

Abstract
Menkes disease is a fatal neurodegenerative disorder of childhood due to the absence or dysfunction of a putative copper-transporting P-type ATPase encoded on the X chromosome. To elucidate the biosynthesis and subcellular localization of this protein, polyclonal antisera were generated against a bacterial fusion protein encoding the 4th to 6th copper-binding domains in the amino terminus of the human Menkes protein. RNA blot analysis revealed abundant Menkes gene expression in several cell lines, and immunoblotting studies utilizing this antiserum readily detected a 178-kDa protein in lysates from these cells. Pulse–chase studies indicate that this protein is synthesized as a single-chain polypeptide which is modified by N-linked glycosylation to a mature endoglycosidase H-resistant form. Sucrose gradient fractionation of HeLa cell lysates followed by immunoblotting of individual fractions with antibodies to proteins of known intracellular location identified the Menkes ATPase in fractions similar to those containing the cation-independent mannose-6-phosphate receptor. Consistent with this observation, confocal immunofluorescence studies of these same cells localized this protein to the trans-Golgi network and a vesicular compartment with no expression in the nucleus or on the plasma membrane. Taken together, these data provide a unique model of copper transport into the secretory pathway of mammalian cells which is compatible with clinical observations in affected patients and with recent data on homologous proteins identified in prokaryotes and yeast.
Menkes disease is a chromosome X-linked disorder of copper metabolism resulting in a loss of developmental milestones, mental retardation, and progressive degeneration of the central nervous system with death in early childhood (1). In addition to these prominent neurologic features, affected patients have a characteristic peculiar appearance of the hair (pili torti), hypopigmentation, vascular complications, and connective tissue abnormalities all secondary to deficiencies in the activity of the copper-dependent enzymes involved in these processes (2). Clinical and pathologic studies in such patients reveal a defect in copper transport across the placenta, the gastrointestinal tract, and the blood–brain barrier resulting in a profound deficiency of copper in affected fetuses and newborn infants (3). The recognition that cultured fibroblasts from patients with Menkes disease accumulate intracellular copper and have impaired copper efflux suggested that the defect in this disorder involves an essential pathway of copper transport (4, 5).
Consistent with these clinical observations, the Menkes disease gene has been cloned and shown to encode a protein with homology to the cation-transporting P-type ATPase family (6, 7, 8). This ATPase family encompasses a large number of polytopic membrane proteins, each of which utilizes the ATP-dependent phosphorylation of an invariant aspartate residue to derive energy for cation transport across a cellular membrane. Recognition that the deduced amino acid sequence of the Menkes gene is homologous to that of known P-type ATPases allowed for the possibility that this protein functions as a copper transporter (9, 10). Support for such a proposed role has been provided by the recent recognition of evolutionarily conserved homologues of the Menkes protein in prokaryotes and yeast, where experimental disruption of the genes encoding these proteins results in phenotypic and biochemical abnormalities due to altered copper metabolism (11, 12, 13).
Although there is as of yet no direct evidence of copper transport by the Menkes protein, recent studies have revealed increased expression of the Menkes protein in Chinese hamster ovary cells resistant to excess copper, the level of expression in such cells correlating with the degree of copper resistance (14). Furthermore, the Wilson disease gene has been cloned and shown to encode a putative P-type ATPase with 55% amino acid identity to the Menkes protein, thus implicating these homologous proteins in each of the known inherited disorders of copper metabolism in humans (15, 16, 17). Taken together, these data support the concept that these proteins function as copper transporters in mammalian cells. Nevertheless, despite these findings there is currently no information on where these proteins are expressed in specific cells, making it difficult to formulate a precise model for the function of these proteins in cellular copper metabolism. In the current study, we have directly addressed this issue by generating polyclonal antisera to the human Menkes protein and using these antisera to characterize the biosynthesis and subcellular localization of this protein in human cell lines.
MATERIALS AND METHODS
Materials.
Hs242T, Daudi, HeLa, H441, and HepG2 cells were obtained from the American Type Culture Collection; Menkes fibroblast cell lines (GM1981, GM0220, and GM3700) were obtained from the Mutant Genetic Cell Repository (Camden, NJ); primary fibroblasts were a gift from Bruce Dowton (Washington University School of Medicine). Murine monoclonal antibodies to the bovine cation-independent mannose-6-phosphate receptor, AP-2, and Lamp-1 were a gift from Stuart Kornfeld (Washington University School of Medicine), a rabbit polyclonal antibody to TAP-1 was a gift from Ted Hansen (Washington University School of Medicine), and murine monoclonal antibodies to γ-adaptin (AP-1) were purchased from Sigma and used according to specifications provided. Texas red-conjugated transferrin was purchased from Molecular Probes.
