

Abstract
The present study characterized conserved residues in a GST (glutathione transferase) in the active-site region that interacts with glutathione. This region of the active site is near the glycine moiety of glutathione and consists of a hydrogen bond network. In the GSTD (Delta class GST) studied, adGSTD4-4, the network consisted of His38, Met39, Asn47, Gln49, His50 and Cys51. In addition to contributing to glutathione binding, this region also had major effects on enzyme catalysis, as shown by changes in kinetic parameters and substrate-specific activity. The results also suggest that the electron distribution of this network plays a role in stabilization of the ionized thiol of glutathione as well as impacting on the catalytic rate-limiting step. This area constitutes a second glutathione active-site network involved in glutathione ionization distinct from a network previously observed interacting with the glutamyl end of glutathione. This second network also appears to be functionally conserved in GSTs. In the present study, His50 is the key basic residue stabilized by this network, as shown by up to a 300-fold decrease in kcat and 5200-fold decrease in kcat/Km for glutathione. Although these network residues have a minor role in structural integrity, the replaced residues induced changes in active-site topography as well as generating positive co-operativity towards glutathione. Moreover, this network at the glycine moiety of GSH (glutathione) also contributed to the ‘base-assisted deprotonation model’ for GSH ionization. Taken together, the results indicate a critical role for the functionally conserved basic residue His50 and this hydrogen bond network in the active site.
Keywords: base-assisted deprotonation model, conserved active-site residue, Delta class GST, enzyme catalysis, glutathione transferase (GST), glycine moiety of glutathione (GSH)
Abbreviations: CDNB, 1-chloro-2,4-dinitrobenzene; DCNB, 1,2-dichloro-4-nitrobenzene; EA, ethacrynic acid; FDNB, 1-fluoro-2,4-dinitrobenzene; G-site, GSH (glutathione)-binding site; GST, glutathione transferase; GSTD, Delta class GST; hGSTP1-1, human GSTP1-1; PNBC, p-nitrobenzyl chloride; PNPBr, p-nitrophenethyl bromide; ZmGST1, Zea mays (maize) GST1
INTRODUCTION
GSTs (glutathione transferases; EC 2.5.1.18) are intracellular proteins, which are widely distributed in Nature, being found in most aerobic eukaryotes and prokaryotes [1,2]. GSTs are polymorphic, with most organisms possessing a genetic capacity to encode multiple isoforms of various classes [3,4]. The enzymes are an integral part of the phase II detoxification mechanism, being involved in xenobiotic metabolism as well as protection against peroxide damage. GSTs catalyse reactions with a very broad range of structurally diverse electrophilic substrates (e.g. alkylhalides, arylhalides, lactones, epoxides, quinones, esters and activated alkenes) [4–6]. This enzyme family therefore displays a wide range of catalytic functions while retaining a high specificity towards the thiol substrate glutathione (GSH). The GSH conjugation by GST increases the solubility of the target molecule, thus facilitating the excretion of the molecule from the organism.
All cytosolic GSTs have very similar three-dimensional structures and the active-site pocket is very similar in the G-site (GSH-binding site) [7–9]. The N-terminal domain of GST adopts a βαβαββα topology that contributes most of the contacts to GSH and is called the G-site. The crystal structures of GSTs show that most of the active-site residues involved in the binding and activation of GSH are found within the N-terminus; hence this region of the protein is highly conserved between GSTs [7,8,10]. The C-terminal domain is all α-helical, providing some of the contacts to the hydrophobic binding site that lies adjacent to the G-site.
Substrate conjugation of electrophilic xenobiotics involves nucleophilic attack at the ionized thiol of GSH in the active site. A conserved tyrosine (for Alpha, Mu and Pi classes) or serine (for Theta and Delta classes) residue within hydrogen-bonding distance of the thiol of GSH has been shown to be of primary importance in facilitating GSH deprotonation as well as stabilization of the ionized GSH by hydrogen-bonding [10–14]. In addition, conserved residues that interact directly with the glutamyl moiety of GSH, for example Ser65, Arg66 and Ile52 in GSTD (Delta class GST) have also been implicated in critical roles in the catalytic mechanism, involvement in GSH binding, GSH thiol ionization, as well as structural integrity [15–17]. Therefore the residues interacting with the other end of GSH, the glycine moiety (His38, Met39, Asn47, Gln49, His50 and Cys51) were of interest (Figure 1) [18]. His38 and His50 interact directly with the glycine carboxylic group of GSH [19]. Both histidines appear to be assisted or stabilized by dipole–dipole interaction with Met39. Moreover, a hydrogen bond network in the G-site near the glycine moiety of GSH, involving several residues (His50, Asn47, Gln49 and Cys51) and GSH, is proposed to be essential to electron distribution for enzyme catalysis. To investigate the roles of these residues, mutagenesis studies were performed and the engineered enzymes were characterized by kinetic constants, substrate specific activity, pKa determination and rate-limiting step determination in addition to physical properties.
Figure 1. Stereoview of conserved G-site residues that directly interact with the glycine moiety of GSH and generate a hydrogen bond network.

His38 and His50 interact directly with the glycine carboxylic group of GSH and are assisted or stabilized by dipole–dipole interaction with Met39. A hydrogen bond network in the G-site is formed by several residues (His50, Asn47, Gln49 and Cys51) and GSH. The previously identified electron-sharing network at the glutamyl end of GSH is shown in grey [16]. The image was produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, CA, U.S.A. (http://www.cgl.ucsf.edu/chimera) (supported by National Institutes of Health grant P41 RR-01081) [18].
