

Abstract
RasGRPs (guanine-nucleotide-releasing proteins) are exchange factors for membrane-bound GTPases. All RasGRP family members contain C1 domains which, in other proteins, bind DAG (diacylglycerol) and thus mediate the proximal signal-transduction events induced by this lipid second messenger. The presence of C1 domains suggests that all RasGRPs could be regulated by membrane translocation driven by C1–DAG interactions. This has been demonstrated for RasGRP1 and RasGRP3, but has not been tested directly for RasGRP2, RasGRP4α and RasGRP4β. Sequence alignments indicate that all RasGRP C1 domains have the potential to bind DAG. In cells, the isolated C1 domains of RasGRP1, RasGRP3 and RasGRP4α co-localize with membranes and relocalize in response to DAG, whereas the C1 domains of RasGRP2 and RasGRP4β do not. Only the C1 domains of RasGRP1, RasGRP3 and RasGRP4α recognize DAG as a ligand within phospholipid vesicles and do so with differential affinities. Other lipid second messengers were screened as ligands for RasGRP C1 domains, but none was found to serve as an alternative to DAG. All of the RasGRP C1 domains bound to vesicles which contained a high concentration of anionic phospholipids, indicating that this could provide a DAG-independent mechanism for membrane binding by C1 domains. This concept was supported by demonstrating that the C1 domain of RasGRP2 could functionally replace the membrane-binding role of the C1 domain within RasGRP1, despite the inability of the RasGRP2 C1 domain to bind DAG. The RasGRP4β C1 domain was non-functional when inserted into either RasGRP1 or RasGRP4, implying that the alternative splicing which produces this C1 domain eliminates its contribution to membrane binding.
Keywords: C1 domain, diacylglycerol (DAG), guanine-nucleotide-exchange factor (GEF), membrane binding, phorbol ester
Abbreviations: DAG, diacylglycerol; DMEM, Dulbecco's modified Eagle's medium; ER, endoplasmic reticulum; GFP, green fluorescent protein; GST, glutathione S-transferase; GRP, guanine-nucleotide-releasing protein; KSR, kinase suppressor of Ras; LPA, lysophosphatidic acid, PA, phosphatidic acid; PC, phosphatidylcholine; PG, phosphatidylglycerol; PKC, protein kinase C; PS, phosphatidylserine, SLV, sucrose-loaded large unilamellar vesicles; TCR, T-cell receptor
INTRODUCTION
DAG (diacylglycerol) is a lipid second messenger that is involved in the transduction of signals from most cell-surface receptors [1–3]. Signal transduction by DAG is mediated by its binding to C1 domains, which are found on a wide variety of kinases, exchange factors and other proteins [4,5]. The binding of DAG to their C1 domains serves to translocate these proteins to membranes, where they can interact with signalling complexes, substrates and regulators [6,7]. In some cases, the binding of C1 domains to DAG within membranes also releases the domains of other signalling proteins, and thus contributes to enzymatic activation as well as translocation to membranes [5,8,9].
C1 domains are functionally heterogeneous in terms of their ligand binding capabilities [5,6]. Most PKC (protein kinase C) family members have a pair of C1 domains, but each of these domains can differ considerably in their affinities for DAG [6]. PKC C1 domains can also differ in their relative affinities for DAG compared with phorbol esters, which are commonly used as artificial ligands for C1 domains [6]. In general, these C1 domains have a higher affinity for phorbol esters, but for some domains, DAG is the preferred ligand [10,11]. Some C1 domains have no detectable affinity for either DAG or phorbol esters, and therefore must have an alternative function [5].
C1 domains are readily identified by sequence analysis owing to their characteristic spacing of two histidine residues and six cysteine residues (Figure 1A). These amino acids co-ordinate two zinc ions, which constrain the domain into a compact structure (Figure 1B). In C1 domains of known structure which bind DAG or phorbol esters, such as the second C1 domain (C1b) of PKCδ [12] or the C1 domain of β2-chimaerin [13], two projecting loops insert into membranes and form a ligand-binding pocket (Figure 1B). This pocket can accommodate either the headgroup of DAG or an analogous segment of phorbol esters. Although the atomic interactions between the C1b domain of PKCδ and phorbol esters have been identified, the precise structural basis for the DAG–C1 domain interaction is unknown. The C1 domains of Raf-1 and KSR (kinase suppressor of Ras), which do not bind DAG or phorbol esters, have radical alterations in loops A and B in comparison with other C1 domain structures [14,15]. They lack the proline residue which dictates the structure of loop A and are missing half of loop B (Figure 1A), and, as a consequence, these two C1 domains lack a ligand-binding pocket.
Figure 1. C1 domain sequences.

(A) The C1 domains of the RasGRPs are compared with four C1 domains of known structure. The C1b domain of PKCδ and the C1 domain of β2-chimaerin (B2-chim.) bind DAG and phorbol esters, while the C1 domains of Raf-1 and KSR do not. The histidine and cysteine residues which co-ordinate zinc are indicated by the shaded bars. Loop A and B residues are italicized. Cationic residues which have the potential to interact with anionic phospholipids within membranes are in boldface. For the C1 domains used in this study, the sequences extending to the C-termini of the expressed proteins are shown, with non-natural amino acids in lower case. The N-termini indicate where the C1 domains were fused to GFP, GST or K-Ras. Numbering of the residues within the C1 domains is shown above the RasGRP1 C1 domain sequence. (B) The C1b domain of PKCδ (PDB code 1PTQ) illustrates the generic structure of a C1 domain that binds DAG or phorbol esters. The zinc atoms are shown as orange balls. Tyr8, Thr12 and Trp22 are highlighted in yellow. Four of the cationic residues implicated in membrane interactions, Lys26, Lys30, Lys41 and Lys45, are highlighted in blue. The image was generated with PyMol (http://pymol.sourceforge.net/). Amino acid residues are indicated using the one-letter code.
In addition to specific ligand binding via the pocket, C1 domains interact with the membrane surface [6]. Hydrophobic residues projecting from or at the base of loops A and B facilitate membrane insertion [6,12,16]. Basic residues distributed on the periphery of the C1 domain surface and at the C-terminus (Figure 1A) provide electrostatic interactions with anionic phospholipids and thus enhance DAG-mediated binding to membranes [12,17–19]. For KSR and Raf-1, these hydrophobic and electrostatic interactions may enable DAG-independent membrane-binding [15,20].
RasGRPs (guanine-nucleotide-releasing proteins) are exchange factors for membrane-bound Ras or Rap GTPases [21]. All RasGRP family members contain C1 domains. This suggests that the activity of all RasGRP family members could be regulated by membrane translocation, which may be mediated by binding of their C1 domains to DAG. Evidence for this hypothesis has been obtained for RasGRP1 and, to a lesser extent, for RasGRP3. Activation of RasGRP1 and RasGRP3 by antigen receptors requires DAG-generating phospholipases Cγ [22–24] and is inhibited by DAG kinases [25–28]. This does not necessarily reflect the involvement of a DAG–C1 domain interaction, because activation of both RasGRP1 and RasGRP3 requires phosphorylation of these proteins by DAG-dependent PKCs [29–33]. However, for RasGRP1, it is clear that a direct DAG–C1 domain interaction contributes to the membrane-localization step of its activation process. Phospholipase Cγ1 is required for TCR (T-cell receptor)-induced translocation of RasGRP1 to membranes, which can be inhibited by DAG kinase [25,34,35], and deletion of the C1 domain eliminates binding of RasGRP1 to membranes [36–38]. The isolated C1 domain of RasGRP1 translocates to membranes in vivo in response to DAG [38,39] and phorbol esters [37,38] and binds directly to both DAG [39] and phorbol esters [19,37,40].
Although direct binding of the C1 domain of RasGRP3 to DAG has not been demonstrated, the domain does bind phorbol esters [19]. The large variations in affinity for DAG compared with phorbol esters of several PKC C1 domains [6] shows that the assumption that phorbol ester binding equates with DAG binding may not be accurate. Antigen-receptor-induced membrane translocation of RasGRP3 also requires its C1 domain and phospholipase C activity [24] and is induced by either DAG or phorbol esters [41]. Therefore the available evidence supports the hypothesis that RasGRP3, like RasGRP1, is regulated in its translocation to the membrane by the interaction of its C1 domain with DAG.
RasGRP4 has several splice variants [42,43]. Two of these splice variants, RasGRP4α and RasGRP4β, have intact C1 domains, although there are an extra five amino acids present within the RasGRP4β C1 domain in comparison with RasGRP4α (Figure 1A). Phorbol ester treatment induces membrane translocation and activation of RasGRP4α [44]. Both RasGRP4α and RasGRP4β synergize with phorbol esters to induce transformation of fibroblasts, while another RasGRP4 splice variant with a truncated C1 domain is unable to transform fibroblasts [42,43]. An isolated form of the C1 domain of RasGRP4α binds directly to phorbol esters within phospholipid vesicles [19], but phorbol ester binding by the RasGRP4β C1 domain or DAG binding by either of the RasGRP4 C1 domains was not examined. These results are compatible with both RasGRP4α and RasGRP4β being activated by membrane translocation driven by the binding of DAG to their C1 domains. However, the evidence for this is not well established at present, particularly for RasGRP4β.
The evidence for regulation of the Rap-specific exchange factor RasGRP2 via a DAG–C1 domain interaction is somewhat contradictory. RasGRP2 is activated by phorbol esters in several cell types [45–48], and the ability of RasGRP2 to enhance TCR-induced adhesion via Rap GTPase activation is dependent on TCR-coupled phospholipases C [49]. Translocation of RasGRP2 to membranes, resulting in the activation of membrane-associated Rap GTPases, can be induced by PMA [45] and a variety of receptors coupling to phospholipases C [48–51]. This tends to result in the assumption that RasGRP2 is equivalent to RasGRP1 and RasGRP, and that RasGRP2 can be regulated by the interaction of its C1 domain with DAG-enriched membranes. However, a synthetic RasGRP2 C1 domain had no detectable affinity for phorbol esters in a vesicle-binding assay [19], in contrast with the C1 domains of RasGRP1, RasGRP3 and RasGRP4α. This result has to be interpreted with caution, because the C1 domains of the peripheral membrane proteins munc-13 and unc-13, which bind phorbol esters when biologically expressed [52,53], failed to bind phorbol esters when synthesized and analysed in the same way as the RasGRP2 C1 domain [19]. Therefore it is possible that the lack of phorbol ester binding reflected misfolding of the synthetic RasGRP2 C1 domain. Binding to DAG was not examined using the synthetic RasGRP2 C1 domain. The localization of full-length RasGRP2 compared with RasGRP1 is different in several cell types [34,36,54]. This suggests that the the localization of both of these RasGRPs is not determined solely by their C1 domains seeking out membranes enriched in DAG. Other domains of both RasGRP1 [39] and RasGRP2 [22,55] can have dominant effects on the protein localization, and thus could obscure equivalent contributions of their C1 domains to localization. It remains unknown whether the C1 domain of RasGRP2 contributes to its localization to membranes, and if the C1 domains do contribute, whether they do so by binding DAG.
