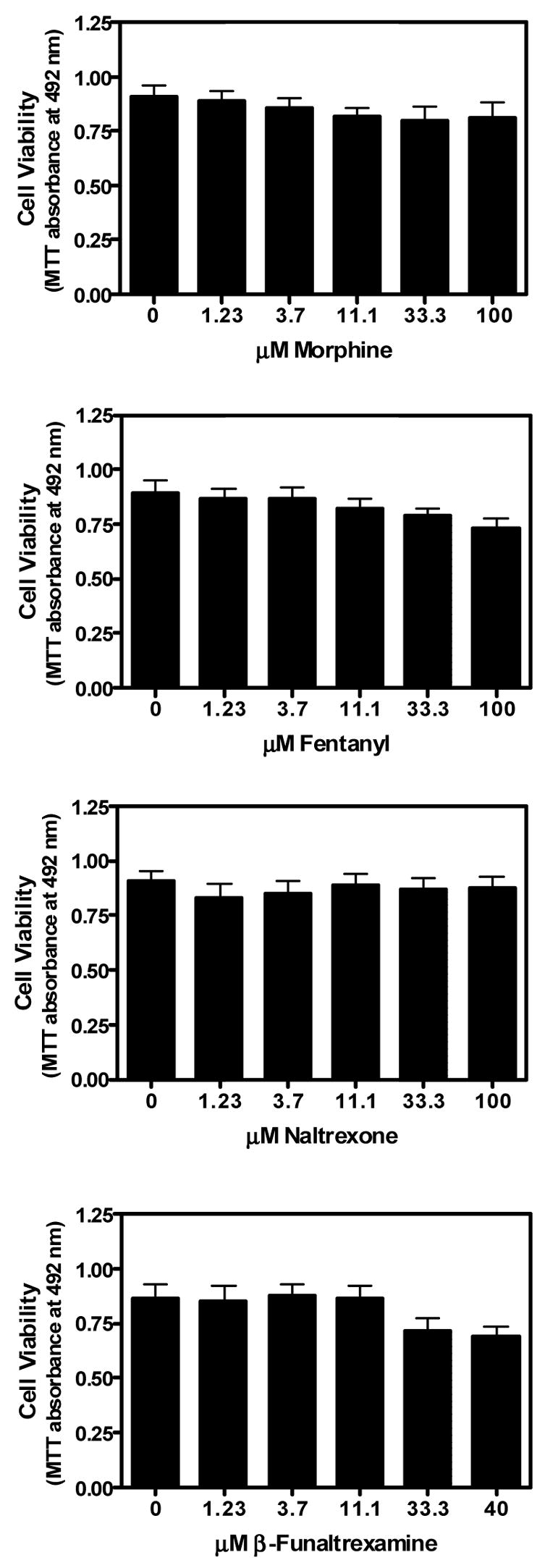
Effects of opioids on viability of TNFα stimulated astroglial cells. A172 cells were exposed for 24 h to human recombinant TNFα (5 ng/ml) in the presence or absence of morphine (1.23–100 μM), fentanyl (1.23–100 μM), naltrexone (1.23–100 μM) or β-funaltrexamine (β-FNA; 1.23–40 μM). Cell viability was determined by the MTT assay. Data represent mean + S.E.M. of duplicate or triplicate measures from 3 independent experiments. One-way analysis of variance (ANOVA) with Neuman-Kuels’ multiple comparisons indicated no significant opioid effects.
At a concentration of 100 μM, the non-specific opioid receptor antagonist, naltrexone, did not prevent either fentanyl- or β-FNA-inhibition of TNFα-induced CXCL10 expression (Fig. 5). In contrast, naltrexone slightly inhibited TNFα-induced CXCL10 expression and actually potentiated 11μM β-FNA-mediated inhibition of CXCL10 expression. Potentiation of β-FNA-mediated inhibition of CXCL10 expression by naltrexone occurred when cells were simultaneously exposed to naltrexone, β-FNA and TNFα for 24 h (Fig. 5). Similarly, potentiation of β-FNA-mediated inhibition of CXCL10 expression by naltrexone occurred in the 30 min pre-exposure paradigm in which cells are exposed to naltrexone and β-FNA for 30 min, followed by washout of the drug, prior to 24 h TNFα exposure (data not shown).
Fig. 5.