MATERIALS AND METHODS
Site directed mutagenesis
The engineered enzymes were generated using Stratagene's QuikChange® site-directed mutagenesis protocol. The primers used were designed based on the sequence of the adGSTD4 wild-type gene (GenBank® accession number AF273040). Full-length DNA sequencing in both directions was performed to confirm the engineered clones.
Protein expression and purification
After transformation of the engineered plasmids into Escherichia coli BL21(DE3)pLysS, protein expression was performed. The recombinant adGSTD4-4 engineered enzymes and wild-type were purified either by glutathione affinity chromatography according to the manufacturer's instructions (Amersham Biosciences) or by cation exchanger (SP-XL) followed by hydrophobic interaction chromatography (phenyl-Sepharose) as described previously [19]. The purified enzymes (in 50 mM potassium phosphate, pH 6.5) were stored in 50% (v/v) glycerol at −20 °C until used. The concentrations of the proteins were determined by Bio-Rad protein reagent (Bio-Rad) by using BSA as the standard protein, and the purity of the proteins was observed by SDS/PAGE.
Kinetic parameters determination
Kinetic experiments were performed as previously described [20]. Kinetic constants were determined by varying the CDNB (1-chloro-2,4-dinitrobenzene) concentration (0.031–3.0 mM) while GSH was held constant at a saturating concentration and by varying GSH concentrations (0.25–20 mM) at a saturating concentration of CDNB. The standard GST assay was performed using 3 mM CDNB and 10 mM GSH for adGSTD4-4 wild-type. The rate of conjugation between GSH and CDNB was monitored by continuously measuring change in absorbance at 340 nm for 1 min using a SpectraMax 250 at 25–27 °C. A molar absorption coefficient (ϵ) of 9.6 mM−1·cm−1 was used to convert the absorbance into moles [21]. Steady-state kinetics followed Michaelis–Menten kinetics except where stated. With evidence of co-operativity upon GSH binding, demonstrated by a sigmoidal curve instead of a hyperbolic curve on a Michaelis–Menten plot, a Hill equation (eqn 1) was used to fit the experimental kinetic data on the plot. K0.5 is the substrate concentration that gives the rate of reaction at half of Vmax, similar to the Km value for non-co-operative binding (h=1).
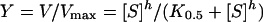 |
(1) |
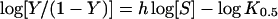 |
(2) |
A sigmoidal Hill equation was practically transformed into a linear rate equation (eqn 2), where Y is the fractional saturation, h is the Hill coefficient, and K0.5 is an averaged binding constant at Y=0.5. A Hill plot, a plot between log[Y/(1–Y)] and log[S], was employed to determine the degree of co-operativity from the slope of the plot that yields the Hill coefficient (h) [22]. Catalytic constant (kcat) and the catalytic efficiency (kcat/Km) were calculated on an active-site basis using the subunit molecular mass of each enzyme. Maximal velocity (Vmax) and Michaelis constant (Km) were determined by nonlinear regression software analysis (GraphPad Prism version 5.00 for Windows; GraphPad Software, San Diego, CA, U.S.A.; http://www.graphpad.com). One-way ANOVA with Dunnett's multiple comparison post test was performed with wild-type as control using GraphPad Prism 5.
Specific activity determination
The specific activities towards several GST substrates were determined as previously described [23]. All measurements were performed at 25–27 °C in 0.1 M potassium phosphate buffer (pH 6.5 or 7.5). The GST activities were measured with glutathione and five hydrophobic substrates: CDNB, DCNB (1,2-dichloro-4-nitrobenzene), EA (ethacrynic acid), PNPBr (p-nitrophenethyl bromide) and PNBC (p-nitrobenzyl chloride). Specific activities were calculated according to the molar absorption coefficient for each substrate [21].
pH dependence of kinetic parameters
The pH dependence of kcat/KmCDNB was obtained by performing kinetic measurements in the following buffers: 0.1 M sodium acetate buffer (from pH 5.0 to 5.5) and 0.1 M potassium phosphate buffer (from pH 6.0 to 8.5). Increments of pH were 0.5 and control experiments showed no discontinuities from buffer types. The pKa values of bound GSH were obtained by fitting the data to the equation y=ylim/(1+10pKa−pH) as previously described [13]. The program GraphPad Prism 5 was used for nonlinear fit of the data to the sigmoidal dose–response (variable slope) equation.
Fluoride/chloride leaving group substitution
A diagnostic test in evaluating the rate-limiting step in nucleophilic aromatic substitution reactions is the effect of different leaving groups on kinetic parameters [13]. The second order kinetic constants at pH 6.5 for the spontaneous reaction of GSH with CDNB and FDNB (1-fluoro-2,4-dinitrobenzene) were determined by kinetic measurement by using CDNB or FDNB as co-substrate as previously described [24]. The ratios of the catalytic constants (kcatFDNB/kcatCDNB) were determined.
Effect of viscosity on the kinetic parameters
The dependence of the steady-state kinetic parameters on relative viscosity was observed by performing kinetics measurements in the presence of various glycerol concentrations. The range of relative viscosities (η/η0) was between 1.53 and 4.43. The slope of the plots for relative catalytic constant (kcat0/kcat) versus relative viscosity (η/η0) was determined. Viscosity values (η) at 25 °C were calculated as described previously [25].
Thermal stability assay
The enzymes at 0.1 mg/ml in 0.1 M phosphate buffer (pH 6.5) containing 1 mM EDTA and 5 mM dithiothreitol were incubated at 45 °C for various times and then activity was measured in the standard GST assay. The data were plotted as log percentage of remaining activity versus pre-incubation time. The half-life (t½) of the enzyme at 45 °C was calculated from the slope of the plot using the equations: slope=k/2.3, k=0.693/t½.