The objective of the present study was to determine the abilities of the C1 domains of RasGRP2, RasGRP4α and RasGRP4β to associate with membranes by interacting with DAG. Our in vivo and in vitro experiments demonstrated that the C1 domain of RasGRP4α binds DAG within membranes, although with a moderately reduced affinity compared with the RasGRP1 C1 domain. In contrast, the C1 domains of RasGRP2 and RasGRP4β did not bind DAG, although they interacted with membrane vesicles enriched in anionic phospholipids in an manner equivalent to that of other C1 domains. From these results, we conclude that RasGRP2 and RasGRP4β cannot be regulated directly by DAG binding to their C1 domains, thus disproving the common assumption that all RasGRPs are regulated by membrane translocation driven by a DAG–C1 domain interaction [45,48,50,51,56,57]. However, our finding that all of the RasGRP C1 domains can interact with anionic phospholipids suggests that this mechanism could contribute to membrane localization of RasGRPs.
MATERIALS AND METHODS
Construction and expression of GFP (green fluorescent protein)–C1 domain fusion proteins
cDNAs encoding the C1 domains were generated by PCR from cDNAs and cloned into the pEGFP-C1 vector (Clontech) to create GFP–C1 domain fusion proteins upon expression. The sequences of the encoded C1 domain peptides are shown in Figure 1(A). All of the C1 domains are murine-derived, with the exception of human Raf-1. The sequence of the C-terminal portion of the GFP peptide in these constructs is DELYKSGLRSLKST, with this sequence being fused to the N-termini of the C1 domain sequences in Figure 1(A). The fusion constructs encoding the GFP–C1 domain were cloned into the retroviral vector pCTV211 [58], converted into retroviral particles by transfection into the BOSC 23 packaging cell line [59] and transduced into NIH 3T3 (A.T.C.C.) or DO11.10 [60] cell lines. Polyclonal populations of transduced cells were obtained by selection using puromycin.
Fluorescence microscopy
Transduced NIH 3T3 cells were cultured on coverslips for 24 h in DMEM (Dulbecco's modified Eagle's medium) containing 4.5 g/l glucose plus 10% (v/v) fetal calf serum, then transferred to the same medium containing either 10% or 0% (v/v) fetal calf serum for an additional 16 h. The cells were treated for 5 min with 2 μM PMA (Sigma) in DMSO, 100 μM dioctanoylglycerol (Sigma) in DMSO, or with DMSO alone as a control. DO11.10 T-cells [60] and WEHI-231 B-cells (A.T.C.C.) were allowed to adhere to poly-L-lysine-coated coverslips and cultured in DMEM supplemented with 10% (v/v) fetal calf serum for 24 h. After fixation with 4% (v/v) formaldehyde in PBS for 2 min, the cells were permeabilized with 0.2% (v/v) Triton X-100 in PBS and stained with an Alexa Fluor® 488-conjugated anti-GFP antibody (Invitrogen) as a secondary antibody. This increases the sensitivity of detection of GFP compared with GFP fluorescence on its own. To highlight Golgi membranes, cells were co-stained with the anti-GFP antibody and a mouse anti-GM130 antibody (BD Biosciences), followed by use of an Alexa Fluor® 647-conjugated anti-(mouse IgG) antibody (Invitrogen) as a secondary antibody. ER (endoplasmic reticulum) was stained by treatment of unfixed cells with glibenclamide BODIPY® (4,4-difluoro-4-bora-3a,4a-diaza-s-indacene)–Texas Red ER Tracker (Invitrogen), followed by fixation using formaldehyde in PBS and subsequent staining with anti-GFP was performed as described above. Images were captured using OpenLab imaging software.
Preparation of cellular membrane and cytosol fractions
Transduced NIH 3T3 cells cultured to 50% confluence were serum-starved for 3.5 h in DMEM containing 1 mg/ml BSA and were then treated for 15 min with DMEM containing 1 mg/ml BSA and 300 μM dioctanoylglycerol, a short-chain DAG. On ice, cells were washed three times with cold PBS, scraped into 0.25 ml of lysis buffer [20 mM Tris (pH 7.4), 2 mM EDTA, 2 mM PMSF, 2.5 μg/ml leupeptin, 2 μg/ml chymostatin, 1 μg/ml antipain, 2 μg/ml pepstatin, 10 μg/ml benzamidine and 10 μg/ml p-aminobenzadine] and lysed by sonicating twice for 30 s in ice at 20% power using a Fisher Model 300 ultrasonic generator. The NaCl concentration was adjusted to 0.15 M, and the lysate was centrifuged at 100000 g for 60 min to separate cytosol from particulate. The resulting pellet was resuspended by two rounds of sonication for 30 s on ice at 20% power in lysis buffer with 0.15 M NaCl and 1% (v/v) Triton X-100, and centrifuged at 100000 g for 60 min to separate detergent-soluble membranes from insoluble particulate matter. Equivalent volumes of each fraction were added to Laemmli sample buffer without 2-mercaptoethanol and were subjected to, without boiling, SDS/PAGE (10% gels). Gels were immediately scanned with a Typhoon 9410 imager (488 nm Blue2 laser, 520 nm BP filter) (Amersham Biosciences) and the fluorescence of GFP–C1 domain bands was quantified using the ImageQuant program. The percentage of the GFP fluorescence in the membrane fraction was calculated as the fluorescence intensity of the band in the detergent-soluble microsome fraction relative to the sum of the intensities of the protein bands in the three subcellular fractions. The recovery in the three subcellular fractions, which was expressed as a percentage of the total-cell lysate fluorescence intensity, was on average 99±9%(S.D.), with a range of 87–116%.
Construction of K-Ras–C1 fusion, RasGRP1Δ–C1 fusion and RasGRP4α–GFP and RasGRP4β–GFP fusion constructs
The K-Ras Q61N Δ173–188 mutant has the C-terminal amino acid sequence KEKMSKDGSTEA (residues not naturally present in K-Ras are italicized). Fusions of K-Ras Q61N Δ173–188 to C1 domains contain the sequence KEKMSKDGST fused to the N-termini of the C1 domain sequences in Figure 1. For the K-Ras/prenylation construct, the basic cluster and prenylation signal of K-Ras, along with an HA (haemagglutinin) epitope tag, was reattached to the K-Ras Q61N Δ173–188 mutant, resulting in the C-terminal sequence KEKMSKDGSTEAYPYDYASGSRKHKEKMSKDGKKKKKKSKTKCVIM. The N-terminus of K-Ras Q61N Δ173–188 was fused to GFP, with the junction sequence being DELYKSGLRSFLLKMTEYKLVVV.
The N-terminal GFP-tagged and C-terminally deleted form of murine RasGRP1 (RasGRP1Δ) was fused to GFP at amino acid 2 of RasGRP1 (GenBank® accession NP_035376). The sequence at this N-terminal fusion site is DELYKSGLRSSAQSE-GTLGKAR with the natural sequence of RasGRP1 in bold. The sequence at the C-terminal fusion junction is YSKLG-ST[C1 domain], with the C1 domain sequences shown in Figure 1.
Murine RasGRP4α and RasGRP4β with N-terminal GFP tags contained the sequence DELYKSGLRSLKSNRKDIKRKS at the GFP-RasGRP4 junction, with the natural sequence of RasGRP4 (starting at amino acid 2) in bold. The encoded RasGRP4α is identical with the database sequence (GenBank® accession AF331457), while the encoded RasGRP4β is identical except for the five-amino-acid insertion in the C1 domain.
NIH 3T3 transformation assays
cDNAs encoding the specified K-Ras fusion constructs were inserted into the retroviral vector pCTV211, converted into retrovirus, transduced into NIH 3T3 cells and selected as described above. Transduced cells were seeded at 10% confluence. For assessment of transformation in 10% serum, the cells were then cultured continuously for 3 days (K-Ras only) or 5–7 days (for RasGRPs) after the cells had formed a monolayer, then imaged. For low-serum transformation assays, the cells were transferred to DMEM containing 0.5% (v/v) fetal calf serum for 1 day, followed by another 2 days in DMEM containing 0.5% (v/v) fetal calf serum only (control) or supplemented with 0.1 μM PMA. The cells were then imaged. The photographed regions were representative of the appearance of the cells across the entire culture dish. To quantify transformation efficiency, NIH 3T3 cultures transduced with the indicated constructs were seeded at a low density. After colonies had grown up from individual cells, they were scored either as transformed or non-transformed cells:
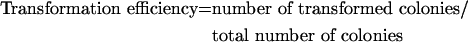 |
More than 30 colonies were scored.
Construction, expression and purification of GST (glutathione S-transferase)–C1 fusion proteins
cDNAs encoding C1 domains were inserted into the GST fusion vector pGEX-2T (Amersham Biosciences), resulting in N-terminal GST fusions containing the junction sequence LVPRGSLKST, with this sequence being fused to the N-termini of the C1 domain sequences in Figure 1(A). The plasmids were transformed into Escherichia coli strain AD202 and cultured in Luria–Bertani medium overnight at 37 °C to saturation. The cell culture was then diluted 40-fold and grown at 37 °C to D600 of 0.8. After 10 min on ice, cells were induced with 0.5 mM IPTG (isopropyl β-D-thiogalactoside) for 5 h at 28 °C. The bacterial cell pellet was resuspended in cold PBS containing 1% (v/v) Triton X-100 and 2 mM PMSF, and the cells were lysed by use of a French press. Insoluble material was removed by centrifugation at 15000 g for 30 min. GST–C1 domains in the soluble fraction were incubated for 2 h at 4 °C with glutathione–Sepharose beads pre-equilibrated in 1% (v/v) Triton X-100 in PBS (Amersham Bioscience) (2 ml of a 50% slurry of glutathione–Sepharose beads/litre of cultured cells). The glutathione–Sepharose beads were collected using a mini-column and washed sequentially with 1% (v/v) Triton X-100 in PBS, PBS, 50 mM Tris/HCl (pH 7.5) and 0.15 M NaCl before elution of the GST fusion protein in 50 mM Tris/HCl (pH 7.5), 0.15 M NaCl and 5 mM glutathione. The fusion protein concentration was quantified by use of a Bradford assay, using ovalbumin as a standard. The GST fusion proteins were divided into portions and stored at −80 °C for use in the in vitro vesicle-binding assay. On average, 40 mg of the GST–C1 fusion protein was obtained per litre of bacterial culture.