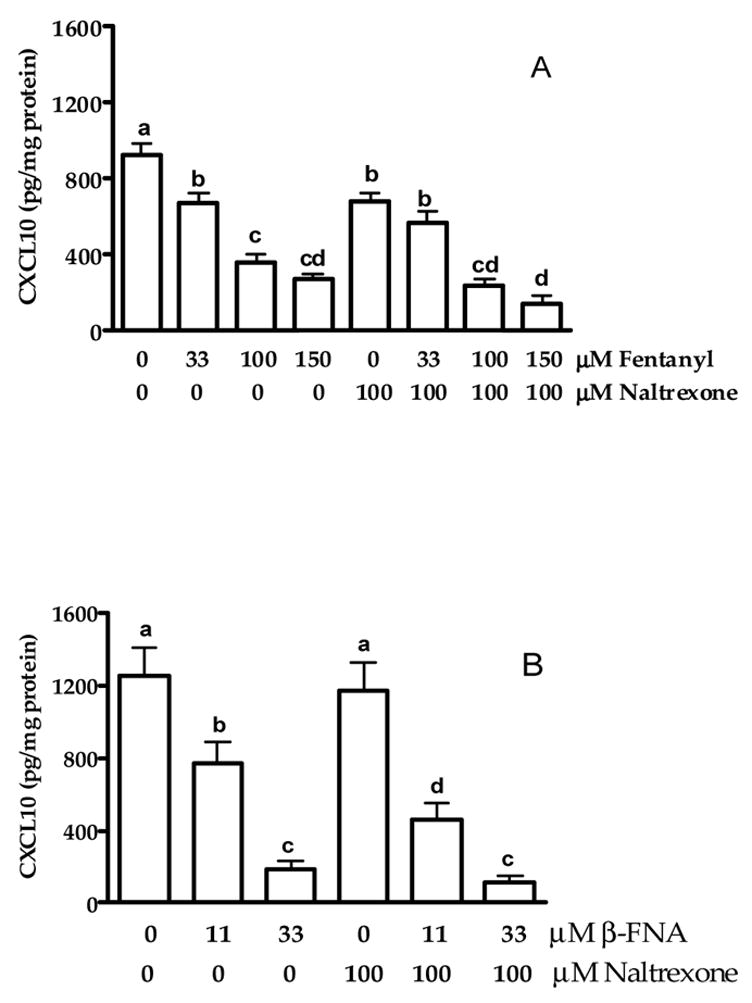
Modulation of (A) fentanyl- and (B) β-FNA-mediated inhibition of TNFα-stimulated CXCL10 protein expression in A172 cells by naltrexone. Astroglial cells were exposed for 24 h to human recombinant TNFα (5 ng/ml) in the presence or absence of fentanyl (33, 100 and 150 μM), β-funaltrexamine (β-FNA; 11 and 33 μM) and naltrexone (100 μM). A standard dual-antibody solid phase immunoassay (ELISA) was used for quantitation of secreted CXCL10 in cell culture supernatants. Data points represent the mean + S.E.M. of triplicate measures from 2 independent experiments (panel A) and quadruplicate measures from 3 independent experiments (panel B). Data points with any common letters above them are not statistically different from each other as determined by one-way analysis of variance (ANOVA) with Neuman-Kuels’ multiple comparisons.
To further assess the inhibitory actions of β-FNA, the time-dependent effects on CXCL10 expression were determined. Pre-exposure to β-FNA for 5 – 60 min, followed by washout of the drug, prior to 24 h TNFα exposure, resulted in a time-dependent decrease in CXCL10 expression (Fig. 6). Exposure to both β-FNA and TNFα for 24 h resulted in > 95% inhibition of CXCL10 expression (Figs. 3C and 6); whereas, 5 min pre-exposure to β-FNA caused minimal inhibition (≈20%) of CXCL10 expression, but only at 100 μM (Fig. 6).
Fig. 6.