Intrinsic fluorescence measurement
There are two tryptophan residues in each monomer of adGSTD4-4: Trp64 and Trp191. The intrinsic fluorescence from Trp64, which is exposed to solvent at the base of the G-site, can be used to monitor the active-site conformation indirectly. The intrinsic fluorescence of adGSTD4-4 was measured in a single-photon counting spectrofluorimeter. Excitation was at 295 nm and emission was scanned from 300 to 450 nm. In these experiments, a number of samples containing 0.1 mg/ml GST in 0.1 M potassium phosphate buffer (pH 6.5) were prepared similarly for the wild-type and engineered enzymes. The wavelength that gives the maximum fluorescence intensity (λmax) and fluorescence intensity at λmax were observed. The experimental data were corrected both for dilution and for inner filter effects.
RESULTS
Enzymatic characterization
Michaelis–Menten analysis was performed using nonlinear regression to determine steady-state kinetic constants (Table 1). The kinetic constants showed that the residue changes of His38, His50, Met39, Asn47, Cys51 and Gln49 had an impact on catalysis. The engineered G-site residue enzymes showed only small changes in KmCDNB, whereas remarkably lower affinities towards GSH were shown (greater KmGSH) compared with wild-type. Positive co-operativity for GSH binding was observed for several engineered enzymes that displayed deviation of steady-state kinetics from a Michaelis–Menten hyperbolic to a sigmoidal curve response. The extent of positive co-operativity is shown by Hill coefficients (h) ranging from 1 (no co-operativity) to 2 (full co-operativity) for a dimeric enzyme with two active sites. Positive co-operativity was observed for H38E, M39A, H50K, C51A, C51D, Q49A and Q49E as determined by Hill coefficients ranging from 1.40 to 1.83, suggesting that the changed residues altered the movement of α2-helix and its flanking region to influence the induced-fit mechanism.
Table 1. Steady-state kinetic constants using GSH and CDNB as GST substrates.
The units of Vmax, kcat, Km and kcat/Km are μmol·min−1·mg of protein−1, s−1, mM and s−1·mM−1 respectively. *These engineered GSTs showed positive co-operativity upon GSH binding with Hill coefficient (h) shown. The value shown is K0.5, obtained from the Hill equation, which is the substrate concentration that gives the rate of reaction at half of Vmax [22]. aSome of these values have been previously reported and are shown for purposes of comparison [19,20]. One-way ANOVA with Dunnett's multiple comparison test was performed with wild-type as control; statistical significance is shown by † for P<0.05, ‡ for P<0.01 and § for P<0.001.
| CDNB | GSH | ||||||
|---|---|---|---|---|---|---|---|
| Enzyme | Vmax | kcat | Km | kcat/Km | Km | kcat/Km | h |
| Wild-typea | 62.45±1.24 | 26.13 | 0.50±0.02 | 51.84 | 0.50±0.10 | 52.06 | 0.91±0.02 |
| H38Aa | 36.4±1.12§ | 15.26 | 1.25±0.12§ | 12.21 | 15.8±1.19§ | 0.97 | − |
| H38Ea | 14.91±0.44§ | 6.23 | 0.92±0.11§ | 6.76 | 8.32±0.22*§ | 0.74 | 1.83±0.01§ |
| H38Fa | 9.71±0.43§ | 4.06 | 0.98±0.09§ | 4.14 | 35.26±0.86§ | 0.12 | − |
| H38D | 4.61±0.12§ | 1.93 | 0.71±0.01† | 2.71 | 15.90±0.21§ | 0.12 | − |
| H38K | 5.54±0.12§ | 2.32 | 0.71±0.08† | 3.27 | 23.50±1.21§ | 0.10 | − |
| M39A | 25.56±0.96§ | 10.70 | 0.79±0.06§ | 13.50 | 6.32±0.34*§ | 1.69 | 1.72±0.12§ |
| M39F | 38.56±1.72§ | 16.14 | 0.50±0.02 | 32.22 | 2.18±0.09† | 5.75 | − |
| N47A | 0.225±0.003§ | 0.09 | 0.67±0.06 | 0.13 | 11.50±0.51§ | 0.01 | − |
| Q49A | 43.28±0.96§ | 18.11 | 0.74±0.06‡ | 24.47 | 5.58±0.46*§ | 3.24 | 1.64±0.04§ |
| Q49E | 49.75±1.36§ | 20.82 | 0.49±0.05 | 42.49 | 2.87±0.09*§ | 7.26 | 1.51±0.03§ |
| H50Aa | 6.46±0.26§ | 2.70 | 1.10±0.15§ | 2.45 | 7.34±0.21§ | 0.37 | − |
| H50Ea | 0.21±0.01§ | 0.09 | 0.81±0.04§ | 0.11 | 12.34±0.36§ | 0.01 | − |
| H50Ya | 0.87±0.02§ | 0.36 | 0.83±0.06§ | 0.43 | 12.57±0.73§ | 0.03 | − |
| H50K | 8.14±0.03§ | 3.40 | 0.69±0.06† | 4.95 | 6.77±0.27*§ | 0.45 | 1.54±0.06§ |
| H50F | 1.44±0.03§ | 0.60 | 0.76±0.01‡ | 0.79 | 6.43±0.38§ | 0.09 | − |
| C51A | 19.64±0.65§ | 8.22 | 0.82±0.04§ | 10.01 | 6.40±0.12*§ | 1.28 | 1.57±0.08§ |
| C51D | 2.16±0.07§ | 0.91 | 1.30±0.08§ | 0.7 | 16.94±0.69*§ | 0.05 | 1.40±0.03§ |
Although Met39 interacts directly with GSH-interacting residues, His38 and His50, the contribution to catalytic function is through the effect on GSH binding. The results suggest that the effect is mostly through a packing rearrangement that includes any perturbation of the orientation of His38 or His50. Rearrangement of active-site residues is elicited by M39A to accommodate the decreased volume as well as the lack of dipole–dipole interaction with the histidines. This condition also appears to engender flexibility to this region, which is shown by positive co-operativity for GSH binding with a Hill coefficient of 1.72±0.12 (Table 1).