Binding of GST–C1 domain fusion proteins to sucrose-loaded phospholipid vesicles
Binding of GST–C1 domain fusion proteins to SLVs (sucrose-loaded large unilamellar vesicles) was measured using a protocol based on one described previously [17,61]. PC (phosphatidylcholine) (1-palmitoyl-2-oleoyl PC), PS (phosphatidylserine) (1,2-dioleoyl PS), PG (phosphatidylglycerol) (1,2-dioleoyl PG), a longchain DAG (1,2-dioleoylglycerol), PA (phosphatidic acid) (1-palmitoyl-2-oleoyl PA), LPA (lysophosphatidic acid) (1-palmitoyl-2-hydroxyglycerophosphate) and ceramide (N-palmitoyl-D-erythro-sphingosine) were all obtained from Avanti Polar Lipids or Northern Lipids, and oleic acid and arachidonic acid were purchased from Sigma. Lipid vesicles were prepared by drying mixtures of lipids in chloroform (including trace amounts of [3H]dipalmitoyl PC for to allow quantification of recovery) on a rotary evaporator, which was resuspended in 20 mM Hepes (pH 7.5) and 170 mM sucrose by vigorous vortex-mixing to a final lipid concentration of 2 mM, followed by five freeze–thaw cycles in liquid nitrogen. These multilamellar vesicles were stored at −20 °C until use. On the day of the experiment, SLVs were prepared by extrusion at room temperature (20 °C) through a 100 nm pore-size membrane using a Lipofast Microextruder (Avestin). Extruded vesicles were diluted 5-fold in 100 mM NaCl and 20 mM Hepes (pH 7.5) and centrifuged at 100000 g for 30 min at 25 °C to dilute the free sucrose. The majority (80%) of the supernatant was removed, and the resulting pellet was resuspended by vigorous vortex-mixing in the residual volume. PMA (in DMSO as a stock) was subsequently added and vortex-mixed vigorously such that the final concentration of DMSO was <2% (v/v). GST–C1 domain fusion proteins (0.38–0.48 μM) were incubated with SLVs (200 μM, except 125 μM in Figure 6A) for 10 min in the presence of 20 mM Hepes (pH 7.5), 100 mM NaCl and 0.05 mg/ml ovalbumin in a volume of 120 μl. Vesicle-bound protein was separated from unbound protein by centrifugation at 100000 g for 30 min at 25 °C. More than 90% of the [3H]dipalmitoyl PC label was present in the pellet fraction. Separate samples of fusion proteins were treated in the same way in the absence of added SLVs to quantify vesicle-independent sedimentation. The quantity of protein in each fraction was determined by analysis of supernatant and pellet fractions by resolution on SDS/PAGE on a Tricine gel system [62]. Gels were stained with SYPRO Orange stain (Invitrogen) according to the manufacturer's instructions, and imaged using a Typhoon 9410 imager [488 nm Blue2 laser, 580 nm BP (band-pass) filter]. The fluorescence intensities of the GST–C1 bands were quantified using the ImageQuant program and calculated as follows:
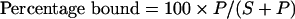 |
where P is the intensity of the fluorescence band as calculated from the pellet fraction and S is the intensity of the fluorescence band from the supernatant fraction. The pellet intensities were corrected for contamination by supernatant, as well as protein that sedimented in the absence of vesicles. The latter correction resulted in some percentage bound values being less than zero. The apparent association constant, Ka=(B/F)×1/L, where B is fraction of protein bound in the pellet, F is the fraction of free protein in the supernatant and L is the molar concentration of accessible lipid (0.5×total lipid concentration, since the C1 domain only binds the outer leaflet).
Figure 6. Lack of binding of RasGRP C1 domains to alternative lipid ligands.

(A) GST fusions of the indicated RasGRP C1 domains were assayed for binding to PC SLVs (white bars); or PC SLVs containing 10 mol% PS (grey bars), 10 mol% PS and 5 mol% DAG (black bars), or 10 mol% PS and 5 mol% PA (hatched bars). The percentage bound values were not corrected for sedimentation in the absence of lipid. Data are means±range for two independent experiments. RG, RasGRP. (B) GST fusions of the C1 domains of RasGRP1 (white bars), RasGRP2 (black bars), and RasGRP4β (hatched bars) were mixed with PC SLVs containing 5 mol% PS, and 5 mol% of oleic acid (OA), arachidonic acid (AA), ceramide (Cer.), sphingosine 1-phosphate (Sph-1P), LPA or 1 mol% DAG. Data are means±range for two independent experiments.
RESULTS
Sequence comparison of RasGRP C1 domains
Modelling of the RasGRP1 C1 domain indicates that the A and B loops can form a ligand-binding pocket that is very similar to that of the C1b domain of PKCδ [63]. The RasGRP3 C1 domain is identical with that of RasGRP1 in loop A, except for an asparagine residue in place of a threonine residue at the base of the loop. Loop B has a isoleucine/valine switch present (Figure 1A). To assess the DAG- or phorbol-ester-binding potentials of the C1 domains of RasGRP2 and the two splice variants of RasGRP4, we compared their ligand-binding loops with those of RasGRP1 and 3, the C1b domain of PKCδ and to the C1 domain of β2-chimaerin (Figure 1A).
There are two atypical residues in loop A of the RasGRP2 C1 domain. RasGRP2 has a serine residue at position 8 compared with tyrosine or phenylalanine in the DAG/phorbol-ester-binding C1 domains. Mutation of Tyr8 to glycine in the PKCδ C1b domain reduced, but did not eliminate, phorbol ester binding [64]. The other atypical residue in loop A of RasGRP2 is Val12, which is threonine or histidine in the DAG- and phorbol-ester-binding C1 domains (Figure 1A). Mutation of Thr12 to valine in PKCδ C1b resulted in a minor reduction in phorbol ester binding of the C1b domain [64], while a similar mutation in the DAG kinase β C1a domain from Ala12 to threonine moderately increased phorbol ester binding [65]. The significant discrepancy in loop B of the RasGRP2 C1 domain is Leu22, which corresponds with tryptophan in the DAG- and phorbol-ester-binding C1 domains (Figure 1). In the C1b domain of PKCβ, switching this residue from Tyr22 to tryptophan increased the affinity for DAG and increased the co-localization with perinuclear membranes [66]. Switching tyrosine to tryptophan at this position has the potential to affect DAG affinity by causing major perturbations in the shape of the ligand-binding pocket [66]. The effect of leucine at position 22 is unknown. While the variations at positions 8, 12 and 22 serve as ‘warning flags’ that the DAG- or phorbol-ester-binding capabilities of the RasGRP2 C1 domain should not be taken for granted, none of these changes by itself is predicted to eliminate binding of either of these ligands.
The RasGRP4α C1 domain has no atypical residues in loops A or B relative to the DAG-/phorbol-ester-binding C1 domains shown in Figure 1(A). Therefore it is expected to bind both DAG and phorbol esters. In RasGRP4β, there is an insertion of five amino acids at the N-terminus of loop B. Although this insertion could radically disrupt the ligand-binding pocket, it is possible that the inserted amino acids project outward, with most of loop B remaining in position to form one side of the ligand-binding pocket. In the latter case, the RasGRP4β C1 domain could retain binding of DAG and/or phorbol esters, or could have an altered ligand specificity relative to the C1 domain of RasGRP4α.
All of the RasGRP C1 domains have cationic residues within their C1 domain and C-terminal to their C1 domain (Figure 1A), which are positioned to interact electrostatically with anionic phospholipids [7]. In conjunction with the hydrophobic residues projecting from loops A and B, these have the potential to either co-operate with DAG to enhance membrane-binding affinity or mediate DAG-independent interactions with membranes.
To summarize, the C1 domain of RasGRP4α is expected to bind both DAG and phorbol esters on the basis of the amino acid sequence similarities in loops A and B to other C1 domains, but the ability of the C1 domains of RasGRP2 and RasGRP4β to bind either of these ligands cannot be predicted with confidence from their sequences. All of the RasGRP C1 domains have the potential to bind membranes independently of DAG or phorbol esters by means of electrostatic and hydrophobic interactions. A combination of in vivo and in vitro experiments are needed to resolve the question of whether all RasGRP C1 domains can mediate the membrane binding required for juxtaposing these exchange factors with their Ras or Rap GTPase substrates and to assess whether this is achieved by direct binding of the C1 domains to DAG.
Only the C1 domains of RasGRP1, RasGRP3 and RasGRP4α co-localize with membranes and translocate in response to DAG or phorbol esters
Fusion constructs of C1 domains with GFP were expressed in NIH 3T3 fibroblasts to compare the localization of the five RasGRP C1 domains to the DAG-binding PKCδ C1b domain with the localization of the Raf-1 C1 domain, which does not bind DAG. The GFP–C1 domain fusions for RasGRPs and PKCδ included clusters of cationic amino acids which are found naturally immediately C-terminal to the C1 domains, as these have been shown to contribute to membrane binding in vivo and in vitro [19,38]. For the Raf-1 C1 domain, the cationic cluster from RasGRP1 was attached to ensure that any deficiencies in membrane binding demonstrated by the Raf-1 C1 domain were not simply due to its lack of a cluster of cationic amino acid residues.
In NIH 3T3 cells cultured in 10% serum, the Raf-1 C1 domain and GFP alone were both distributed throughout the nucleus and cytoplasm (Figure 2A). In contrast, the PKCδ C1b domain and the RasGRP1 C1 domain were excluded from the nucleus and were concentrated in the perinuclear cytoplasm, where they co-localized with internal membranes. This was shown by staining of GFP–RasGRP1 C1 domain-expressing cells with ER tracker (glibenclamide), which binds to sulfonylurea receptors in the ER [67] or by fluorescence using an antibody to the Golgi-associated protein GM130 [68] (Figure 2B). The C1 domain of RasGRP3 was also largely co-localized with internal membranes, while the C1 domain of RasGRP4α was similarly localized, but was accumulated away from internal membranes and partially localized to the nucleus. The C1 domains of RasGRP2 and RasGRP4β were radically different in their localization from RasGRP1 or RasGRP3 C1 domains. The Ras GRP1 and Ras GRP3 C1 domains were distributed throughout the cytoplasm and nucleus, as was seen for the Raf-1 C1 domain and GFP alone. A similar pattern of C1 domain localization was seen in the T-cell line DO11.10 and the B-cell line WEHI-231, where the C1 domains of RasGRP1 and RasGRP3 strongly co-localized with internal membranes, the C1 domains of RasGRP2 and RasGRP4β being dispersed throughout the cells, and the C1 domain of RasGRP4α being partially excluded from the nucleus, but only weakly co-localized with internal membranes (Figure 3).