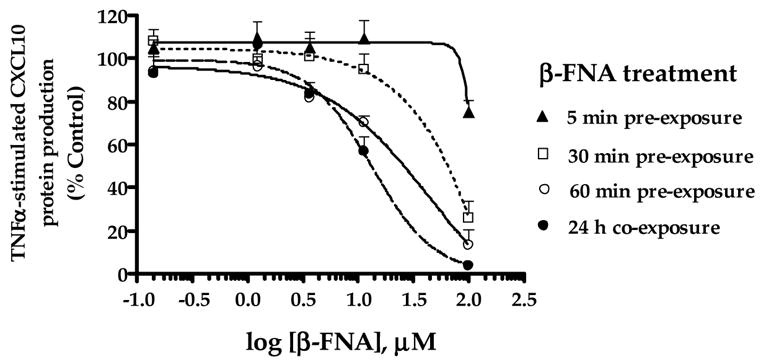
Time-course effects of β-FNA on TNFα-stimulated CXCL10 protein expression in A172 cells. Astroglial cells were either co-exposed to β-funaltrexamine (β-FNA; 0.14–100 μM) and human recombinant TNFα (5 ng/ml) for 24 h (●) or pre-exposed to β-FNA for 5 (▲), 30 (□) or 60 (○) min, followed by washout of the drug, and subsequent exposure to TNFα for 24 h. A standard dual-antibody solid phase immunoassay (ELISA) was used for quantitation of secreted CXCL10 in cell culture supernatants. Data for each experiment were transformed to percent control (TNFα stimulation in the absence of β-FNA). Data points represent the mean + S.E.M. of duplicate measures from 3 independent experiments. Curves are resultant from nonlinear regression analysis in which the Hill slope was not constrained.
3.4. Effect of opioids on NF-κB activation
Nuclear translocation of the p65 subunit was assessed as a measure of NF-κB activation (Fig. 7). Very low levels of p65 protein were present in the nucleus of unstimulated (not exposed to TNFα) cells. In contrast, exposure to TNFα for 0.5 h resulted in a marked increase in p65 accumulation in the nucleus. When cells were stimulated with TNFα in the presence 33 μM β-FNA (a concentration that produces remarkable inhibition of stimulated CXCL10 expression) there was an apparent decrease in nuclear p65 levels. Similarly, at this same concentration (33 μM) fentanyl and to a greater degree, morphine seemed to prevent p65 nuclear translocation. However, statistical analyses did not reveal any differences among the opioid treated groups. There was no evidence of a naltrexone effect on p65 nuclear translocation. It should be noted that both the gel image and the graphic representation of the data suggest that when compared to control (TNFα-stimulated) cells, β-FNA reduces TNFα-induced p65 nuclear translocation. Yet, statistical conclusions can not be made given that the data are normalized to these control data. Additionally, combined effects of SN50 and opioids on CXCL10 expression were assessed. As noted in Fig. 8, SN50-inhibition of CXCL10 is not potentiated by either 11 μM β-FNA or 100 μM fentanyl.
Fig. 7.
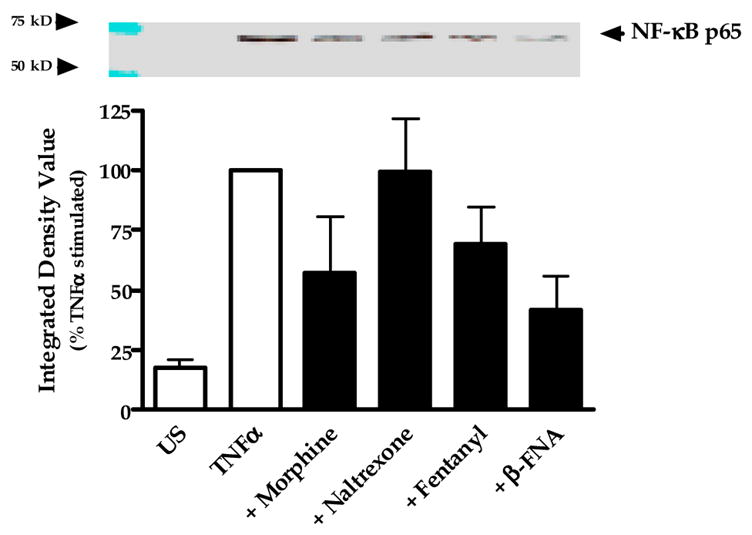
Modulation of NF-κB p65 nuclear translocation by opioids. Astroglial cells were exposed for 0.5 h to human recombinant TNFα (5 ng/ml) in the presence or absence of 33 μM morphine, naltrexone, fentanyl or β-funaltrexamine. Nuclear fractions were collected and NF-κB p65 protein levels assessed by immunoblot analysis of 10 μg total protein. β-tubulin levels were measured and used as loading controls. Images of the blots were digitally captured, and then integrated density values (IDV) were obtained using ImageJ software (NIH). β-tubulin band densities were used to normalize the densities of the NF-κB bands. Data points represent the mean + S.E.M. of the IDV obtained from 4 independent experiments (normalized for each experiment as percent stimulated control); the blot is representative of the 4 independent experiments.