The GSH interaction was decreased for all His38 engineered enzymes as shown by an increase in KmGSH of 17–71-fold. In addition, positive co-operativity for GSH was observed for H38E with a Hill coefficient (h) of 1.83±0.01.
There is a hydrogen bond network in the GST active site that is adjacent to the GSH glycine moiety. This network is formed by several residues, Asn47, Gln49 and Cys51, as well as His50, with the latter directly interacting with GSH. His50 replacements not only decreased binding affinity to GSH (ranging from 13- to 25-fold) but also had an impact on catalytic rates as shown by kcat values that decreased from 13 to 0.3% of the wild-type enzyme kcat values. The N47A change displayed several large effects on the enzyme, one on enzyme catalysis, with the enzyme having only 0.3% kcat of the wild-type, and another on GSH binding with a 23-fold increase in KmGSH. Gln49 and Cys51 interaction is through their main-chain nitrogens and oxygens. Changes in these residues showed intermediate effects on catalysis except for C51D, which had a kcat only 3.5% of wild-type. However, the changes in these residues all showed a major impact on GSH binding with increased KmGSH values from 6- to 34-fold. The results suggest that a packing rearrangement occurred. This rearrangement also enabled subunit–subunit communication as shown by the observed positive co-operativity upon GSH binding (Table 1).
Substrate specific activity was affected for all the engineered enzymes to varying degrees (Table 2). For example, most of the enzymes exhibited less effect on activity for EA. This suggests that the binding mode and the orientation of this substrate are different from the other substrates. Both GSH and hydrophobic substrate-binding sites are located in the same active-site pocket of GSTs, therefore it is not surprising that changes in the G-site residues can perturb the hydrophobic substrate site.
Table 2. Substrate-specific activity of the engineered enzymes compared to the wild-type.
The substrates used were 3 mM CDNB, 1 mM DCNB, 0.1 mM PNPBr, 0.1 mM PNBC and 0.2 mM EA. The reactions were performed at a constant GSH concentration (appropriate for each enzyme). aSome of these values have been previously reported and are shown for purposes of comparison [19,20]. One-way ANOVA with Dunnett's multiple comparison test was performed with wild-type as control; statistical significance is shown by † for P<0.05, ‡ for P<0.01 and § for P<0.001.
| Specific activity (μmol·min−1·mg−1) | |||||
|---|---|---|---|---|---|
| Enzyme | CDNB | DCNB | EA | PNPBr | PNBC |
| Wild-typea | 52.50±0.52 | 0.035±0.006 | 0.286±0.062 | 0.074±0.012 | 0.064±0.002 |
| H38Aa | 21.70±0.20§ | 0.037±0.001 | 0.146±0.016§ | 0.040±0.004§ | 0.013±0.002§ |
| H38Ea | 11.21±0.24§ | <0.001 | 0.191±0.026§ | <0.008 | <0.005 |
| H38Fa | 3.76±0.09§ | <0.001 | 0.230±0.019† | <0.005 | <0.003 |
| H38D | 2.33±0.07§ | <0.001 | 0.111±0.011§ | <0.001 | <0.003 |
| H38K | 2.02±0.07§ | <0.001 | 0.165±0.007§ | <0.003 | <0.002 |
| M39A | 18.84±0.66§ | 0.012±0.001§ | 0.183±0.011§ | <0.005 | <0.003 |
| M39F | 28.67±0.71§ | 0.043±0.002† | 0.166±0.010§ | 0.029±0.008§ | 0.065±0.009 |
| N47A | 0.13±0.01§ | <0.001 | 0.079±0.001§ | <0.001 | <0.001 |
| Q49A | 33.36±0.92§ | 0.023±0.002‡ | 0.244±0.012 | 0.050±0.002§ | 0.044±0.001‡ |
| Q49E | 41.76±0.60§ | 0.032±0.001 | 0.244±0.020 | 0.020±0.001§ | 0.048±0.003‡ |
| H50Aa | 3.80±0.10§ | 0.015±0.007§ | 0.146±0.050§ | 0.012±0.002§ | 0.016±0.007§ |
| H50Ea | 0.15±0.01§ | <0.001 | 0.026±0.004§ | <0.005 | <0.005 |
| H50Ya | 0.47±0.01§ | <0.001 | 0.064±0.008§ | <0.008 | <0.005 |
| H50K | 6.28±0.04§ | 0.007±0.001§ | 0.053±0.020§ | <0.007 | <0.004 |
| H50F | 0.93±0.03§ | <0.001 | 0.077±0.005§ | <0.001 | <0.001 |
| C51A | 15.44±0.58§ | 0.009±0.001§ | 0.046±0.001§ | 0.019±0.001§ | <0.001 |
| C51D | 1.24±0.03§ | <0.001 | 0.080±0.004§ | <0.002 | <0.001 |
It has been proposed that many steps occur in the GST catalytic mechanism, including GSH ionization through thiol deprotonation, substrate conjugation by nucleophilic attack of the thiolate at the electrophilic centre, product formation and product release from the active site [13,26–28]. The overall velocity of the enzyme-catalysed reaction is affected by most of the engineered residues, although to different extents. Therefore several steps in the catalytic pathway were studied to determine the roles of the engineered residues.