Figure 2. Only the C1 domains of RasGRP1, RasGRP3 and RasGRP4α co-localize with membranes and translocate in response to DAG or phorbol esters.

(A) GFP fusions of the indicated C1 domains, or GFP alone, were expressed in NIH 3T3 cells. After culture in DMEM containing 10 or 0% (v/v) calf serum with or without dioctanoylglycerol (indicated as DAG in the Figure) or PMA, the cells were fixed and imaged by fluorescence microscopy. (B) NIH 3T3 cells expressing the GFP–RasGRP1 C1 domain fusion were stained with either ER Tracker (to mark ER) or anti-GM130 antibody (to mark Golgi membranes) as described in the Materials and methods section. Individual cells showing fluorescence from the GFP-tagged RasGRP1 C1 domain and either ER Tracker staining or GM130 staining are shown.
Figure 3. Distribution of C1 domains in DO11.10 T-cells and WEHI-231 B-cells.

(A, C) GFP fusions of the indicated C1 domains, or GFP alone, were expressed in DO11.10 T-cells (A) or WEHI-231 B-cells (C). After culture in DMEM containing 10% (v/v) fetal calf serum, the cells were fixed and imaged. (B, D) DO11.10 T-cells (B) or WEHI-231 B-cells (D) expressing the GFP–RasGRP1 C1 domain fusion were stained with either ER Tracker (to mark ER) or anti-GM130 antibody (to mark Golgi membranes) as described in the Materials and methods section. After culture in DMEM containing 10% (v/v) fetal calf serum, the cells were fixed and imaged.
Culturing NIH 3T3 cells in medium lacking serum resulted in a considerable disturbance in the distribution of the RasGRP4α C1 domain from perinuclear membranes into the nucleus (Figure 2A). Serum starvation had no noticeable effect on the other C1 domains. Relative to the RasGRP1 and RasGRP3 C1 domains, the RasGRP4α C1 domain appeared to be more dependent on serum stimulation to maintain its localization at the internal membrane, as the domain was less co-localized upon starvation. However, it is clearly distinguished from the C1 domains of RasGRP2 and RasGRP4β which lack co-localization with membranes.
Treatment of the serum-starved NIH 3T3 cells with dioctanoylglycerol, a short-chain DAG, induced accumulation of the C1 domains of PKCδ and RasGRP1, RasGRP3 and RasGRP4α in the nuclear envelope, while retaining their localization in the perinuclear region (Figure 2A). This membrane-selective relocalization presumably reflects accumulation of exogenous DAG at the nuclear envelope. Exogenous DAG could also have accumulated at the ER and Golgi, but with no discernible effect on the C1 domains, because they were already located at these sites, potentially by the presence of endogenous DAG. PMA also resulted in relocalization of the C1 domains of RasGRP1, RasGRP3, RasGRP4α, and PKCδ. PMA had no observable effect on the localization of the C1 domains of Raf-1, RasGRP2 or RasGRP4β (Figure 2A), nor did DAG when serum-starved cells were incubated at either 100 μM for 5 min (Figure 2A) or 300 μM for 5 or 15 min (results not shown).
A differential response of the C1 domains of RasGRP1 and RasGRP4α compared with RasGRP2 and RasGRP4β was observed when DAG-induced membrane binding was assessed by fractionation of NIH 3T3 cells. GFP-tagged C1 domains were quantified in cell lysates, cytosol, detergent-solubilized particulate fraction (membrane fraction) and a detergent-insoluble particulate fraction. The percentage of each C1 domain in the membrane fraction is shown in Figure 4. In serum-starved cells, less than 10% of each C1 domain was present in the membrane fraction. After DAG treatment, 35–40% of the C1 domains of PKC, RasGRP1 and RasGRP4α were membrane-associated, whereas the RasGRP2 and RasGRP4β C1 domains, like the Raf-1 C1 domain, did not show significant DAG-induced changes in membrane binding.
Figure 4. DAG induces translocation of the C1 domains of RasGRP1, RasGRP3 and RasGRP4α to membranes.

NIH 3T3 cells expressing the indicated GFP-tagged C1 domains were cultured in serum-free medium for 3.5 h, then treated for 15 min with DMSO (white bars) or 300 μM dioctanoylglycerol (indicated as DAG in the Figure, grey bars). Cells were fractionated and the GFP–C1 proteins in the membrane and non-membrane fractions were quantified as described in the Materials and methods section. Results are means±S.E.M. for two independent experiments.
The C1 domains of RasGRP1 and RasGRP4α bind directly to DAG within phospholipid membranes, while the C1 domains of RasGRP2 and RasGRP4β do not
To determine whether the differences in membrane localization of the RasGRP C1 domains reflected differences in their abilities to directly bind DAG or phorbol esters within phospholipid bilayers, we investigated their binding to unilamellar phospholipid vesicles in vitro. C1 domains were expressed in E. coli as GST fusion proteins and purified by affinity chromatography on glutathione–agarose beads. The GST–C1 fusion proteins resolved as expected by SDS/PAGE (single 34 kDa species), with the exception of the GST–RasGRP3 C1 construct, which had a significant amount of a smaller 26 kDa GST species present, indicating partial premature termination of translation near the GST/C1 junction had occurred.
Binding of the C1 domains to phospholipid bilayers was assessed by their co-sedimentation with SLVs composed of PC supplemented with 5 mol% of the anionic phospholipid PS, either alone or in combination with 5 mol% 1,2-dioleoylglycerol (a long-chain DAG) or 1 mol% PMA. The PKCδ C1b and Raf-1 C1 domains were used as positive and negative controls to test this system (Figure 5A). The PKCδ C1b domain bound poorly to the PC/5% PS vesicles and very well to vesicles containing DAG or PMA. In contrast, the Raf-1 C1 domain had no significant vesicle binding in the absence or presence of DAG or PMA.
Figure 5. Binding of the C1 domains of RasGRP1, RasGRP3 and RasGRP4α to DAG or PMA within membrane vesicles.

(A) GST fusions of the indicated C1 domains were mixed with SLVs composed of PC and 5 mol% PS (open bars) or 5 mol% PS and 5 mol% 1,2-dioleoylglycerol (black bars) or 5 mol% PS and 1 mol% PMA (hatched bars). Binding was assessed by co-sedimentation as described in the Materials and methods section. Some values are less than 0% because of correction for the amount of protein which sedimented in the absence of vesicles. Data are means±range for two independent experiments. (B) Binding of GST fusions of the RasGRP1 (□), RasGRP4α (Δ), and PKCδ C1 (◊) domains to PC SLVs containing 10 mol% PS with various mol% DAG. Data are means±range for two independent experiments. (C) Binding of GST fusions of the RasGRP1, RasGRP4α and PKCδ C1 domains to PC SLVs containing 2 mol% DAG and either no anionic lipid (open bars), 5 mol% PS (black bars), 2.5 mol% PS and 2.5 mol% PG (hatched bars), or 5 mol% PG (cross-hatched bars). Data are means±range for two independent experiments. RG, RasGRP.
The RasGRP1 C1 domain behaved similarly to the PKCδ C1b domain, as the addition of either DAG or PMA induced nearly complete binding to the vesicles (Figure 5A). The RasGRP3 and RasGRP4α C1 domains were similar in binding preferences to the C1 domain of RasGRP1. Nearly complete binding occurred to the PMA-containing vesicles, but this binding affinity was reduced for DAG-containing vesicles (Figure 5A). The C1 domains of RasGRP2 and RasGRP4β had minimal vesicle binding and this was not altered by the addition of either DAG or PMA.
These vesicle-binding properties mirror the results obtained from the in vivo experiments and confirm that direct binding to DAG and PMA is restricted to the C1 domains of RasGRP1, RasGRP3 and RasGRP4α. This initial vesicle-binding experiment also indicated that there could be quantitative heterogeneity among these three DAG-binding C1 domains. However, the lower DAG binding of the RasGRP3 C1 domain seen in Figure 5(A) may be artifactual, because it was produced as a mixture of full-length and truncated GST–C1 fusion proteins. It is known that GST can dimerize, and this would have resulted in some GST binding to GST–C1, resulting in heterodimers which would have reduced avidity for membranes relative to GST–C1 homodimers, owing to a reduction in the number of DAG-binding sites within the heterodimer compared with the homodimer. Considering this anomaly, the RasGRP3 C1 domain was not used in subsequent experiments aimed at quantifying differences in DAG-dependent and DAG-independent membrane binding.
The C1 domains of RasGRP1 and RasGRP4α have different affinities for DAG
Binding of the RasGRP1 C1 domain was compared with the binding of the RasGRP4α C1 domain to PC/10 mol% PS vesicles, in the presence of DAG concentrations ranging from 0 to 2 mol% (Figure 5B). Binding of the RasGRP4α C1 domain was less sensitive to DAG at low concentrations and reached saturation at 2 mol% DAG, compared with 1 mol% DAG for the RasGRP1 C1 domain. The binding curve for the PKCδ C1b domain was an intermediate curve between those for RasGRP1 and for RasGRP4α at low DAG concentrations; saturation was reached at a lower concentration of DAG than was required for the RasGRP4α C1 domain. From these binding curves we calculated the apparent affinity constants for membranes containing 10 mol% PS and 0.2 mol% DAG. The values are 21000, 7500 and 3800 M−1 for the RasGRP1, PKCδ and RasGRP4α C1 domains respectively. Figure 5(B) also indicated that the RasGRP1 C1 domain binds more strongly to 10 mol% PS vesicles containing no DAG, in comparison with the RasGRP4α or PKCδ C1b domains.
The role of anionic phospholipid in the response to DAG is illustrated in Figure 5(C). Binding of the C1 domains of PKCδ, RasGRP1 or RasGRP4α to vesicles containing 2 mol% DAG was enhanced by the presence of either PS or PG (anionic phospholipids) at 5 mol%. Of the three, the RasGRP4α C1 domain showed the weakest binding and was the most dependent on the presence of the anionic phospholipid. Membrane binding of the RasGRP C1 domains does not specifically require the PS headgroup. Instead, they appear to respond to the negative charge on the membrane surface, which is the case for the PKCδ C1b domain as well as other PKC C1 domains [17].
PA, LPA, ceramide, fatty acids and sphingosine 1-phosphate are not alternative ligands for RasGRP2 or RasGRP4β
The RasGRP2 and 4β C1 domains do not bind to DAG, but their sequences are compatible with their having a modified pocket structure that could bind to an alternative ligand. To address the hypothesis that the C1 domains of RasGRP2 and RasGRP4β are specialized to recognize lipid second messengers other than DAG, we tested candidate lipid ligands which have small headgroups which are likely to be suitable for occupying the pocket and which act as signalling molecules.