4. Discussion
To our knowledge, this is the first report describing the effects of morphine or fentanyl on CXCL10 expression in astroglia. In recent years, there has been considerable interest in understanding the effects of opioids (primarily morphine) on chemokine expression and function in astroglia (El-Hage et al., 2005; El-Hage et al., 2006; Mahajan et al., 2005; Mahajan et al., 2002). For instance, morphine (≥ 1 nM for 24 h) reduced constitutive expression of IL-8 (or CXCL2) and macrophage inhibitory protein 1β (MIP-1β or CCL4) genes and proteins in normal human astrocytes (Mahajan et al., 2002). Suppression of constitutive chemokine expression by morphine was reversed by β-FNA, indicating a MOR-dependent mechanism (Mahajan et al., 2002). More recently, this same group (Mahajan et al., 2005) showed that MIP-1β and monocyte chemoattractant protein 1 (MCP-1 or CCL2) gene expression is down-regulated by morphine. Additionally, the non-selective opioid receptor antagonist, naloxone blocked morphine-induced inhibition of chemokine expression in these cells, suggesting an opioid receptor (likely MOR)-dependent mechanism.
Other investigators have also provided evidence of MOR-independent actions of morphine on astroglial chemokines expression. For instance, the HIV-1 Tat protein (Tat1-72) increases MCP-1 and RANTES expression and morphine synergistically enhances Tat1-72-induced expression of these chemokines (El-Hage et al., 2005). Interestingly, β-FNA reversed the synergistic action of morphine on Tat1-72-induced chemokine expression but did not significantly reduce the stimulatory actions of morphine alone. Together these data indicate that the potentiation of Tat1-72-induction of MCP-1 and RANTES by morphine is MOR-dependent, yet stimulation of these chemokines by morphine alone may be, in part, via MOR-independent actions. Therefore, with regard to morphine and chemokine expression in astrocytes there appear to be substantial species- and chemokine-specific effects.
Previously, we found that CXCL10 protein expression in A172 cells is strongly induced by either LPS (1 μg/ml) + IL-1β (10 ng/ml) or a cytokine mixture containing IL-1β (5 ng/ml) + TNFα (30 ng/ml) + IFN-γ (100 ng/ml) (Davis and Syapin 2004b; unpublished data). In the present study we determined that TNFα alone, at a considerably lower concentration (5 ng/ml), significantly induced CXCL10 protein expression in A172 cells. Other studies have found that opioids, such as morphine, suppress the expression of astroglial chemokines, including IL-8, MCP-1 and MIP-1β (Mahajan et al., 2005; Mahajan et al., 2002). In the present study, morphine did not modulate CXCL10 protein expression whereas the more potent MOR agonist, fentanyl, suppressed TNFα-induced CXCL10 expression in A172 cells. In addition to the observation that morphine (≤150 μM) failed to alter CXCL10 protein expression, there are several other factors to suggest that this reduction in CXCL10 expression may not be MOR-dependent. For instance, the concentration required for fentanyl inhibition of CXCL10 expression is relatively high and the highly MOR-specific antagonist, β-FNA did not reverse the effects of fentanyl. Additionally, the non-selective opioid receptor antagonist naltrexone did not block fentanyl-induced effects on CXCL10 expression. Given that β-FNA irreversibly binds to MOR, we predicted that if β-FNA was acting in a MOR-dependent manner, inhibition of CXCL10 expression would result even when β-FNA was added for only 5–60 min and subsequently removed prior to TNFα exposure. However, we observed a time- and dose-dependent effect of β-FNA on CXCL10 expression. One explanation for these findings is that β-FNA is acting through an alternate receptor and/or is modulating an intracellular target. Presumably, it would require more time (or higher concentrations) for β-FNA to reach intracellular targets. Failure of naltrexone to reverse β-FNA effects further suggests that β-FNA is acting independent of opioid receptors. Furthermore, naltrexone potentiated β-FNA-mediated inhibition of CXCL10 expression; yet by itself, naltrexone (100 μM) only minimally reduced CXCL10 expression.