RNA Analysis.
Cells were grown to confluence and RNA was isolated by CsCl density gradient centrifugation after dissolution in guanidinium isothiocyanate (18). RNA samples were electrophoresed in 0.8% agarose/2.2 M formaldehyde gels, transferred to nylon membranes, and hybridized at 58.5°C, using a 420-bp 32P-labeled complementary RNA (cRNA) probe encoding amino acids 486–621 of the human Menkes ATPase. This region of the Menkes cDNA is 48% homologous to the human Wilson cDNA nucleotide sequence and does not cross-react with the Wilson mRNA on RNA blots under these conditions (data not shown). RNA blots were subsequently washed and exposed to film as described (19).
Menkes Antibody Production.
Oligonucleotide primers were synthesized and used to amplify a human Menkes cDNA encoding amino acids 343–641 as described previously (17). The resulting cDNA fragment was ligated into pGEX2T, and Escherichia coli DH5α cells harboring the expression plasmid were grown to an optical density of 0.6 at 600 nm and induced with isopropyl 1-thio-β-d-galactopyranoside. Cultures were harvested by centrifugation, resuspended in phosphate-buffered saline (PBS) containing 1% (vol/vol) Triton X-100, 1 μM pepstatin, 2.5 μg/ml leupeptin, and 0.2 mM phenylmethylsulfonyl fluoride (PMSF), and disrupted by sonication, and after centrifugation the supernatant was incubated with glutathione-agarose beads. Bound glutathione S-transferase (GST) fusion protein was eluted with 5 mM reduced glutathione in 50 mM Tris·HCl, pH 8.0, dialyzed, resuspended, and cleaved with thrombin (2 mg of protein per unit of thrombin) followed by removal of GST by glutathione-agarose affinity chromatography (20). New Zealand White rabbits were immunized with 200 μg of purified thrombin-cleaved Menkes protein by intradermal injection after emulsification of the antigen in Freund’s complete adjuvant. To ensure specificity of the Menkes antibody and to generate reagents for characterization of the Wilson disease protein, oligonucleotides were designed and utilized to amplify the homologous region of the Wilson protein encoding amino acids 325–635. This region was similarly subcloned in the pGEX2T vector, and thrombin-cleaved protein was used to immunize rabbits as described above. These regions were chosen because of limited homology in these two proteins in the regions between these putative copper-binding domains.
Immunoblotting and Immunoprecipitation.
Cells were grown to confluence, lysed in 0.25% Nonidet P-40/100 mM Tris·HCl, pH 8.0, at 4°C for 30 min, and proteins were separated by SDS/PAGE under reducing conditions followed by electrophoretic transfer to nitrocellulose membranes. Membranes were blocked with 5% nonfat dry milk and developed using the ECL (chemiluminescence) reagent (Amersham) according to the manufacturer’s protocol. In some experiments purified Menkes or Wilson antibodies were preincubated with an excess of the corresponding GST-fusion protein at 4°C for 4 h prior to use. Cells were metabolically labeled by supplementation of media with [35S]methionine and [35S]cysteine at 500 μCi/ml (1 μCi = 37 kBq) for specific pulse periods followed by a chase period in serum-free media for times indicated. Biosynthetic studies and immunoprecipitations were performed as described (21). In some experiments immune complexes bound to beads coated with staphylococcal protein A were boiled in 100 mM sodium acetate at pH 5.5, incubated with endo-β-N-acetylglycosaminidase (endo F) or endo-β-N-acetylglucosaminidase (endo H) at 37°C for 16 h, and analyzed by SDS/PAGE (21).
Cell Extracts and Gradient Fractionation.
Cells were lysed in 10 mM KCl/10 mM Tris·HCl, pH 7.4, containing 1 mM PMSF, 2 mM EDTA, and 1 μg/ml each of pepstatin, leupeptin, and aprotinin. Following lysis, cells were given 20 strokes with a tight-fitting glass Dounce homogenizer, and the postnuclear supernatant was isolated by centrifugation at 500 × g for 5 min. This supernatant was then subjected to isopycnic centrifugation in preformed 12-ml linear continuous sucrose gradients (0.7–1.5 M) in a Beckman SW41 rotor at 300,000 × g for 4 h at 4°C (22). Fractions were removed, subjected to electrophoresis in SDS/5–12% polyacrylamide gradient gels, and analyzed by immunoblotting as described above.
Immunofluorescence.