PA was of particular interest as a ligand because it can be generated from DAG by DAG kinases, which have been functionally linked to RasGRPs [25–27,69]. However, PA at 5 mol% did not promote vesicle binding of any of the C1 domains, under conditions in which 5 mol% DAG promoted nearly complete binding of the C1 domains of RasGRP1, RasGRP4α and PKCδ (Figure 6A). Other signalling lipids with larger headgroups, LPA and sphingosine 1-phosphate, also failed to promote binding of the C1 domains of RasGRP1, RasGRP2 or RasGRP4β (Figure 6B). Ceramide is structurally similar to DAG and has been postulated to be a potential ligand for the non-DAG-binding C1 domains of Raf-1, PKCs and DAG kinases, although a direct interaction with these or any other C1 domain has not been demonstrated [70–72]. Ceramide at 5 mol% was also ineffective as a ligand for the C1 domains of RasGRP1, RasGRP2 or RasGRP4β (Figure 6B). Fatty acids, particularly polyunsaturated species such as arachidonic acid, have been reported to influence the activity of some PKC isoforms [73]. Arachidonic acid results in a C1b-dependent redistribution of PKCϵ in CHO (Chinese-hamster ovary) cells [70], which implied that it could be a ligand for other C1 domains. Arachidonic acid and another fatty acid, oleic acid, both induced a minor increase in membrane binding of the C1 domains of RasGRP1 and RasGRP2 (Figure 6B). However, this is likely to be as the result of an increased membrane negative charge provided by these anionic lipids (see below).
High concentrations of anionic phospholipids enable membrane binding by RasGRP C1 domains in the absence of DAG
Electrostatic interactions with anionic phospholipids have the potential to provide a DAG-independent mechnism for membrane binding by C1 domains. The anionic-lipid-dependence for membrane binding in the absence of DAG was tested, and the results are shown in Figure 7(A). All RasGRP C1 domains bound vesicles in an anionic-lipid-dependent manner. GST alone did not bind these vesicles, regardless of the anionic lipid composition (results not shown). All C1 domains were >60% bound to vesicles containing 40 mol% PS, but the sensitivity to lower PS concentrations was variable. The RasGRP1 C1 domain was the most PS-sensitive of all the C1 domains tested, with considerable binding to vesicles containing just 10 mol% PS and only minor increases in binding at higher PS concentrations. Along with the PKCδ C1b domain, the RasGRP4β C1 domain had the weakest binding response, requiring greater than 20 mol% PS for a similar level of binding to other C1 domains tested. The C1 domains of RasGRP2, RasGRP4α and Raf-1 had intermediate dependencies on PS, with transitions from low to high binding occurring between 10 and 20 mol% PS.
Figure 7. RasGRP C1 domains bind to vesicles enriched in anionic phospholipids.

(A) Dependence of C1 domain binding on the mol% of PS. GST fusions of the indicated C1 domains were mixed with SLVs composed of PC and the indicated amounts of PS. Data are means±range for two independent experiments. The curves were generated by the sigmoidal dose-dependence variable slope option of GraphPad Prism, with the exception of the curve for the RasGRP1 C1 domain, which was drawn by hand to give a sigmoidal curve fitting the known percentage bound value of 20% at 5 mol% PS (Figure 5A). (B) C1 domain binding to anionic phospholipids is not headgroup-specific. GST fusions of the indicated C1 domains were assayed for binding to PC SLVs (open bars), 70 mol% PC and 30 mol% PS (black bars), 30 mol% PG (hatched bars), or 30 mol% PA (cross-hatched bars). Data are means± range for two independent experiments. RG, RasGRP.
In addition to their charges, the specific structures of anionic phospholipid headgroups could influence C1 domain binding to membranes. This is of considerable biological interest because the C1b domain of PKCδ has a selective affinity for PS, which contributes to its preferential localization at the PS-enriched plasma membrane [11,74]. We observed a selectivity for PS by the C1b domain of PKCδ (Figure 7B). The RasGRP C1 domains, like the Raf-1 C1 domain, showed no significant preference for PS compared with PG or PA and are therefore likely to interact with anionic phospholipids strictly through an electrostatic interaction rather than by specific headgroup recognition. The data in Figure 7(B) also show weaker binding of RasGRP4β and PKCδ C1 domains to anionic lipids compared with the other C1 domains.
These experiments demonstrated that all RasGRP C1 domains can bind to membranes in the absence of DAG if anionic phospholipids are present at sufficient concentrations. Because anionic phospholipid concentrations range between 10 and 20% in cellular membranes [75], this property of RasGRP C1 domains may be of a biological significance and, in particular, could contribute to the differential targeting of RasGRPs to specific membranes within cells. In the following experiments, we tested the hypothesis that the C1 domains of RasGRP2 and RasGRP4β could contribute to membrane binding in vivo, despite being unable to bind to DAG.
Only the DAG-binding C1 domains of RasGRPs can complement a membrane-binding-deficiency mutation in K-Ras
Complementation of a membrane-localization-defective mutant of K-Ras has been used previously to demonstrate the ability of the RasGRP1 C1 domain to confer membrane binding [38]. We used this approach to test the abilities of the other RasGRP C1 domains to bind membranes in vivo, to determine whether membrane binding could occur even when the C1 domain was unable to bind specifically to DAG.
In NIH 3T3 cells, constitutive signalling from the mutationally-activated Q61N form of K-Ras induces oncogenic transformation, detectable by cell contraction from the substratum, high refractility and loss of contact inhibition. Signal transduction by K-Ras is entirely dependent on its membrane localization, which is naturally provided by prenylation at the C-terminus, in combination with a polybasic cluster of amino acids (Figure 8A). As a result, the Q61N Δ173–188 K-Ras double mutant (K-RasΔ), which has a deletion of the C-terminal basic cluster and prenylation signal, was non-transforming. Reattachment of the basic cluster and prenylation signal to K-RasΔ resulted in restoration of transformation, which was evident even when the cells were cultured in only 0.5% serum (Figure 8B).
Figure 8. Only the C1 domains of RasGRP1, RasGRP3 and RasGRP4α provide serum- or phorbol-ester-dependent complementation of a membrane-binding-deficient K-Ras mutant.

(A) Structures of K-Ras proteins. K-RasΔ is shorthand for K-Ras Q61N Δ173–188. K-RasΔ+pren is K-Ras Q61N Δ173–188 with the K-Ras basic cluster+prenylation signal reattached. K-RasΔ+C1 is fusion of a C1 domain to the C-terminus of K-Ras Q61N Δ173–188. (B) Transformation assays. The indicated constructs were expressed in NIH 3T3 cells. After culture in medium containing 10% (v/v) fetal calf serum, or 0.5% (v/v) fetal calf serum with or without PMA, the cell cultures were photographed to distinguish those forming a non-refractile contact-inhibited monolayer (non-transformed) from those exhibiting contraction from the substratum, high refractility and loss of contact inhibition (transformed via K-Ras activation).
Attachment of the RasGRP1 C1 domain to the C-terminus of K-RasΔ resulted in transformation when the cells were cultured in 10% serum, but not when they were cultured in 0.5% serum. The serum-independent transformation by prenylated K-Ras, compared with the serum-dependent transformation by K-Ras–RasGRP1 C1 domain may reflect the depletion of a serum-induced C1 ligand, presumably DAG. This interpretation is supported by the observation that transformation via the K-Ras–RasGRP1 C1 fusion protein did occur when the low-serum medium was supplemented with a C1 domain ligand, PMA. Equivalent experiments using DAG supplementation were deemed impractical, because DAG is rapidly metabolized.
The C1 domains of RasGRP3 and RasGRP4α demonstrated equivalent behaviour to that of the RasGRP1 C1 domain in conferring serum-dependent or PMA-dependent complementation of the membrane-binding mutation in K-RasΔ (Figure 8B). In contrast, the fusion of K-RasΔ to the C1 domains of RasGRP2 or 4β were non-transforming under any conditions. This experiment confirmed that stable membrane binding and responsiveness to PMA are restricted to the C1 domains of RasGRP1, RasGRP3 and RasGRP4α, and demonstrated that the C1 domains of RasGRP2 and RasGRP4β are incapable of conferring membrane binding to K-Ras at the level required to trigger NIH 3T3 cell transformation.
Despite lacking discernible membrane localization in vivo, the C1 domain of RasGRP2 can functionally replace the C1 domain of RasGRP1
Expression of RasGRP1 also induced transformation of NIH 3T3 cells via its stimulation of GTP loading of Ras GTPases [38]. Because all Ras GTPases are membrane-localized, RasGRP1 presumably has to interact with membranes to be active, although this interaction could be weak and transient. Deletion of its C-terminal region, including the C1 domain, completely eliminated membrane localization and transforming activity of RasGRP1, while reattachment of just the C1 domain fully restored membrane localization and transforming activity [38]. Along with the observation that the C1 domain can be functionally replaced by a membrane-localization signal, this demonstrated that transformation by RasGRP1 is dependent on the ability of its C1 domain to confer membrane localization [38]. This enabled the functional replacement of the C1 domain of RasGRP1 to serve as an indicator of the ability of another C1 domain to confer membrane binding sufficiently in order to support Ras activation by RasGRP1.
Similarly to their effects on K-Ras, the C1 domains of RasGRP3 and RasGRP4α were also able to restore transformation to the deleted form of RasGRP1 (RasGRP1Δ), while the C1 domain of RasGRP4β did not (Figures 9A and 9B). Unexpectedly, the C1 domain of RasGRP2 was as effective as the DAG-binding C1 domains of RasGRP1, RasGRP3 or RasGRP4α in restoring the transforming activity of RasGRP1Δ. The localization of RasGRP1Δ fused to the RasGRP1 or RasGRP3 C1 domains was very similar to the localization of the isolated C1 domains, being concentrated in the regions occupied by the ER and the Golgi (Figure 9B). In contrast, the RasGRP1Δ fusions to the C1 domains of RasGRP2, RasGRP4α and RasGRP4β were much more diffusely distributed, such that they were not easily distinguishable from GFP alone. Therefore the abilities of the RasGRP2 and RasGRP4α C1 domains to functionally replace the C1 domain within RasGRP1 occured despite their inabilities to mimic the observable membrane localization properties of the RasGRP1 C1 domain. It is evident that, although membrane localization of RasGRPs is essential for accessing membrane-bound Ras and Rap substrates, this can occur at a low and transient level which is not observable by microscopy. Membrane binding by the RasGRP4α C1 domain appears to be partially destabilized by the attachment of the RasGRP1Δ protein, but a weak interaction with DAG in membranes may explain the ability of this C1 domain to restore transforming activity to RasGRP1Δ. However, the experiments demonstrating a lack of DAG binding by the RasGRP2 C1 domain, implying that the RasGRP1Δ+RasGRP2 C1 domain fusion protein must be activated by another mechanism. This possibly reflects the ability of the RasGRP2 C1 domain to bind weakly to membranes via anionic phospholipids.