Interestingly, we have demonstrated that it is not the interaction of fentanyl or β-FNA with MOR that is causing the inhibition of TNFα-induced CXCL10 expression. This is very interesting given that fentanyl is typically described as a MOR agonist and β-FNA is best known as a MOR antagonist. Of particular interest is the finding that β-FNA inhibited TNFα-induced CXCL10 expression more potently than fentanyl. The anti-inflammatory actions of β-FNA have not been previously characterized and the mechanism responsible for the MOR independent actions of β-FNA (and fentanyl) have yet to be identified. However, others have found unexpected effects of β-FNA on chemokine (IL-8 and MIP-1β) expression in human astrocytes (Mahajan et al., 2002). More specifically, these investigators reported that in addition to blocking morphine-suppression of constitutive chemokine expression, β-FNA alone up-regulated chemokine expression in these cells (Mahajan et al., 2002). These researchers postulated that β-FNA may be activating at another type of opioid family receptor such as the nociceptin (ORL) receptor or another as yet unidentified site. For instance, depending on the receptor type, β-FNA can exhibit agonist or antagonist activity (Holtzman, 1997; Sayre et al., 1984). Astroglia express all three opioid receptor types (μ, δ and κ) and these receptors are differentially expressed among brain regions (Ruzicka et al., 1995; Stiene-Martin et al., 1998). Also, age-related and cell-cycle-dependent variations in opioid phenotype can be observed at the cellular level (Stiene-Martin et al., 1998). However, morphine functions predominantly through MOR (Baumhaker et al., 1993; Stefano et al., 1996). MOR is expressed on human astroglia, yet in normal human astrocytes and the U87 human astroglial cell line, expression levels are relatively low (Mahajan et al., 2005; Mahajan et al., 2002). MOR expression is up-regulated in human astroglial cells following morphine exposure (Mahajan et al., 2005; Mahajan et al., 2002). We recently determined that expression of the human mu-opioid receptor (hMOR) mRNA is up-regulated in A172 cells following TNFα exposure (unpublished data). TNFα also induces MOR expression in various cell types including peripheral immune effector cells, microvascular endothelial cells and SH SY5Y neuroblastoma cells (Borner et al., 2004; Kraus et al., 2003). Further characterization of opioid receptor expression in A172 cells is currently being investigated in our laboratory.
The anti-inflammatory actions of β-FNA identified in this study also support and add to the findings of others (Liu et al., 2000; Liu and Hong, 2003) which have identified MOR-independent, anti-inflammatory actions of naloxone (a non-specific, opioid receptor antagonist). For example, addition of lipopolysaccharide (LPS)-stimulated microglia to rat mesencephalic mixed neuronglia cultures resulted in pronounced neuronal toxicity (Liu et al., 2000). However, co-exposure of these cultures to naloxone provided neuroprotection, reportedly by blocking LPS-binding to microglia (Liu et al., 2000). Additionally, the inactive stereoisomer, (+)-naloxone, was equally effective as (−)-naloxone (Liu et al., 2000). In a more recent study, this same group determined that Aβ (1–42)-induced neurotoxicity was reduced by naloxone in an opioid receptor-independent mechanism (Liu et al., 2002). In this study it was determined that the neuroprotective effects of naloxone were due to reduced microglial superoxide production (Liu et al., 2002). Together, these insights support a potential role of incorporating opioid agents in the treatment of neuroinflammation associated with neurodegenerative diseases such as Alzheimer’s, Parkinson’s, HIV dementia and ischemic stroke. However, more mechanistic information is necessary before this therapeutic potential can be fully realized.
The therapeutic relevance of attenuating the transcription factor NF-κB, with respect to neuroinflammation and neuropathogenesis, has increased in recent years. For example, the potential benefit of down regulating NF-κB activation and CXCL10 expression in astroglial cells on recovery from spinal cord injury was recently highlighted (Brambilla et al., 2005). Use of the functionally distinct inhibitors of NF-κB activation provided data to suggest that TNFα-induced CXCL10 protein expression is dependent in part, on proteasome degradation, NF-κB p50 mediated nuclear translocation and NF-κB p65-DNA binding. That MG-132 is a more potent inhibitor of CXCL10 expression compared to helenalin, suggests that NF-κB activation in A172 cells is more sensitive to disruption of proteasome degradation than blockade of p65-DNA binding. It should be noted that as an inhibitor of proteasome degradation, MG-132 may also modulate other pathways that utilize the 26S proteasome. Importantly though, we have confirmed by immunoblot analysis that MG-132 reduces p65 translocation into the nucleus of cytokine-stimulated A172 cells (unpublished data).