For indirect immunofluorescence, cells were plated on glass coverslips, allowed to grow for 36 h, fixed in 4% paraformaldehyde in PBS for 20 min, quenched with 0.1% ethanolamine, and permeabilized in 0.2% Triton-X 100 in PBS for 10 min. Nonspecific binding was blocked by incubation in 3% BSA in PBS for 30 min, followed by incubation with primary antibodies and secondary antibodies conjugated with fluorescein isothiocyanate (FITC) or tetramethylrhodamine isothiocyanate (TRITC). After staining, coverslips were mounted in Mowiol 4-88 (Sigma) and analyzed using an Olympus BX-60 microscope. To obtain through-focus images of cells these same slides were analyzed with a laser scanning confocal microscope (Molecular Dynamics) equipped with a Nikon planapo (63×) oil-immersion lens (numerical aperture = 1.4). Argon/krypton laser emission wavelengths were 488 nm for FITC and 508 nm for TRITC.
RESULTS
To identify cell lines expressing the Menkes protein, RNA blot analysis was performed utilizing a cRNA probe encoding a portion of the human Menkes gene. Consistent with previously reported findings, a single 8-kb mRNA was detected in RNA from primary fibroblasts (control), and this was either absent or altered in size in fibroblast cell lines from patients with Menkes disease (Fig. 1A, lanes 1–5). Similar analysis performed on RNA isolated from human cell lines revealed an identically sized 8-kb mRNA in Daudi, HeLa, and H441 cells. A similar transcript was also faintly present in the human hepatoma-derived cell line HepG2 (Fig. 1A, lanes 6–9). On the basis of these observations cell lysates were prepared from a series of human cell lines and analyzed by immunoblotting with an antibody specific to the human Menkes ATPase. As can be seen in Fig. 1B, a single 178-kDa band was observed in HeLa cell lysates when this antibody was used. A similarly sized band was also detected in a lung cell line (Hs242T) and primary cells (Con) but was not present in cell lysates from fibroblasts of a patient with Menkes disease or HepG2 cells (Fig. 1B, lanes 2 and 5). As the Menkes and Wilson proteins are highly homologous in sequence, the specificity of these findings was further confirmed by analysis of these same cell lysates by immunoblotting with an antibody to the Wilson protein. As can be seen in Fig. 1B (lanes 6–10) a single 165-kDa band consistent with the Wilson protein was observed only in lysates from HepG2 cells (Fig. 1B, lanes 3 and 4).
Figure 1.
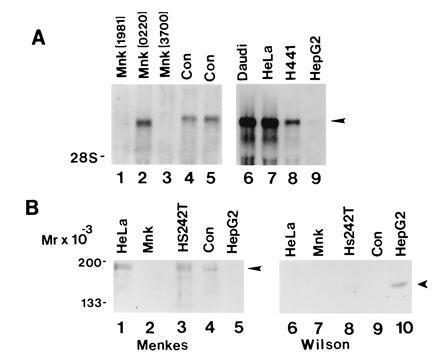
(A) RNA blot analysis of Menkes mRNA in cell lines from Menkes patients (lanes 1–3), control primary fibroblasts (lanes 4 and 5), and human cell lines (lanes 6–9). Ten micrograms of total RNA was isolated, electrophoresed, transferred to nylon, and hybridized with a human Menkes [32P]cRNA probe. (B) Immunoblot of cell lysates with Menkes (lanes 1–5) or Wilson (lanes 6–10) antibody. Protein was loaded at 75 μg per lane, and after SDS/10% PAGE proteins were transferred to nitrocellulose and analyzed by ECL (Amersham).
To further characterize the Menkes protein observed above, HeLa cells were pulse-labeled for 1 h with [35S]methionine and [35S]cysteine, and cell lysates were immnuoprecipitated and subjected to SDS/PAGE. In this analysis a single 178-kDa band was detected that was not observed when preimmune serum or antisera preincubated with Menkes fusion protein were used (Fig. 2, lanes 1–3). Analysis of the Menkes protein by both immunoblotting and immunoprecipitation revealed a broad band on SDS/PAGE suggestive of post-translational modification. The derived amino acid sequence of the Menkes protein contains several potential consensus sites for N-linked glycosylation and, consistent with this, preincubation of HeLa cells with tunicamycin prior to immunoprecipitation resulted in a reduction in size of the detected Menkes protein (Fig. 2, lanes 4 and 5). To determine the degree of modification of the asparagine-linked carbohydrate, digestion was performed with endo H and endo F. Following a 1-h metabolic pulse, the immunoprecipitated Menkes protein was found to be insensitive to endo H but reduced in size by endo F to that observed following preincubation of cells with tunicamycin (Fig. 2B, lanes 6–9). The faint larger-sized bands observed in these experiments (lane 8) were nonspecific, as indicated by identically sized bands following immunoprecipitation with preimmune sera (lane 1).