Figure 9. The C1 domains of RasGRP1, RasGRP2, RasGRP3 and RasGRP4α are functional within RasGRPs, whereas the C1 domain of RasGRP4β is not.

(A) Structure of RasGRP1 compared with the deleted form of RasGRP1 used to test functionality of attached C1 domains. The GEF (guanine-nucleotide-exchange factor) domains, EF (EF hand domains) and C1 domains are shown, along with the N-terminal GFP tag. (B) NIH 3T3 cells expressing the indicated RasGRP1Δ+C1 fusion proteins or expressing GFP alone as a control, were assessed for oncogenic transformation (low-magnification pictures of cell cultures, left-hand panel, with high refractility and loss of contact inhibition indicating transformation), and for localization of the GFP-tagged proteins (higher-magnification fluorescence microscopy of typical individual cells, right-hand panel). The efficiency of transformation of each construct was determined as described in the Materials and methods section. (C) Structures of GFP-tagged RasGRP4α and β. The bar represents the five-amino-acid insertion in the C1 domain. (D) NIH 3T3 cells were transduced with retroviral vectors expressing the GFP-tagged RasGRP4 constructs and imaged by fluorescence microscopy.
The C1 domain of RasGRP4β was unable to activate RasGRP1Δ (Figure 9B), thus distinguishing it from the RasGRP2 C1 domain. It is possible that the RasGRP4β C1 domain makes a functionally significant contribution to membrane localization, but that this is effective only in co-operation with other domains of RasGRP4. To test this, we compared the localization of RasGRP4α with RasGRP4β in NIH 3T3 cells as these two proteins differ only by the five-amino-acid insertion in the C1 domain of RasGRP4β (Figure 9C). RasGRP4α was excluded from the nucleus and partially concentrated in the perinuclear region occupied by the ER (Figure 9D), which was similar to the distribution of the isolated C1 domain of RasGRP4α (Figure 2A). In contrast, RasGRP4β was distributed throughout the cell (Figure 9D), equivalent to the distribution of GFP alone or the isolated C1 domain of RasGRP4β (Figure 2A). These results indicated that the C1 domain is the primary determinant of RasGRP4α membrane localization in NIH 3T3 cells, and that the five-amino-acid insertion in the RasGRP4β C1 domain renders it non-functional as a membrane-localization domain.
DISCUSSION
The presence of C1 domains in all RasGRP proteins, followed by the convincing demonstration that RasGRP1 is regulated by DAG binding directly to its C1 domain, has fostered the assumption that all RasGRPs are regulated in the same way as is RasGRP1 [48,50,51,56,57]. We have directly addressed the question of whether all RasGRP C1 domains are functionally equivalent by several independent experimental approaches: fluorescence microscopy and cell fractionation of the distribution of GFP-tagged C1 domains, direct lipid vesicle-binding assays, and PMA- and serum-dependent complementation by C1 domains of K-Ras defective in its membrane binding. The results demonstrated that the RasGRP C1 domains can be divided into two distinct classes. The translocation of the C1 domains of RasGRP 1, RasGRP3, and RasGRP4α to membranes can be driven by their binding to DAG or its functional analogue PMA, although the RasGRP4α C1 domain has a reduced affinity for DAG in vitro and shows less intense co-localization with membranes in vivo. In contrast, the C1 domains of RasGRP2 and RasGRP4β were unable to bind DAG or PMA and were not detected to co-localize with membranes in vivo. Previous reports of phorbol ester-induced or PLC-dependent activation of RasGRP2 or 4β, which were interpreted as evidence for direct binding of their C1 domains to phorbol esters or DAG [43,45–49], may have instead reflected the involvement of DAG or phorbol-ester-dependent PKCs in the activation of these two RasGRPs.
In the RasGRP2 C1 domain, the combined effects of the alterations at positions 8, 12 and 22 in comparison with other RasGRPs may have altered the structure of the ligand-binding pocket sufficiently to prevent DAG or phorbol-ester binding. The position 8 alteration is of particular interest, because mutation of this residue from tyrosine to serine (the residue in RasGRP2) eliminated the localization of RasGRP1 to internal membranes [36], which could reflect a loss of DAG binding. Our data demonstrated that the five-amino-acid insertion at the base of the B loop of the ligand-binding pocket of the RasGRP4β C1 domain is sufficient to eliminate DAG binding and phorbol ester binding, and also eliminates membrane binding as detected by microscopy or complementation of the membrane-binding-defective mutants of K-Ras or RasGRP1. The alternative splicing event affecting this C1 domain apparently provides a mechanism for generating two functionally distinct forms of RasGRP4, one with and one without the capability of being activated by DAG-mediated translocation to membranes.
RasGRP2 and RasGRP4β presumably underwent opportunistic evolution away from DAG-mediated regulation following the expansion of the RasGRP family by gene duplication and acquisition of alternative splicing. Conservation of the basic structure of these two C1 domains and their retention of membrane binding via anionic phospholipids suggests that they still play significant roles in the interactions of RasGRP2 and RasGRP4β with membranes. We tested the hypothesis that these C1 domains have evolved to recognize different lipid signal transducers, but none of the C1 domains examined had specific binding to PA, LPA, ceramide, sphingosine 1-phosphate, oleic acid or arachidonic acid in the vesicle-binding assay. An alternative hypothesis is that the C1 domains of RasGRP2 and RasGRP4β, as well as those of the other RasGRPs, provide a weak membrane-binding site via their electrostatic interactions with anionic phospholipids. The surface of the PKCδ C1b domain contains basic residues which are positioned to interact with an anionic membrane surface [12,63]. The RasGRP C1 domains contain these and additional basic residues positioned appropriately for interaction with anionic surfaces, particularly at positions 10 and 32 (Figure 1). The RasGRP C1 domains are also bordered at their C-termini by a three- or four-residue basic patch (Figure 1A) which enhances binding of the RasGRP1 and RasGRP3 C1 domains to PMA/PS micelles [19]. Our analyses have demonstrated that all C1 domains bound phospholipid vesicles in proportion to the anionic lipid content and that this binding is insensitive to the specific headgroup and can occur in the absence of DAG. In the case of a DAG-binding C1 domain, the weak electrostatic interaction with negatively charged phospholipids may facilitate a two-dimensional search for DAG on the membrane surface, as well as reinforcing the membrane-binding strength of the ligated C1 domain. For the C1 domains of RasGRP2 and RasGRP4β, a DAG-independent electrostatic interaction with membranes is evidently insufficient to dictate strong membrane binding in vivo, since RasGRP2 and RasGRP4β did not noticeably co-localize with membranes or complement the membrane-binding-defective K-Ras mutant. However, the RasGRP2 C1 domain can functionally replace the C1 domain within RasGRP1. As the C1 domain is essential and sufficient for RasGRP1 membrane localization, leading to Ras GTP loading [38], this implies that the RasGRP2 C1 domain can provide membrane binding to a physiologically significant extent despite its lack of detectable DAG binding and lack of membrane localization observable by microscopy. It appears that the association of RasGRP1 with membranes can be quite weak and yet still be sufficient for Ras activation, as detected by transformation of NIH 3T3 cells. In contrast, K-Ras may have to be more stably bound to membranes in order to transduce signals sufficient to maintain transformation of NIH 3T3 cells.
In addition to the RasGRP2 C1 domain, there are other examples of C1 domains which do not bind DAG and do not provide microscopy-detectable binding to membranes on their own, but are required for efficient localization to membranes. The Raf-1 C1 domain is required to stabilize membrane binding via the adjacent Ras-binding domain [20], while the C1 domain of KSR makes an essential contribution to mediate constitutive localization to internal membranes and is needed for cytokine-induced translocation to the plasma membrane [15]. The inability of the C1 domain of RasGRP4β to confer transforming activity on either RasGRP1 or RasGRP4α may be due to its lower binding to anionic phospholipids relative to the C1 domain of RasGRP2 (Figure 7). It is possible that, under some circumstances (although not in NIH 3T3 cells), this C1 domain can also make physiologically significant contributions to membrane binding by RasGRP4β. However, it could be that the alternative splicing event which converts RasGRP4α into RasGRP4β has the purpose of eliminating any contribution of the C1 domain to RasGRP4 activation.
Acknowledgments
This work was supported by grants from the Canadian Institutes for Health Research to R. C. and R. K.