Regarding the role of NF-κB p65 in the anti-inflammatory effects of fentanyl and β-FNA, findings in this study indicate that the effects of these opioid compounds on TNFα induced CXCL10 expression are not directly related to decreased NF-κB p65 nuclear translocation. Further analysis is needed to fully appreciate the importance of p65 (or other NF-κB molecules) in fentanyl and β-FNA modulation of CXCL10 expression. Additionally, further investigation of the potential role of other opioid receptor types in β-FNA mediated inhibition of CXCL10 expression in A172 cells is warranted. It will also be important to identify and characterize the effects of opioids on CXCL10 expression and other inflammatory molecules in normal human astrocytes and in vivo models of neuropathogenesis. A better understanding of the mechanism governing the MOR-independent, anti-inflammatory actions of opioid agents, and especially the safe and non-addictive opioid antagonist, β-FNA, may result in important treatments for neuroinflammation.
Acknowledgments
This work supported in part by NIH grants AA 014955 (RLD) and DA 012448 (CWS). We are grateful to Melissa Herron and Shekher Mohan for their technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bajetto A, Bonavia R, Barbero S, Schettini G. J Neurochem. 2002;82:1311–1329. doi: 10.1046/j.1471-4159.2002.01091.x. [DOI] [PubMed] [Google Scholar]
- Baumhaker Y, Gafni M, Keren O, Sarne Y. Mol Pharmacol. 1993;44:461–467. [PubMed] [Google Scholar]
- Beech JS, Reckless J, Mosedale DE, Grainger DJ, Williams SC, Menon DK. J Cereb Blood Flow Metab. 2001;21:683–689. doi: 10.1097/00004647-200106000-00006. [DOI] [PubMed] [Google Scholar]
- Belperio JA, Keane MP, Arenberg DA, Addison CL, Ehlert JE, Burdick MD, Strieter RM. J Leukoc Biol. 2000;68:1–8. [PubMed] [Google Scholar]
- Borner C, Kraus J, Schroder H, Ammer H, Hollt V. Mol Pharmacol. 2004;66:1719–1726. doi: 10.1124/mol.104.003806. [DOI] [PubMed] [Google Scholar]
- Brambilla R, Bracchi-Ricard V, Hu WH, Frydel B, Bramwell A, Karmally S, Green EJ, Bethea JR. J Exp Med. 2005;202:145–156. doi: 10.1084/jem.20041918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael J, DeGraff WG, Gazdar AF, Minna JD, Mitchell JB. Cancer Research. 1987;47:936–942. [PubMed] [Google Scholar]
- Cinque P, Bestetti A, Marenzi R, Sala S, Gisslen M, Hagberg L, Price RW. J Neuroimmunol. 2005;168:154–163. doi: 10.1016/j.jneuroim.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Davis RL, Dertien J, Syapin PJ. Alcohol Clin Exp Res. 2002;26:1404–1411. doi: 10.1097/01.ALC.0000030841.92766.80. [DOI] [PubMed] [Google Scholar]
- Davis RL, Syapin PJ. Alcohol. 2004a;32:195–202. doi: 10.1016/j.alcohol.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Davis RL, Syapin PJ. Neurosci Lett. 2004b;362:220–225. doi: 10.1016/j.neulet.2004.03.015. [DOI] [PubMed] [Google Scholar]
- Davis RL, Syapin PJ. Neurosci Lett. 2004c;371:128–132. doi: 10.1016/j.neulet.2004.08.051. [DOI] [PubMed] [Google Scholar]