Figure 2.

Immunoprecipitation of Menkes ATPase from HeLa cell lysates. Following a 1-h pulse-label with [35S]methionine and [35S]cysteine, cells were lysed and immunoprecipitated with preimmune sera (lane 1), Menkes antibody (lanes 2, 4, and 6–9), or Menkes antibody preincubated with Menkes fusion protein (lanes 3 and 5). For some experiments cells were pretreated with tunicamycin (4 μg/ml) for 2 h prior to immunoprecipitation (lanes 4, 5, and 9). Immunoprecipitated Menkes protein was digested with endo H or endo F (lanes 7 and 8) as described. Samples were electrophoresed on SDS/5-12% polyacrylamide gradient gels and subjected to fluorography.
The endoglycosidase studies suggested that the Menkes protein contains mature complex N-linked carbohydrate, a modification which requires transit through the medial Golgi complex. As study of this post-translational modification can assist in determining protein localization, the kinetics of this process was next examined by pulse–chase analysis. Following an initial 10-min pulse period, a single 170-kDa Menkes-specific band was identified by immunoprecipitation, and this band was converted to the 178-kDa form during the chase period (Fig. 3A). This intracellular 178-kDa form was never observed following immunoprecipitation of cell media (data not shown). When the Menkes protein was subjected to endoglycosidase digestion during these studies a characteristic pattern of carbohydrate maturation was observed (Fig. 3B). Following the 10-min pulse, the Menkes protein was sensitive to both endo H and endo F, corresponding to a high-mannose glycosylated precursor (Fig. 3B, lanes 1–3). Following a 1-h chase, the mature 178-kDa protein was converted into the complex glycosylated endo H-resistant form (Fig. 3B, lanes 4 and 5). As anticipated, digestion of this product with endo F resulted in the appearance of a band identical in size to that observed during the initial pulse period (Fig. 3B, lane 6).
Figure 3.
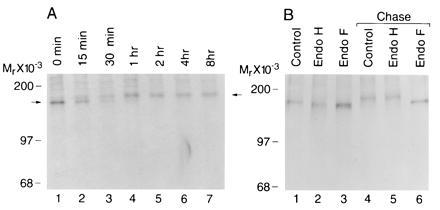
(A) Pulse–chase labeling of biosynthesized Menkes ATPase in HeLa cells. Cultures were pulsed for 10 min with [35S]methionine and [35S]cysteine at 500 μCi/ml and then chased for indicated times in media containing excess methionine and cysteine. Immunoprecipitates of cell lysates were analyzed by SDS/10% PAGE and fluorography. (B) HeLa cells were pulse-labeled for 10 min (lanes 1–3) followed by a 1-h chase (lanes 4–6). Cell lysates were immunoprecipitated with Menkes antibody and incubated without enzyme (lanes 1 and 4) or digested with endo H (lanes 2 and 5) or endo F (lanes 3 and 6).
These biosynthetic studies reveal that the Menkes protein transits through the secretory pathway at least to the medial Golgi compartment. However, these kinetic studies do not provide information about the steady-state location of this protein, and thus to address this issue in greater detail isopycnic sucrose gradient centrifugation was performed. Following ultracentrifugation of HeLa cell lysates, isolated fractions were separated by SDS/PAGE and analyzed by immunoblotting. The results of such an experiment are shown in Fig. 4, where the Menkes protein is detected in the later fractions of the gradient (lanes 7–11). This band was specifically blocked by preincubation of the antibody with Menkes fusion protein (data not shown). To approximate the location of specific organelles in these fractions, immunoblotting was performed using antibodies which recognize proteins of known subcellular location. In this study, TAP-1, a protein which functions in transport of peptides into the endoplasmic reticulum and is confined to this compartment and the cis-Golgi compartment (23), was detected in the earliest fractions of this same gradient (Fig. 4, lanes 1–5). In contrast, both γ-adaptin (AP-1), which is localized to the trans-Golgi network, and the cation-independent mannose-6 phosphate receptor (MPR), which is localized in the trans-Golgi network and late endosomes (24, 25), were detected in fractions which overlap with the Menkes protein (Fig. 4, lanes 8–11). The mannose-6-phosphate receptor is also located on the plasma membrane, and a small amount of this protein was detected in the last fraction of this gradient (Fig. 4, lane 12), whereas the Menkes protein was not observed in this fraction of the gradient.