pH dependence of kinetic constants
The pKa values of ionized GSH (enzyme-bound GSH) were calculated from plots of kcat/KmCDNB versus pH for the wild-type and engineered enzymes (Table 3; Figure 2). The apparent pKa for wild-type is approx. 6.0. The pKa values of ionized GSH in the engineered enzymes varied, with the greatest effect shown by H50A which increased it by almost 1 pH unit. GSH ionization is considered to be an important step in the catalytic mechanism of GST that generates the thiolate anion (intermediate deprotonated form of GSH) for conjugation with the electrophilic substrate. This kinetically relevant ionization of GSH has been shown to be reflected in pH dependence for kcat/KmCDNB [13]. Several reports have shown that residues located near the cysteine thiol and glutamyl α-carboxylate of GSH contribute to promoting and stabilizing the anionic glutathione thiol group [13,27–31]. However, this is the first report of involvement of the GSH glycine moiety in GSH ionization.
Table 3. The pKa values of ionized GSH in the active sites of wild-type and engineered enzymes.
The effect of pH on kcat/kmCDNB was obtained by measuring kinetic constants using various pH buffers of 0.1 M sodium acetate buffers (from pH 5.0 to 5.5) and 0.1 M potassium phosphate buffer (from pH 6.0 to 8.5). The pKa values of ionized GSH (GSH-bound enzyme) were determined. n.d., Not determined (low activity precluded measurement). One-way ANOVA with Dunnett's multiple comparison test was performed with wild-type as control; statistical significance is shown by § for P<0.001. See Figure 2 for a plot of the experimental data.
| Enzyme | pKa (ionized GSH) |
|---|---|
| Wild-type | 6.00±0.05 |
| H38A | 6.10±0.02 |
| M39A | 6.52±0.05§ |
| N47A | n.d. |
| Q49A | 6.23±0.05§ |
| H50A | 6.92±0.06§ |
| C51A | 6.22±0.03§ |
| C51D | 6.42±0.03§ |
Figure 2. Plot of the data to determine the pKa values of ionized GSH in the active sites of wild-type and engineered enzymes.

The effect of pH on kcat/kmCDNB was obtained by measuring kinetic constants using various pH buffers of 0.1 M sodium acetate buffers (from pH 5.0 to 5.5) and 0.1 M potassium phosphate buffer (from pH 6.0 to 8.5). The pKa values of ionized GSH (GSH-bound enzyme) were determined (see Table 3). The results are presented as means±S.D. from at least three independent experiments.
Meisenheimer complex formation
Meisenheimer complex or σ-complex intermediate, is generated during a nucleophilic aromatic substitution reaction. Leaving-group effects on enzyme catalysis can be used to determine whether the rate-limiting step of the reaction is σ-complex formation. The substitution of a more electronegative leaving group concomitant with an increase in the rate constant of the spontaneous reaction with glutathione is a signal that the σ-complex formation is a rate-limiting step [13]. Therefore the influence on the catalytic constant of the chlorine leaving group of CDNB, replaced with fluorine, was examined (Table 4).
Table 4. Effect of fluoride/chloride leaving group substitution on the rate of catalysis.
The ratio of catalytic rates for the conjugation reaction catalysed by wild-type and engineered GSTs using GSH and CDNB or FDNB as co-substrates. One-way ANOVA with Dunnett's multiple comparison test was performed with wild-type as control; statistical significance is shown by † for P<0.05, ‡ for P<0.01 and § for P<0.001.
| Leaving group effect | ||
|---|---|---|
| Enzyme | kcatFDNB/kcatCDNB | (kcat/Km)FDNB/(kcat/Km)CDNB |
| Wild-type | 1.76±0.01 | 20.59±1.02 |
| H38A | 3.11±0.21 | 14.12±0.84† |
| M39A | 8.29±0.95§ | 32.45±3.72§ |
| N47A | 18.09±0.90§ | 29.65±1.32‡ |
| Q49A | 2.63±0.08 | 26.29±1.77† |
| Q49E | 1.50±0.08 | 24.95±1.21 |
| H50A | 18.57±0.41§ | 16.55±1.59 |
| C51A | 2.93±0.05 | 30.31±2.85§ |
| C51D | 26.75±0.99§ | 44.71±4.15§ |
Although the wild-type enzyme was insensitive, several of the engineered enzymes demonstrated sensitivity to the halogen leaving group, particularly H50A, C51D and N47A, with increased ratio values of 19-, 27- and 18-fold respectively. The catalytic efficiency (kcat/Km) of the engineered enzymes showed a different sensitivity to the nature of the leaving group, suggesting that an alteration of the relative catalytic-centre activity is a consequence of changes in the rate of σ-complex formation rather than changes in binding affinity towards the different substrate leaving groups. The results suggest that the process of σ-complex intermediate formation is affected by the disruption of the hydrogen bond network to GSH which affected the overall velocity of the enzyme-catalysed reaction. It is possible that the residue changes caused incorrect orientation of the GSH thiol or of the His50 side chain, which then disturbs the conjugation process of the thiolate anion with the electrophilic substrate. Additionally, the results strongly support that the hydrogen bond network contributes to both the GSH ionization process and σ-complex intermediate formation. This contribution appears to stabilize the His50 residue as shown by the changes in pKa and the greater effect of the leaving group on the catalytic constants of C51D but not C51A.