- El-Hage N, Gurwell JA, Singh IN, Knapp PE, Nath A, Hauser KF. Glia. 2005;50:91–106. doi: 10.1002/glia.20148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hage N, Wu G, Wang J, Ambati J, Knapp PE, Reed JL, Bruce-Keller AJ, Hauser KF. Glia. 2006;53:132–146. doi: 10.1002/glia.20262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn G, Maru S, Loughlin J, Romero IA, Male D. J Neuroimmunol. 2003;136:84–93. doi: 10.1016/s0165-5728(03)00009-2. [DOI] [PubMed] [Google Scholar]
- Glaser J, Gonzalez R, Perreau VM, Cotman CW, Keirstead HS. J Neurosci Res. 2004;77:701–708. doi: 10.1002/jnr.20204. [DOI] [PubMed] [Google Scholar]
- Gonzalez R, Glaser J, Liu MT, Lane TE, Keirstead HS. Exp Neurol. 2003;184:456–463. doi: 10.1016/s0014-4886(03)00257-7. [DOI] [PubMed] [Google Scholar]
- Guthikonda P, Baker J, Mattson DH. J Neuroimmunol. 1998;82:133–139. doi: 10.1016/s0165-5728(97)00172-0. [DOI] [PubMed] [Google Scholar]
- Holtzman SG. Pharmacol Biochem Behav. 1997;57:771–777. doi: 10.1016/s0091-3057(96)00395-4. [DOI] [PubMed] [Google Scholar]
- Kadiu I, Glanzer JG, Kipnis J, Gendelman HE, Thomas MP. Neurotox Res. 2005;8:25–50. doi: 10.1007/BF03033818. [DOI] [PubMed] [Google Scholar]
- Khurdayan VK, Buch S, El-Hage N, Lutz SE, Goebel SM, Singh IN, Knapp PE, Turchan-Cholewo J, Nath A, Hauser KF. Eur J Neurosci. 2004;19:3171–3182. doi: 10.1111/j.0953-816X.2004.03461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb SA, Sporer B, Lahrtz F, Koedel U, Pfister HW, Fontana A. J Neuroimmunol. 1999;93:172–181. doi: 10.1016/s0165-5728(98)00223-9. [DOI] [PubMed] [Google Scholar]
- Kraus J, Borner C, Giannini E, Hollt V. Mol Pharmacol. 2003;64:876–884. doi: 10.1124/mol.64.4.876. [DOI] [PubMed] [Google Scholar]
- Kubota N, Kiuchi Y, Nemoto M, Oyamada H, Ohno M, Funahashi H, Shioda S, Oguchi K. Eur J Pharmacol. 2001;417:69–76. doi: 10.1016/s0014-2999(01)00906-2. [DOI] [PubMed] [Google Scholar]
- Lee DH, Goldberg AL. J Biol Chem. 1996;271:27280–27284. doi: 10.1074/jbc.271.44.27280. [DOI] [PubMed] [Google Scholar]
- Lee J, Kim MS, Park C, Jung EB, Choi DH, Kim TY, Moon SK, Park R. Immunopharmacol Immunotoxicol. 2004;26:17–28. doi: 10.1081/iph-120029941. [DOI] [PubMed] [Google Scholar]
- Liao SL, Chen WY, Raung SL, Chen CJ. Neurosci Lett. 2003;345:169–172. doi: 10.1016/s0304-3940(03)00540-8. [DOI] [PubMed] [Google Scholar]
- Liu B, Du L, Hong JS. J Pharmacol Exp Ther. 2000;293:607–617. [PubMed] [Google Scholar]
- Liu B, Hong JS. J Pharmacol Exp Ther. 2003;304:1–7. doi: 10.1124/jpet.102.035048. [DOI] [PubMed] [Google Scholar]
- Liu Y, Qin L, Wilson BC, An L, Hong JS, Liu B. J Pharmacol Exp Ther. 2002;302:1212–1219. doi: 10.1124/jpet.102.035956. [DOI] [PubMed] [Google Scholar]
- Luster AD, Unkeless JC, Ravetch JV. Nature. 1985;315:672–676. doi: 10.1038/315672a0. [DOI] [PubMed] [Google Scholar]
- Lyss G, Knorre A, Schmidt TJ, Pahl HL, Merfort I. J Biol Chem. 1998;273:33508–33516. doi: 10.1074/jbc.273.50.33508. [DOI] [PubMed] [Google Scholar]
- Mahajan SD, Schwartz SA, Aalinkeel R, Chawda RP, Sykes DE, Nair MP. Clin Immunol. 2005;115:323–332. doi: 10.1016/j.clim.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Mahajan SD, Schwartz SA, Shanahan TC, Chawda RP, Nair MP. J Immunol. 2002;169:3589–3599. doi: 10.4049/jimmunol.169.7.3589. [DOI] [PubMed] [Google Scholar]