Figure 4.
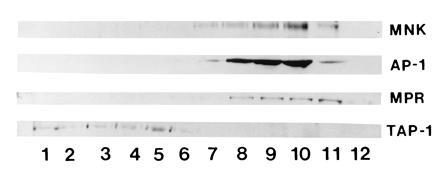
Immunoblot analysis of HeLa cell lysates following isopycnic centrifugation in a continuous linear sucrose gradient. A 75-μg sample of protein from each fraction was separated by SDS/10% PAGE, transferred to nitrocellulose, and immunoblotted with Menkes antibody or antibodies to TAP-1, γ-adaptin (AP-1), or the cation-independent mannose-6-phosphate receptor (MPR).
The results of the biosynthetic and fractionation studies were consistent and revealed that the Menkes protein is localized to an intracellular compartment in the later portion of the sorting or secretory pathway of the cell. Considerable overlap amongst fractions is observed in fractionation experiments, and thus in an attempt to more directly characterize the specific compartments containing the Menkes protein indirect immunofluorescence was performed with the Menkes antibody and defined organelle markers. In these studies the Menkes protein was consistently detected concentrated in a perinuclear location, with some signal also present in variable amounts in a vesicular distribution throughout the cytoplasm (Fig. 5 a, d, g, and j). No specific signal was observed when this antibody was preincubated with the Menkes fusion protein prior to use, and no Menkes-specific signal was detected in cell lines from patients with known deletions of the Menkes gene (data not shown). When double-label immunofluorescence studies were performed, we consistently observed overlap of the Menkes protein with γ- adaptin (AP-1), a known trans-Golgi network resident protein (Fig. 5 a–c). In addition, the Menkes signal was also observed to partially overlap in the trans-Golgi network and a vesicular compartment with the mannose-6-phosphate receptor (Fig. 5 d–f). In contrast, no overlap was observed when we used antibodies localizing the lysosomal (lamp-1; Fig. 5 g–i) or plasma membrane (AP-Z, Fig. 5 j–l) compartments.
Figure 5.
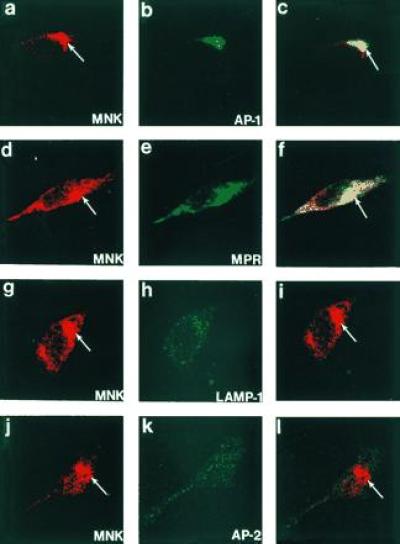
Confocal immunofluorescence localization of the Menkes ATPase in HeLa cells. Cells were processed for indirect immunofluorescence and analyzed after incubation with antibodies to the Menkes ATPase (a, d, g, and j) and γ-adaptin (AP-1) (b), the cation-independent mannose-6-phosphate receptor (MPR) (e), lamp-1 (h), and AP-2 (k). The combined confocal image from each double-labeling is shown in c, f, i, and l. (×400.)
DISCUSSION
Taken together, the results of these biosynthetic, fractionation, and immunofluorescence studies indicate that the Menkes protein is an intracellular 178-kDa protein which is predominantly resident in the trans-Golgi network. These data suggest that the primary role of this protein is to transport copper from the cytosol into the secretory compartment of the cell for incorporation into newly synthesized cuproproteins as well as for export from the cell. The delivery of copper into this portion of the sorting pathway would exclude this metal from the early secretory pathway, where preservation of the redox state is essential for disulfide bond formation and subsequent folding and vectorial secretion of nascent polypeptides (26, 27, 28). These data are also consistent with a proposed model for the localization of a homologous copper-transporting ATPase (CCC2) in Saccharomyces cerevisiae and with earlier data on the site of copper incorporation into apoceruloplasmin in hepatocytes (13, 21). These later observations suggest that the data observed here may provide a generalizable model for the localization and function of copper-transporting ATPases in mammalian cells.