Viscosity effect on kinetic constants
The viscosity effect was studied to determine whether the rate-limiting step of the reaction is physical or chemical. The slope of the reciprocal plot of the inverse relative catalytic constant (kcat0/kcat) versus the relative medium viscosity (η/η0) was determined (Table 5). A slope near unity gives a proportional decrease in rate constant with increasing viscosity of the solution and shows a physical step is rate-determining, whereas a slope of zero indicates that a chemical reaction step is rate limiting [32,33].
Table 5. Viscosity effect on kinetic constants and free energy changes of wild-type and engineered enzymes.
The effect of viscosity on kinetic constants was assayed by using 0.1 M potassium phosphate buffer (pH 6.5) with various glycerol concentrations. The slope of a reciprocal plot of the relative catalytic constant (kcat0/kcat) versus relative viscosity (η/η0) was determined. n.d., Not determined (low activity precluded measurement); ΔΔG (the difference in the free energy changes for the formation of the transition states in the wild-type and engineered enzymes) is calculated from the equation: ΔΔG=−RT ln(kcat/KmCDNB)engineered/(kcat/KmCDNB)wild-type. One-way ANOVA with Dunnett's multiple comparison test was performed with wild-type as control; statistical significance is shown by § for P<0.001.
| Enzyme | Slope | ΔΔG (kJ/mol) |
|---|---|---|
| Wild-type | 0.959±0.036 | − |
| H38A | 0.485±0.019§ | 3.58±0.033 |
| M39A | 0.430±0.031§ | 3.33±0.132 |
| N47A | n.d. | 14.8±0.222 |
| Q49A | 0.797±0.036§ | 1.86±0.105 |
| H50A | 0.005±0.001§ | 7.56±0.095 |
| C51A | 1.11±0.010§ | 4.08±0.125 |
| C51D | 0.051±0.007§ | 10.7±0.201 |
Wild-type enzyme displayed a linear dependence with a slope of approx. 1.0, suggesting that a physical step of the reaction that includes product release and/or structural transition is rate limiting. The engineered enzymes exhibited viscosity effects on kcat to different degrees. H50A and C51D were viscosity-independent with a slope approaching zero. These engineered enzymes changed the rate-limiting step of the reaction to a chemical step that includes a GSH ionization step and σ-complex formation as described above, whereas partial dependence on a diffusion barrier and other viscosity-dependent motions were observed for the remaining enzymes that displayed viscosity effects with intermediate values (0<slope<1).
Also displayed in Table 5 is ΔΔG, which is shown to illustrate the differences in the free energy changes for the formation of the transition states in the wild-type and engineered enzymes, as calculated at 25 °C from the equation below [34]:
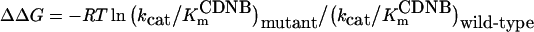 |
H50A, N47A and C51D have a greater ΔΔG (7.56, 14.8 and 10.7 kJ/mol respectively) compared with other engineered enzymes (ranging from 1.860 to 4.075 kJ/mol) indicating that upon disruption of the hydrogen bond network, the enzymes require more energy than the wild-type enzyme to form and stabilize the transition state.
Characterization of physical properties
The stability of the proteins was determined in comparison with the adGSTD4-4 wild-type (Table 6). In general, the engineered His38, His50, Asn47, Cys51 and Gln49 proteins exhibited comparable stabilities to the wild-type, indicating a minor role of these residues in structural maintenance. However, H38E increased stability of the enzyme 2.6-fold, suggesting that a conformational change occurred which is also supported by the observed positive co-operativity. Met39 appears to play a role in structural integrity as shown by 3-fold decreased enzyme stability for both the alanine and phenylalanine changes. However, the initial folding processes of the enzymes were observed to yield comparable refolding rates to the wild-type (results not shown). These results suggest that the Met39 dipole–dipole interaction and positioning of His38 and His50 in a suitable conformation have an impact not only on the kinetic properties but also on enzyme stability.
Table 6. Thermal stability of wild-type and engineered adGSTD4-4 at 45 °C.
The remaining GST activity was measured after incubating the enzyme at various time points at 45 °C. n.d., Not determined (low activity precluded measurement). aSome of these values have been previously reported and are shown for purposes of comparison [19,20]. One-way ANOVA with Dunnett's multiple comparison test was performed with wild-type as control; statistical significance is shown by ‡ for P<0.01 and § for P<0.001.
| Enzyme | Half-life at 45 °C (min) |
|---|---|
| Wild-typea | 15.32±0.31 |
| H38Aa | 15.33±0.88§ |
| H38Ea | 40.17±1.26§ |
| H38Fa | 19.33±0.59§ |
| H38D | 15.18±1.67 |
| H38K | 14.07±0.73 |
| M39A | 4.71±0.16§ |
| M39F | 4.72±0.23§ |
| N47A | n.d. |
| Q49A | 15.19±1.06§ |
| Q49E | 25.19±1.37§ |
| H50Aa | 25.81±1.99§ |
| H50Ea | 15.31±0.54 |
| H50Ya | 20.79±0.34§ |
| H50K | 11.99±0.51§ |
| H50F | 27.07±0.82§ |
| C51A | 14.86±0.64 |
| C51D | 18.18±1.01‡ |
The intrinsic fluorescence of tryptophan was used as an indicator of changes in tertiary structure (Figure 3). The λmax of tryptophan fluorescence in the engineered enzymes was slightly different from wild-type, with a red shift in the range of 1–4 nm indicating a different polarity in tryptophan environments. In addition, differences in fluorescence intensity between wild-type and engineered enzymes were observed. This result suggests that there are minor conformational disturbances in the active-site topography, which affect orientations of tryptophan and/or its neighbouring residues thereby modulating the quenching and tryptophan exposure to the electrophilic environment. For example, the active-site topology changes that generated the positive co-operativity in H50K also yielded the lowest fluorescence intensity among the His50 engineered enzymes (∼44% of wild-type). Moreover, the conformational change induced by glutamic acid residue replacement of His38 also gave a 2-fold increase in enzyme stability, increased intrinsic fluorescence intensity to 197% of wild-type, modulated GSH binding and enzyme catalysis as well as yielding the observed positive co-operativity towards GSH.