References
- 1.Gomez-Fernandez J. C., Torrecillas A., Corbalan-Garcia S. Diacylglycerols as activators of protein kinase C. Mol. Membr. Biol. 2004;21:339–349. doi: 10.1080/09687860400010508. [DOI] [PubMed] [Google Scholar]
- 2.Brose N., Betz A., Wegmeyer H. Divergent and convergent signaling by the diacylglycerol second messenger pathway in mammals. Curr. Opin. Neurobiol. 2004;14:328–340. doi: 10.1016/j.conb.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 3.Carrasco S., Merida I. Diacylglycerol, when simplicity becomes complex. Trends Biochem. Sci. 2006;32:27–36. doi: 10.1016/j.tibs.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 4.Hurley J. H., Newton A. C., Parker P. J., Blumberg P. M., Nishizuka Y. Taxonomy and function of C1 protein kinase C homology domains. Protein Sci. 1997;6:477–480.. doi: 10.1002/pro.5560060228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Colon-Gonzalez F., Kazanietz M. G. C1 domains exposed: from diacylglycerol binding to protein–protein interactions. Biochim. Biophys. Acta. 2006;1761:827–837. doi: 10.1016/j.bbalip.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 6.Cho W., Stahelin R. V. Membrane–protein interactions in cell signaling and membrane trafficking. Annu. Rev. Biophys. Biomol. Struct. 2005;34:119–151. doi: 10.1146/annurev.biophys.33.110502.133337. [DOI] [PubMed] [Google Scholar]
- 7.Hurley J. H. Membrane-binding domains. Biochim. Biophys. Acta. 2006;1761:805–811. doi: 10.1016/j.bbalip.2006.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hall C., Lim L., Leung T. C1, see them all. Trends Biochem. Sci. 2005;30:169–171. doi: 10.1016/j.tibs.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 9.Newton A. C. Protein kinase C: structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem. Rev. 2001;101:2353–2364. doi: 10.1021/cr0002801. [DOI] [PubMed] [Google Scholar]
- 10.Ananthanarayanan B., Stahelin R. V., Digman M. A., Cho W. Activation mechanisms of conventional protein kinase C isoforms are determined by the ligand affinity and conformational flexibility of their C1 domains. J. Biol. Chem. 2003;278:46886–46894. doi: 10.1074/jbc.M307853200. [DOI] [PubMed] [Google Scholar]
- 11.Stahelin R. V., Digman M. A., Medkova M., Ananthanarayanan B., Rafter J. D., Melowic H. R., Cho W. Mechanism of diacylglycerol-induced membrane targeting and activation of protein kinase Cδ. J. Biol. Chem. 2004;279:29501–29512. doi: 10.1074/jbc.M403191200. [DOI] [PubMed] [Google Scholar]
- 12.Zhang G., Kazanietz M. G., Blumberg P. M., Hurley J. H. Crystal structure of the Cys2 activator-binding domain of protein kinase Cδ in complex with phorbol ester. Cell. 1995;81:917–924. doi: 10.1016/0092-8674(95)90011-x. [DOI] [PubMed] [Google Scholar]
- 13.Canagarajah B., Leskow F. C., Ho J. Y., Mischak H., Saidi L. F., Kazanietz M. G., Hurley J. H. Structural mechanism for lipid activation of the Rac-specific GAP, β2-chimaerin. Cell. 2004;119:407–418. doi: 10.1016/j.cell.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 14.Mott H. R., Carpenter J. W., Zhong S., Ghosh S., Bell R. M., Campbell S. L. The solution structure of the Raf-1 cysteine-rich domain: a novel ras and phospholipid binding site. Proc. Natl. Acad. Sci. U.S.A. 1996;93:8312–8317. doi: 10.1073/pnas.93.16.8312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou M., Horita D. A., Waugh D. S., Byrd R. A., Morrison D. K. Solution structure and functional analysis of the cysteine-rich C1 domain of kinase suppressor of Ras (KSR) J. Mol. Biol. 2002;315:435–446. doi: 10.1006/jmbi.2001.5263. [DOI] [PubMed] [Google Scholar]
- 16.Wang Q. J., Fang T. W., Nacro K., Marquez V. E., Wang S., Blumberg P. M. Role of hydrophobic residues in the C1b domain of protein kinase Cδ on ligand and phospholipid interactions. J. Biol. Chem. 2001;276:19580–19587. doi: 10.1074/jbc.M010089200. [DOI] [PubMed] [Google Scholar]
- 17.Johnson J. E., Giorgione J., Newton A. C. The C1 and C2 domains of protein kinase C are independent membrane targeting modules, with specificity for phosphatidylserine conferred by the C1 domain. Biochemistry. 2000;39:11360–11369. doi: 10.1021/bi000902c. [DOI] [PubMed] [Google Scholar]
- 18.Medkova M., Cho W. Interplay of C1 and C2 domains of protein kinase C-α in its membrane-binding and activation. J. Biol. Chem. 1999;274:19852–19861. doi: 10.1074/jbc.274.28.19852. [DOI] [PubMed] [Google Scholar]
- 19.Irie K., Masuda A., Shindo M., Nakagawa Y., Ohigashi H. Tumor promoter binding of the protein kinase C C1 homology domain peptides of RasGRPs, chimaerins, and Unc13s. Bioorg. Med. Chem. 2004;12:4575–4583. doi: 10.1016/j.bmc.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 20.Bondeva T., Balla A., Varnai P., Balla T. Structural determinants of Ras–Raf interaction analyzed in live cells. Mol. Biol. Cell. 2002;13:2323–2333. doi: 10.1091/mbc.E02-01-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stone J. C. Regulation of Ras in lymphocytes: get a GRP. Biochem. Soc. Trans. 2006;34:858–861. doi: 10.1042/BST0340858. [DOI] [PubMed] [Google Scholar]
- 22.Caloca M. J., Zugaza J. L., Matallanas D., Crespo P., Bustelo X. R. Vav mediates Ras stimulation by direct activation of the GDP/GTP exchange factor Ras GRP1. EMBO J. 2003;22:3326–3336. doi: 10.1093/emboj/cdg316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zugaza J. L., Caloca M. J., Bustelo X. R. Inverted signaling hierarchy between RAS and RAC in T-lymphocytes. Oncogene. 2004;23:5823–5833. doi: 10.1038/sj.onc.1207768. [DOI] [PubMed] [Google Scholar]
- 24.Oh-hora M., Johmura S., Hashimoto A., Hikida M., Kurosaki T. Requirement for Ras guanine nucleotide releasing protein 3 in coupling phospholipase C-γ2 to Ras in B cell receptor signaling. J. Exp. Med. 2003;198:1841–1851. doi: 10.1084/jem.20031547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanjuan M. A., Pradet-Balade B., Jones D. R., Martinez A. C., Stone J. C., Garcia-Sanz J. A., Merida I. T cell activation in vivo targets diacylglycerol kinase α to the membrane: a novel mechanism for Ras attenuation. J. Immunol. 2003;170:2877–2883. doi: 10.4049/jimmunol.170.6.2877. [DOI] [PubMed] [Google Scholar]
- 26.Topham M. K., Prescott S. M. Diacylglycerol kinase ζ regulates Ras activation by a novel mechanism. J. Cell Biol. 2001;152:1135–1143. doi: 10.1083/jcb.152.6.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zha Y., Marks R., Ho A. W., Peterson A. C., Janardhan S., Brown I., Praveen K., Stang S., Stone J. C., Gajewski T. F. T cell anergy is reversed by active Ras and is regulated by diacylglycerol kinase-α. Nat. Immunol. 2006;7:1166–1173. doi: 10.1038/ni1394. [DOI] [PubMed] [Google Scholar]
- 28.Jones D. R., Sanjuan M. A., Stone J. C., Merida I. Expression of a catalytically inactive form of diacylglycerol kinase α induces sustained signaling through RasGRP. FASEB J. 2002;16:595–597. doi: 10.1096/fj.01-0762fje. [DOI] [PubMed] [Google Scholar]
- 29.Teixeira C., Stang S. L., Zheng Y., Beswick N. S., Stone J. C. Integration of DAG signaling systems mediated by PKC-dependent phosphorylation of RasGRP3. Blood. 2003;102:1414–1420. doi: 10.1182/blood-2002-11-3621. [DOI] [PubMed] [Google Scholar]
- 30.Zheng Y., Liu H., Coughlin J., Zheng J., Li L., Stone J. C. Phosphorylation of RasGRP3 on threonine 133 provides a mechanistic link between PKC and Ras signaling systems in B cells. Blood. 2005;105:3648–3654. doi: 10.1182/blood-2004-10-3916. [DOI] [PubMed] [Google Scholar]
- 31.Aiba Y., Oh-hora M., Kiyonaka S., Kimura Y., Hijikata A., Mori Y., Kurosaki T. Activation of RasGRP3 by phosphorylation of Thr-133 is required for B cell receptor-mediated Ras activation. Proc. Natl. Acad. Sci. U.S.A. 2004;101:16612–16617. doi: 10.1073/pnas.0407468101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roose J. P., Mollenauer M., Gupta V. A., Stone J., Weiss A. A diacylglycerol-protein kinase C-RasGRP1 pathway directs Ras activation upon antigen receptor stimulation of T cells. Mol. Cell. Biol. 2005;25:4426–4441. doi: 10.1128/MCB.25.11.4426-4441.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brodie C., Steinhart R., Kazimirsky G., Rubinfeld H., Hyman T., Ayres J. N., Hur G. M., Toth A., Yang D., Garfield S. H., et al. PKCδ associates with and is involved in the phosphorylation of RasGRP3 in response to phorbol esters. Mol. Pharmacol. 2004;66:76–84. doi: 10.1124/mol.66.1.76. [DOI] [PubMed] [Google Scholar]
- 34.Bivona T. G., Perez De Castro I., Ahearn I. M., Grana T. M., Chiu V. K., Lockyer P. J., Cullen P. J., Pellicer A., Cox A. D., Philips M. R. Phospholipase Cγ activates Ras on the Golgi apparatus by means of RasGRP1. Nature. 2003;424:694–698. doi: 10.1038/nature01806. [DOI] [PubMed] [Google Scholar]
- 35.Reynolds L. F., de Bettignies C., Norton T., Beeser A., Chernoff J., Tybulewicz V. L. Vav1 transduces T cell receptor signals to the activation of the Ras/ERK pathway via LAT, Sos, and RasGRP1. J. Biol. Chem. 2004;279:18239–18246. doi: 10.1074/jbc.M400257200. [DOI] [PubMed] [Google Scholar]
- 36.Caloca M. J., Zugaza J. L., Bustelo X. R. Exchange factors of the RasGRP family mediate Ras activation in the Golgi. J. Biol. Chem. 2003;278:33465–33473. doi: 10.1074/jbc.M302807200. [DOI] [PubMed] [Google Scholar]
- 37.Ebinu J. O., Bottorff D. A., Chan E. Y., Stang S. L., Dunn R. J., Stone J. C. RasGRP, a Ras guanyl nucleotide-releasing protein with calcium- and diacylglycerol-binding motifs. Science. 1998;280:1082–1086. doi: 10.1126/science.280.5366.1082. [DOI] [PubMed] [Google Scholar]
- 38.Tognon C. E., Kirk H. E., Passmore L. A., Whitehead I. P., Der C. J., Kay R. J. Regulation of RasGRP via a phorbol ester-responsive C1 domain. Mol. Cell. Biol. 1998;18:6995–7008. doi: 10.1128/mcb.18.12.6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carrasco S., Merida I. Diacylglycerol-dependent binding recruits PKCθ and RasGRP1 C1 domains to specific subcellular localizations in living T lymphocytes. Mol. Biol. Cell. 2004;15:2932–2942. doi: 10.1091/mbc.E03-11-0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lorenzo P. S., Beheshti M., Pettit G. R., Stone J. C., Blumberg P. M. The guanine nucleotide exchange factor RasGRP is a high-affinity target for diacylglycerol and phorbol esters. Mol. Pharmacol. 2000;57:840–846. [PubMed] [Google Scholar]