Transport of proteins through the secretory pathway is thought to occur by default, with localization to a given compartment being accomplished by recognition of specific signals for retention. Previous studies with chimeric and mutant proteins have suggested that specific transmembrane regions within a protein may determine localization to the trans-Golgi/trans-Golgi network (29, 30). In addition, recent studies with alternative forms of β-1,4-galactosyltransferase as well as deletion mutants of the asialoglycoprotein receptor suggest that modulation of Golgi retention by cytoplasmic domains may also be a mechanism for fine-tuning intracellular distribution of specific proteins (31, 32). Although the data in the current study do not provide insight into the mechanism of retention of the Menkes protein in this compartment, the consistent observation that a variable amount of this protein is also localized further along the sorting pathway suggests that such mechanisms may be utilized to alter the location of this protein, perhaps in response to specific metabolic demands of the cell.
The data in this paper suggest that the antiserum utilized in this study specifically recognizes the human Menkes protein, and this is supported by the results shown with the Wilson protein antibody (Fig. 1B) which detects a 165-kDa protein in liver and hepatoma cells (Y.Y. and J.D.G., unpublished data) and by recent findings on the size of the homologous protein in hamster cell lysates (14). Nevertheless, several experimental caveats must be considered. Although no Menkes protein was detected by immunofluorescence on the plasma membrane, it is possible that the specific domain recognized by this antibody is not accessible at the cell surface. To address this concern several different methods of cell permeabilization were utilized which preserved the ability to detect the transferrin receptor on the cell surface, and in all cases no Menkes signal was observed in this region. Consistent with this, immunoprecipitation of iodinated cell surface proteins from intact cells did not detect the Menkes protein (data not shown). To exclude the possibility that the Menkes protein is transiently expressed on the cell surface and cycles back to the Golgi following endocytosis, pulse–chase studies were performed with excess Menkes antibody in the media. In all such experiments the subsequent isolation of immune complexes with protein A failed to detect any Menkes protein, suggesting that under the conditions studied the Menkes protein was not present on the plasma membrane (data not shown). In addition, pretreatment of cells with cycloheximide, which blocks protein synthesis but not transport, did not alter the location of the Menkes protein, arguing against sorting to the plasma membrane with only transient accumulation in the trans-Golgi network (data not shown). While it remains possible that the distribution of this protein, although identical in all cells examined thus far, may differ in vivo, the current data argue against localization on the plasma membrane.
Consistent with the data reported here, the copper enzymes that are affected in Menkes disease are those such as lysyl oxidase and dopamine-β-hydroxylase that are synthesized within the secretory pathway (2, 3). The observation that the activity of cytochrome oxidase, a mitochondrial enzyme, is deficient in brain tissue from such patients would also be consistent with the proposed model, as the failure to export copper across the blood–brain barrier results in generalized copper deficiency within specific regions of the brain. In this case, the loss of cytochrome oxidase activity is not the direct result of a failure of copper delivery to the mitochondria but rather is a secondary result of cellular copper deficiency. Such a model is compatible with a failure to detect the Menkes protein in mitochondria by biochemical or immunofluorescence studies and with recent studies which have identified unique proteins essential for the delivery of cytosolic copper to this and other cellular organelles (33, 34). At present the data leave open the mechanism for copper export from the cell, although recent studies clearly indicate that this process is essential for copper homeostasis and survival (35). The detection of Menkes protein in a vesicular compartment which overlaps with the cation-independent mannose-6-phosphate receptor suggests that this protein may concentrate copper in vesicles, which then proceed on to the lysosome or plasma membrane for copper export, while the protein itself is recycled back to the trans-Golgi network. As this overlap is only partial (Fig. 5f), this also suggests the existence of a unique vesicular compartment for copper export from the cell. This mechanism would then permit direct participation of the Menkes protein in copper export but also would be consistent with our observations that no Menkes protein is detected on the plasma membrane or in the lysosomal compartment. This model then implies additional steps in cellular copper export which remain to be elucidated.
The ability to detect the human Menkes protein also has practical implications for the diagnosis and treatment of patients with Menkes disease. The structure of the human Menkes gene has been characterized, and molecular genetic analysis of affected patients reveals that at least 30% of individuals with Menkes disease have deletions or mutations which should eliminate expression of this gene (3, 36, 37). The availability of specific antibodies may therefore be useful for diagnosis in circumstances where clinical material is limited or the lack of family data precludes focused molecular analysis. This may be especially important for prenatal diagnosis, as early initiation of copper replacement is currently the only treatment. Likewise, specific antibodies may prove useful in mildly affected patients with splicing mutations, to determine the absolute amount of protein necessary to preserve neurologic function (3, 38). Finally, controversy has arisen regarding the effectiveness of copper-histidine therapy in Menkes patients in whom specific mutations predict a total loss of protein (39, 40). A careful analysis of such patients with Menkes-specific antibodies will help to resolve this issue by direct determination of the effects of such mutations on the production of Menkes protein.