- Myers RR, Campana WM, Shubayev VI. Drug Discov Today. 2006;11:8–20. doi: 10.1016/S1359-6446(05)03637-8. [DOI] [PubMed] [Google Scholar]
- Pahan K, Sheikh FG, Liu X, Hilger S, McKinney M, Petro TM. J Biol Chem. 2001;276:7899–7905. doi: 10.1074/jbc.M008262200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peart JN, Gross ER, Gross GJ. Vascul Pharmacol. 2005;42:211–218. doi: 10.1016/j.vph.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Poluektova L, Meyer V, Walters L, Paez X, Gendelman HE. Glia. 2005;52:344–353. doi: 10.1002/glia.20253. [DOI] [PubMed] [Google Scholar]
- Rambhia S, Mantione KJ, Stefano GB, Cadet P. Med Sci Monit. 2005;11:BR386–396. [PubMed] [Google Scholar]
- Ruzicka BB, Fox CA, Thompson RC, Meng F, Watson SJ, Akil H. Brain Res Mol Brain Res. 1995;34:209–220. doi: 10.1016/0169-328x(95)00165-o. [DOI] [PubMed] [Google Scholar]
- Sanchez AC, Davis RL, Syapin PJ. J Neurochem. 2003;86:1379–1390. doi: 10.1046/j.1471-4159.2003.01943.x. [DOI] [PubMed] [Google Scholar]
- Sayre LM, Larson DL, Takemori AE, Portoghese PS. J Med Chem. 1984;27:1325–1335. doi: 10.1021/jm00376a018. [DOI] [PubMed] [Google Scholar]
- Sorensen TL. Curr Neurovasc Res. 2004;1:183–190. doi: 10.2174/1567202043480143. [DOI] [PubMed] [Google Scholar]
- Stefano GB, Scharrer B, Smith EM, Hughes TK, Jr, Magazine HI, Bilfinger TV, Hartman AR, Fricchione GL, Liu Y, Makman MH. Crit Rev Immunol. 1996;16:109–144. doi: 10.1615/critrevimmunol.v16.i2.10. [DOI] [PubMed] [Google Scholar]
- Stiene-Martin A, Zhou R, Hauser KF. Glia. 1998;22:249–259. [PMC free article] [PubMed] [Google Scholar]
- Taub DD, Lloyd AR, Conlon K, Wang JM, Ortaldo JR, Harada A, Matsushima K, Kelvin DJ, Oppenheim JJ. J Exp Med. 1993;177:1809–1814. doi: 10.1084/jem.177.6.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toborek M, Lee YW, Flora G, Pu H, Andras IE, Wylegala E, Hennig B, Nath A. Cell Mol Neurobiol. 2005;25:181–199. doi: 10.1007/s10571-004-1383-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlodavsky E, Palzur E, Soustiel JF. Neuropathol Appl Neurobiol. 2006;32:40–50. doi: 10.1111/j.1365-2990.2005.00698.x. [DOI] [PubMed] [Google Scholar]
- Wang X, Ellison JA, Siren AL, Lysko PG, Yue TL, Barone FC, Shatzman A, Feuerstein GZ. J Neurochem. 1998;71:1194–1204. doi: 10.1046/j.1471-4159.1998.71031194.x. [DOI] [PubMed] [Google Scholar]
- Wang X, Feuerstein GZ, Xu L, Wang H, Schumacher WA, Ogletree ML, Taub R, Duan JJ, Decicco CP, Liu RQ. Mol Pharmacol. 2004;65:890–896. doi: 10.1124/mol.65.4.890. [DOI] [PubMed] [Google Scholar]
- Wang X, Li X, Schmidt DB, Foley JJ, Barone FC, Ames RS, Sarau HM. Mol Pharmacol. 2000;57:1190–1198. [PubMed] [Google Scholar]
- Xia MQ, Bacskai BJ, Knowles RB, Qin SX, Hyman BT. J Neuroimmunol. 2000;108:227–235. doi: 10.1016/s0165-5728(00)00285-x. [DOI] [PubMed] [Google Scholar]
- Yang YT, Ju TC, Yang DI. J Neurochem. 2005;93:513–525. doi: 10.1111/j.1471-4159.2005.03032.x. [DOI] [PubMed] [Google Scholar]
- Zaheer A, Zhong W, Lim R. Neurochem Res. 1995;20:1457–1463. doi: 10.1007/BF00970594. [DOI] [PubMed] [Google Scholar]