- 41.Lorenzo P. S., Kung J. W., Bottorff D. A., Garfield S. H., Stone J. C., Blumberg P. M. Phorbol esters modulate the Ras exchange factor RasGRP3. Cancer Res. 2001;61:943–949. [PubMed] [Google Scholar]
- 42.Li L., Yang Y., Wong G. W., Stevens R. L. Mast cells in airway hyporesponsive C3H/HeJ mice express a unique isoform of the signaling protein Ras guanine nucleotide releasing protein 4 that is unresponsive to diacylglycerol and phorbol esters. J. Immunol. 2003;171:390–397. doi: 10.4049/jimmunol.171.1.390. [DOI] [PubMed] [Google Scholar]
- 43.Yang Y., Li L., Wong G. W., Krilis S. A., Madhusudhan M. S., Sali A., Stevens R. L. RasGRP4, a new mast cell-restricted Ras guanine nucleotide-releasing protein with calcium- and diacylglycerol-binding motifs. Identification of defective variants of this signaling protein in asthma, mastocytosis, and mast cell leukemia patients and demonstration of the importance of RasGRP4 in mast cell development and function. J. Biol. Chem. 2002;277:25756–25774. doi: 10.1074/jbc.M202575200. [DOI] [PubMed] [Google Scholar]
- 44.Reuther G. W., Lambert Q. T., Rebhun J. F., Caligiuri M. A., Quilliam L. A., Der C. J. RasGRP4 is a novel Ras activator isolated from acute myeloid leukemia. J. Biol. Chem. 2002;277:30508–30514. doi: 10.1074/jbc.M111330200. [DOI] [PubMed] [Google Scholar]
- 45.Clyde-Smith J., Silins G., Gartside M., Grimmond S., Etheridge M., Apolloni A., Hayward N., Hancock J. F. Characterization of RasGRP2, a plasma membrane-targeted, dual specificity Ras/Rap exchange factor. J. Biol. Chem. 2000;275:32260–32267. doi: 10.1074/jbc.M006087200. [DOI] [PubMed] [Google Scholar]
- 46.Kawasaki H., Springett G. M., Toki S., Canales J. J., Harlan P., Blumenstiel J. P., Chen E. J., Bany I. A., Mochizuki N., Ashbacher A., et al. A Rap guanine nucleotide exchange factor enriched highly in the basal ganglia. Proc. Natl. Acad. Sci. U.S.A. 1998;95:13278–13283. doi: 10.1073/pnas.95.22.13278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dupuy A. J., Morgan K., von Lintig F. C., Shen H., Acar H., Hasz D. E., Jenkins N. A., Copeland N. G., Boss G. R., Largaespada D. A. Activation of the Rap1 guanine nucleotide exchange gene, CalDAG-GEF I, in BXH-2 murine myeloid leukemia. J. Biol. Chem. 2001;276:11804–11811. doi: 10.1074/jbc.M008970200. [DOI] [PubMed] [Google Scholar]
- 48.Crittenden J. R., Bergmeier W., Zhang Y., Piffath C. L., Liang Y., Wagner D. D., Housman D. E., Graybiel A. M. CalDAG-GEFI integrates signaling for platelet aggregation and thrombus formation. Nat. Med. 2004;10:982–986. doi: 10.1038/nm1098. [DOI] [PubMed] [Google Scholar]
- 49.Katagiri K., Shimonaka M., Kinashi T. Rap1-mediated lymphocyte function-associated antigen-1 activation by the T cell antigen receptor is dependent on phospholipase C-γ1. J. Biol. Chem. 2004;279:11875–11881. doi: 10.1074/jbc.M310717200. [DOI] [PubMed] [Google Scholar]
- 50.Guo F. F., Kumahara E., Saffen D. A CalDAG-GEFI/Rap1/B-Raf cassette couples M1 muscarinic acetylcholine receptors to the activation of ERK1/2. J. Biol. Chem. 2001;276:25568–25581. doi: 10.1074/jbc.M101277200. [DOI] [PubMed] [Google Scholar]
- 51.Eto K., Murphy R., Kerrigan S. W., Bertoni A., Stuhlmann H., Nakano T., Leavitt A. D., Shattil S. J. Megakaryocytes derived from embryonic stem cells implicate CalDAG-GEFI in integrin signaling. Proc. Natl. Acad. Sci. U.S.A. 2002;99:12819–12824. doi: 10.1073/pnas.202380099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Betz A., Ashery U., Rickmann M., Augustin I., Neher E., Sudhof T. C., Rettig J., Brose N. Munc13-Munc11 is a presynaptic phorbol ester receptor that enhances neurotransmitter release. Neuron. 1998;21:123–136. doi: 10.1016/s0896-6273(00)80520-6. [DOI] [PubMed] [Google Scholar]
- 53.Kazanietz M. G., Lewin N. E., Bruns J. D., Blumberg P. M. Characterization of the cysteine-rich region of the Caenorhabditis elegans protein Unc-13 as a high affinity phorbol ester receptor. Analysis of ligand-binding interactions, lipid cofactor requirements, and inhibitor sensitivity. J. Biol. Chem. 1995;270:10777–10783. doi: 10.1074/jbc.270.18.10777. [DOI] [PubMed] [Google Scholar]
- 54.Perez de Castro I., Bivona T. G., Philips M. R., Pellicer A. Ras activation in Jurkat T cells following low-grade stimulation of the T-cell receptor is specific to N-Ras and occurs only on the Golgi apparatus. Mol. Cell. Biol. 2004;24:3485–3496. doi: 10.1128/MCB.24.8.3485-3496.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Caloca M. J., Zugaza J. L., Vicente-Manzanares M., Sanchez-Madrid F., Bustelo X. R. F-actin-dependent translocation of the Rap1 GDP/GTP exchange factor RasGRP2. J. Biol. Chem. 2004;279:20435–20446. doi: 10.1074/jbc.M313013200. [DOI] [PubMed] [Google Scholar]
- 56.Bunney T. D., Katan M. Phospholipase C ϵ: linking second messengers and small GTPases. Trends Cell Biol. 2006;16:640–648. doi: 10.1016/j.tcb.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 57.Mitin N., Rossman K. L., Der C. J. Signaling interplay in Ras superfamily function. Curr. Biol. 2005;15:R563–R574. doi: 10.1016/j.cub.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 58.Guilbault B., Kay R. J. RasGRP1 sensitizes an immature B cell line to antigen receptor-induced apoptosis. J. Biol. Chem. 2004;279:19523–19530. doi: 10.1074/jbc.M314273200. [DOI] [PubMed] [Google Scholar]
- 59.Pear W. S., Nolan G. P., Scott M. L., Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. U.S.A. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Haskins K., Kubo R., White J., Pigeon M., Kappler J., Marrack P. The major histocompatibility complex-restricted antigen receptor on T cells. I. Isolation with a monoclonal antibody. J. Exp. Med. 1983;157:1149–1169. doi: 10.1084/jem.157.4.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Buser C. A., McLaughlin S. Ultracentrifugation technique for measuring the binding of peptides and proteins to sucrose-loaded phospholipid vesicles. Methods Mol. Biol. 1998;84:267–281. doi: 10.1385/0-89603-488-7:267. [DOI] [PubMed] [Google Scholar]
- 62.Schagger H., von Jagow G. Tricine–sodium dodecyl sulfate–polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 63.Rong S. B., Enyedy I. J., Qiao L., Zhao L., Ma D., Pearce L. L., Lorenzo P. S., Stone J. C., Blumberg P. M., Wang S., Kozikowski A. P. Structural basis of RasGRP binding to high-affinity PKC ligands. J. Med. Chem. 2002;45:853–860. doi: 10.1021/jm010422z. [DOI] [PubMed] [Google Scholar]
- 64.Kazanietz M. G., Wang S., Milne G. W., Lewin N. E., Liu H. L., Blumberg P. M. Residues in the second cysteine-rich region of protein kinase Cδ relevant to phorbol ester binding as revealed by site-directed mutagenesis. J. Biol. Chem. 1995;270:21852–21859. doi: 10.1074/jbc.270.37.21852. [DOI] [PubMed] [Google Scholar]
- 65.Shindo M., Irie K., Masuda A., Ohigashi H., Shirai Y., Miyasaka K., Saito N. Synthesis and phorbol ester binding of the cysteine-rich domains of diacylglycerol kinase (DGK) isozymes. DGKγ and DGKβ are new targets of tumor-promoting phorbol esters. J. Biol. Chem. 2003;278:18448–18454. doi: 10.1074/jbc.M300400200. [DOI] [PubMed] [Google Scholar]
- 66.Dries D. R., Gallegos L. L., Newton A. C. A single residue in the C1 domain sensitizes novel protein kinase C isoforms to cellular diacylglycerol production. J. Biol. Chem. 2007;282:826–830. doi: 10.1074/jbc.C600268200. [DOI] [PubMed] [Google Scholar]
- 67.Hambrock A., Loffler-Walz C., Quast U. Glibenclamide binding to sulphonylurea receptor subtypes: dependence on adenine nucleotides. Br. J. Pharmacol. 2002;136:995–1004. doi: 10.1038/sj.bjp.0704801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nakamura N., Rabouille C., Watson R., Nilsson T., Hui N., Slusarewicz P., Kreis T. E., Warren G. Characterization of a cis-Golgi matrix protein, GM130. J. Cell Biol. 1995;131:1715–1726. doi: 10.1083/jcb.131.6.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Regier D. S., Higbee J., Lund K. M., Sakane F., Prescott S. M., Topham M. K. Diacylglycerol kinase ι regulates Ras guanyl-releasing protein 3 and inhibits Rap1 signaling. Proc. Natl. Acad. Sci. U.S.A. 2005;102:7595–7600. doi: 10.1073/pnas.0500663102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kashiwagi K., Shirai Y., Kuriyama M., Sakai N., Saito N. Importance of C1B domain for lipid messenger-induced targeting of protein kinase C. J. Biol. Chem. 2002;277:18037–18045. doi: 10.1074/jbc.M111761200. [DOI] [PubMed] [Google Scholar]
- 71.van Blitterswijk W. J. Hypothesis: ceramide conditionally activates atypical protein kinases C, Raf-1 and KSR through binding to their cysteine-rich domains. Biochem. J. 1998;331:679–680. [PMC free article] [PubMed] [Google Scholar]
- 72.van Blitterswijk W. J., van der Luit A. H., Veldman R. J., Verheij M., Borst J. Ceramide: second messenger or modulator of membrane structure and dynamics? Biochem. J. 2003;369:199–211. doi: 10.1042/BJ20021528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Khan W. A., Blobe G. C., Hannun Y. A. Arachidonic acid and free fatty acids as second messengers and the role of protein kinase C. Cell. Signalling. 1995;7:171–184. doi: 10.1016/0898-6568(94)00089-t. [DOI] [PubMed] [Google Scholar]
- 74.Stahelin R. V., Digman M. A., Medkova M., Ananthanarayanan B., Melowic H. R., Rafter J. D., Cho W. Diacylglycerol-induced membrane targeting and activation of protein kinase Cϵ: mechanistic differences between protein kinases Cδ and Cϵ. J. Biol. Chem. 2005;280:19784–19793. doi: 10.1074/jbc.M411285200. [DOI] [PubMed] [Google Scholar]
- 75.Daum G. Lipids of mitochondria. Biochim. Biophys. Acta. 1985;822:1–42. doi: 10.1016/0304-4157(85)90002-4. [DOI] [PubMed] [Google Scholar]