Figure 3. The maximum emission wavelength (λmax) and intrinsic fluorescence intensity at λmax of tryptophan fluorescence of wild-type and the engineered enzymes.

Excitation was at 295 nm and emission was scanned from 300 to 450 nm. Samples (n=3) contained 0.1 mg/ml protein in 0.1 M potassium phosphate buffer (pH 6.5). Percent intensity change compared with wild-type enzyme was measured at fluorescence λmax averaged over three scans corrected for dilution and inner filter effects. a.u., arbitrary units.
DISCUSSION
The G-site residues interacting directly with cysteine and glutamyl moieties of GSH have been shown in several studies to contribute to GST catalytic mechanisms, including GSH binding, catalysis, GSH ionization, as well as rate-limiting step determination [10–14]. In the present study, we have examined the significant contributions of GST residues that interact with the GSH glycine moiety as well as identifying a hydrogen bond network. The network consists of His38, Met39, Asn47, Gln49, His50 and Cys51 and contributes to catalysis through multiple processes including GSH ionization, nucleophilic substitution, product formation and product dissociation.
The kinetic studies demonstrated that the engineered residues greatly affected the enzyme's ability to interact with GSH. Moreover, the engineered enzymes of His38, Met39, His50, Cys51 and Gln49 have shown strong positive co-operativity upon binding of GSH. However, the positive co-operativity was not observed for the engineered enzymes of residues interacting with the other moieties of GSH [11–13,15]. Therefore it can be suggested that the residues interacting with the GSH glycine moiety are not only involved in GSH interaction but also control the motion of a flexible region of GST. Upon binding of GSH in the active site, these GST residues induce an active-site conformational change for the induced-fit mechanism. These residues are on the loops on either side of the α2-helix which connects the helix to the β2-sheet and β3-sheet. These loops serve as hinges for the movement of the α2-helix.
In the present study, changes of the active-site residues generated positive co-operativity between two subunits, a finding similar to that obtained in previous studies in which the subunit interface had been changed or where changes in highly flexible regions, for example Gly41, Cys47 and Lys54 of hGSTP1-1 (human GSTP1-1) [33,35] or Asn49 and Gln53 of maize GST1 [36,37], had been made. It has been proposed that the conformational transitions generate two different binding modes upon GSH binding: low-affinity and high-affinity conformations, which are related to the positive co-operativity observed [5,38,39]. The binding of GSH to the first active site stabilizes the low-affinity conformation of the enzyme which then becomes a high-affinity conformational state. Positive co-operativity upon GSH binding observed in His38, Met39, His50, Cys51 and Gln49 engineered enzymes may be relevant to this conformational transition concept. These residues are located in a highly flexible region, therefore the residue changes would alter the flexibility of the α2-helix to fit GSH in the active site, which may then generate two different conformational transition states upon GSH binding.
Both His38 and His50 interact directly with the glycine carboxylate of GSH. The functional groups at positions 38 and 50 are significant in size, volume and polarity for GSH binding and enzyme catalysis. However, His50 contributes more to the GSH activation process and enzyme catalysis in which the full function is achieved by synergistic action with the hydrogen bond network residues (Asn47, Gln49 and Cys51). The disruption of the hydrogen bond network, for the His50, Asn47 and Cys51 engineered enzymes, showed progressively decreased kcat values to more than 90%. Moreover, several aspects of the GST catalytic mechanism were altered by H50A. H50A decreased the enzyme's ability to lower the pKa of the GSH thiol group up to 1 pH unit; an effect this large has been observed for mutations of the conserved serine/tyrosine residue interacting with the GSH thiol group [13,40–42]. Additionally, the rate-limiting step of the H50A is fully switched from a physical step to a chemical step, as determined by fluoride/chloride leaving group studies and viscosity effect on the kinetic constants. As the important His50 is still present, lesser effects were observed for the Gln49 and Cys51 engineered enzymes for minor disruptions of the stabilizing hydrogen bond network.
The results strongly support that His50 is a key residue in the hydrogen bond network and functions as a proton acceptor as well as controlling the electron distribution in the active site to promote ionization and stabilization of the GSH thiolate anion. The His50 residue contributes to precise residue and substrate orientations, GSH ionization, σ-complex intermediate formation and the release of product from the active site. The regulation of the electrostatic field in the active site by positively charged residues has also been reported in several studies [15,43,44].
The changes in catalytic efficiency are also related to differences in free energy changes for the formation of transition states in the engineered and wild-type enzymes, as determined by ΔΔG [34]. The greater value of ΔΔG refers to a decrease in the stabilization of the transition state compared with wild-type in which the enzyme utilizes more energy to stabilize its transition state. Moreover, the stabilization of the transition state contributes to multiple mechanisms in enzyme-catalysed reactions. ΔΔG values are increased up to 7.562, 14.84 and 10.666 kJ/mol for H50A, N47A and C51D respectively in which the hydrogen bond network is strongly disrupted, indicative of the incomplete pre-organized environment for enhancing catalysis along the reaction pathway of the engineered enzymes [45]. Therefore it can be noted that the network of hydrogen bonds is also required for the organization inside the enzyme molecule to provide stabilization of the transition state.