Acknowledgments
This manuscript is dedicated to the memory of Prof. Kiichi Yamaguchi. We thank Steve Kayler, John Menkes, and Sey Packman for helpful discussions about Menkes disease; Jane Gitschier, Julian Mercer, Richard Klausner, and Daniel Yuan for valuable insights and sharing information prior to publication; Stuart Kornfeld, Alan Schwartz, and Ger Strous for technical advice and critical review of the data; and Stuart Kornfeld and Ted Hansen for gifts of antibodies. This work was supported by funds from National Institutes of Health Grant DK44464 (J.D.G.).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: GST, glutathione S-transferase; PMSF, phenylmethylsulfonyl fluoride; FITC, fluorescein isothiocyanate; TRITC, tetramethylrhodamine isothiocyanate; endo H, endo-β-N-acetylglucosaminidase H; endo F, endo-β-N-acetylglycosaminidase F; BSA, bovine serum albumin.
References
- 1.Menkes J H, Alter M, Steigleder G K, Weakley D R, Sung J H. Pediatrics. 1962;29:764–769. [PubMed] [Google Scholar]
- 2.Kaler S G. Adv Pediatrics. 1994;41:263–304. [PubMed] [Google Scholar]
- 3.Vulpe C D, Packman S. Annu Rev Nutr. 1995;15:293–322. doi: 10.1146/annurev.nu.15.070195.001453. [DOI] [PubMed] [Google Scholar]
- 4.Goka T J, Stevenson R E, Hefferan P M, Howell R R. Proc Natl Acad Sci USA. 1976;73:604–606. doi: 10.1073/pnas.73.2.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Camakaris J, Danks D M, Ackland L, Cartwright E, Borger P, Cotton R G H. Biochem Genet. 1980;18:117–131. doi: 10.1007/BF00504364. [DOI] [PubMed] [Google Scholar]
- 6.Vulpe C, Levinson B, Whitney S, Packman S, Gitschier J. Nat Genet. 1993;3:7–13. doi: 10.1038/ng0193-7. [DOI] [PubMed] [Google Scholar]
- 7.Chelly J, Tümer Z, Tønneson T, Petterson A, Ishikawa-Brush Y, Tommerup N, Horn N, Monaco A P. Nat Genet. 1993;3:14–19. doi: 10.1038/ng0193-14. [DOI] [PubMed] [Google Scholar]
- 8.Mercer J F B, Livingston J, Hall B, Paynter J A, Begy C, Chandrasekharappa S, Lockhart P, Grimes A, Bhave M, Siemieniak D, Glover T W. Nat Genet. 1993;3:20–25. doi: 10.1038/ng0193-20. [DOI] [PubMed] [Google Scholar]
- 9.Silver S, Nucifora G, Phung L T. Mol Microbiol. 1993;10:7–12. doi: 10.1111/j.1365-2958.1993.tb00898.x. [DOI] [PubMed] [Google Scholar]
- 10.Bull P C, Cox D W. Trends Genet. 1994;10:246–252. doi: 10.1016/0168-9525(94)90172-4. [DOI] [PubMed] [Google Scholar]
- 11.Odermatt A, Suter H, Krapf R, Solioz M. J Biol Chem. 1993;268:12775–12779. [PubMed] [Google Scholar]
- 12.Phung L T, Ajlani G, Haselkorn R. Proc Natl Acad Sci USA. 1994;91:9651–9654. doi: 10.1073/pnas.91.20.9651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan S D, Stearman R, Dancis A, Dunn T, Beeler T, Klausner R D. Proc Natl Acad Sci USA. 1995;92:2632–3636. doi: 10.1073/pnas.92.7.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Camakaris J, Petris M, Bailey L, Shen P, Lockhart P, Glover T W, Barcroft C L, Patton J, Mercer J F B. Hum Mol Genet. 1995;4:2117–2123. doi: 10.1093/hmg/4.11.2117. [DOI] [PubMed] [Google Scholar]
- 15.Bull P C, Thomas G R, Rommens J M, Forbes J R, Cox D W. Nat Genet. 1993;5:327–337. doi: 10.1038/ng1293-327. [DOI] [PubMed] [Google Scholar]
- 16.Tanzi R E, Petrukhin K, Chernov I, Pellequer J L, Wasco W, Ross B, Romano D M, Parano E, Pavone L, Brzustowicz L M, Devoto M, Peppercorn J, Bush A J, Sternlieb I, Pirastu M, Gusella J F, Evgrafov O, Penchasqadeh G K, Honig B, Edelman I S, Soares M B, Scheinberg I H, Gilliam T C. Nat Genet. 1993;5:344–350. doi: 10.1038/ng1293-344. [DOI] [PubMed] [Google Scholar]
- 17.Yamaguchi Y, Heiny M E, Gitlin J D. Biochem Biophys Res Commun. 1993;197:271–277. doi: 10.1006/bbrc.1993.2471. [DOI] [PubMed] [Google Scholar]
- 18.Chirgwin J M, Przybyla A E, MacDonald R J, Rutter W J. Biochemistry. 1979;18:5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- 19.Hackett B P, Gitlin J D. Proc Natl Acad Sci USA. 1992;89:9079–9083. doi: 10.1073/pnas.89.19.9079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith D B, Johnson K S. Gene. 1988;64:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 21.Sato M, Gitlin J D. J Biol Chem. 1991;266:5128–5134. [PubMed] [Google Scholar]
- 22.Strous G J, Berger E G. J Biol Chem. 1982;257:7623–7628. [PubMed] [Google Scholar]
- 23.Townsend A, Trowsdale J. Semin Cell Biol. 1993;4:53–61. doi: 10.1006/scel.1993.1007. [DOI] [PubMed] [Google Scholar]
- 24.Griffiths G, Hoflack B, Simmons K, Mellman I, Kornfeld S. Cell. 1988;52:329–341. doi: 10.1016/s0092-8674(88)80026-6. [DOI] [PubMed] [Google Scholar]
- 25.Stoorvogel W, Gueze H J, Griffith J M, Schwartz A L, Strous G J. J Cell Biol. 1989;108:2137–2148. doi: 10.1083/jcb.108.6.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hwang, C., Sinslay, A. J. & Lodish, H. F. Science 257, 1496–1502. [DOI] [PubMed]
- 27.Lodish H L, Kong N. J Biol Chem. 1993;268:20598–20605. [PubMed] [Google Scholar]
- 28.Losch A, Koch-Brandt C. J Biol Chem. 1995;270:11543–11548. doi: 10.1074/jbc.270.19.11543. [DOI] [PubMed] [Google Scholar]
- 29.Guan J L, Rose J K. Cell. 1984;37:779–787. doi: 10.1016/0092-8674(84)90413-6. [DOI] [PubMed] [Google Scholar]
- 30.Low S H, Tang B L, Wong S H, Hong W. J Biol Chem. 1994;269:1985–1994. [PubMed] [Google Scholar]
- 31.Lopez L C, Youakim A, Evans S, Shur B D. J Biol Chem. 1991;266:15984–15991. [PubMed] [Google Scholar]
- 32.Wahlberg J M, Geffen R, Reymond F, Simmen T, Spies M. J Cell Biol. 1995;130:285–297. doi: 10.1083/jcb.130.2.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Glerum D M, Shtanko A, Tzagoloff A. J Biol Chem. 1996;271:14504–14509. doi: 10.1074/jbc.271.24.14504. [DOI] [PubMed] [Google Scholar]
- 34.Lin S J, Culotta V C. Proc Natl Acad Sci USA. 1995;92:3784–3788. doi: 10.1073/pnas.92.9.3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kelly E J, Palmiter R J. Nat Genet. 1996;13:219–222. doi: 10.1038/ng0696-219. [DOI] [PubMed] [Google Scholar]
- 36.Dierick H A, Ambrosini L, Spencer J, Glover T W, Mercer J F B. Genomics. 1995;28:462–469. doi: 10.1006/geno.1995.1175. [DOI] [PubMed] [Google Scholar]
- 37.Tümer Z, Vural B, Tønneson T, Chelly J, Monaco A P, Horn N. Genomics. 1995;26:437–442. doi: 10.1016/0888-7543(95)80160-n. [DOI] [PubMed] [Google Scholar]
- 38.Kaler S G, Gallo L K, Proud V K, Percy A K, Mark Y, Segal N A, Goldstein D S, Holmes C S, Gahl W A. Nat Genet. 1994;8:195–202. doi: 10.1038/ng1094-195. [DOI] [PubMed] [Google Scholar]
- 39.Tümer Z, Horn N, Tønneson T, Christodoulou J, Clark J T R, Sarkar B. Nat Genet. 1996;12:11–13. doi: 10.1038/ng0196-11. [DOI] [PubMed] [Google Scholar]
- 40.Kaler S G. Nat Genet. 1996;13:21–22. doi: 10.1038/ng0596-21. [DOI] [PubMed] [Google Scholar]