The positively charged residue position 50 appears to be highly conserved or functionally conserved across GST classes by histidine, lysine or arginine located at the equivalent structural position. This conserved residue is in hydrogen bonding distance of the glycine carboxylate moiety of GSH which is stabilized by a hydrogen bond network of surrounding residues (Figure 4) for example Lys40 in PtGSTU1 [Pinus tabulaeformis (Chinese hard pine) GSTU1], Lys41 in ZmGST1 [Zea mays (maize) GST1], Lys44 in hGSTP1-1, Lys41 in hGSTT2-2, Lys59 in hGSTO1-1, Lys49 in hGSTM2-2, Lys42 in squid GSTS1-1, Lys45 in Sj26GST [26 kDa GST from Schistosoma japonicum (oriental blood-fluke)] and Arg45 in hGSTA1-1. However, the feature of two positively charged histidines, His38 and His50, interacting with the glycine carboxylate moiety of GSH is a unique trait for insect GSTDs. His38 is responsible for GSH binding, whereas His50 contributes to several steps of the enzyme-catalysed reaction from GSH interaction to the release of product from the active site.
Figure 4. Conserved G-site residues that directly interact with the glycine moiety of GSH and generate a hydrogen bond network in several GST classes.

The conserved G-site residues are shown by stick models, while line models represent the hydrogen bond network residues at the glycine moiety of GSH. GSH or GSH analogues are illustrated in ball-and-stick; GSH (a, b, c, d, e, f), lactoylglutathione (g), iodobenzylglutathione (h) and S-hexyl-GSH (i). Green dotted lines represent hydrogen-bond interactions. PDB code numbers: hGSTP1-1 (PDB id 8GSS), hGSTA1-1 (PDB id 1PKW), hGSTM2-2 (PDB id 1XW5), hGSTO1-1 (PDB id 1EEM), hGSTT2-2 (PDB id 1LJR), Lucilia GST [7], ZmGST1 (PDB id 1AXD), squid GSTS1-1 (PDB id 2GSQ) and TaGSTU4 [Triticum aestivum (wheat) GSTU4; PDB id 1GWC]. The Figure was created with Accelrys DS ViewerPro 5.0.
For the GSH activation mechanism of Alpha, Mu and Pi GST isoenzymes, it has been described that the thiol proton is quantitatively released into solution after the thiolate anion is formed [46,47]. In contrast with GSTD, which behaves differently, the thiol proton is captured by at least an internal base residue at high pH value [46,47]. The present study shows that His50 is also a candidate for the thiol proton acceptor in addition to the residues at the γ-glutamyl portion of GSH reported previously [15,31]. Moreover, the overall velocity of His50 engineered enzymes is progressively restricted; therefore it can be assumed that the thiol proton released must be accepted first by the active site residues, e.g. His50, before a new cycle of the reaction can be initiated.
The ‘base-assisted deprotonation model’ is an alternative model that describes the mechanism of GST enzymes in the GSH ionization process. This model has been implicated in several studies of the glutamyl α-carboxylate of GSH acting as a catalytic base involved in the thiol proton acceptance from the GSH thiol group [15,16,31,46]. In the present study, we addressed the contribution of the glycine moiety of GSH for GSH ionization in which this end of GSH is involved in proton acceptance by providing a counter ion from the charged His50 which is stabilized by the hydrogen bond network.
In conclusion, the present study revealed a critical role for residues located at the glycine moiety of GSH in catalytic rate determination. This area constitutes a second G-site network involved in GSH ionization distinct from the network previously reported interacting with the glutamyl end of GSH [16,17]. This second network also appears to be functionally conserved in GSTs (Figure 4). In the present study, we showed that His50 is a central residue in the hydrogen bond network to GSH, with the protonated imidazole ring of His50 being stabilized by the network. His50 plays important roles in several processes of the enzyme mechanism. Moreover, this network at the glycine moiety of GSH also contributes to the ‘base-assisted deprotonation model’ for GSH ionization.
Acknowledgments
This work was funded by the TRF (Thailand Research Fund). A. V. was supported by a Royal Golden Jubilee Ph.D. Scholarship.
References
- 1.Sheehan D., Meade G., Foley V. M., Dowd C. A. Structure, function and evolution of glutathione transferases: implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem. J. 2001;360:1–16. doi: 10.1042/0264-6021:3600001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ketterer B. A bird's eye view of the glutathione transferase field. Chem. Biol. Interact. 2001;138:27–42. doi: 10.1016/s0009-2797(01)00277-0. [DOI] [PubMed] [Google Scholar]
- 3.Mannervik B., Awasthi Y. C., Board P. G., Hayes J. D., Di Ilio C., Ketterer B., Listowsky I., Morgenstern R., Muramatsu M., Pearson W. R., et al. Nomenclature for human glutathione transferases. Biochem. J. 1992;282:305–306. doi: 10.1042/bj2820305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hayes J. D., Flanagan J. U., Jowsey I. R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 5.Mannervik B., Danielson U. H. Glutathione transferases – structure and catalytic activity. CRC Crit. Rev. Biochem. 1988;23:283–337. doi: 10.3109/10409238809088226. [DOI] [PubMed] [Google Scholar]
- 6.Armstrong R. N. Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem. Res. Toxicol. 1997;10:2–18. doi: 10.1021/tx960072x. [DOI] [PubMed] [Google Scholar]
- 7.Wilce M. C. J., Board P. G., Feil S. C., Parker M. W. Crystal structure of a theta-class glutathione transferase. EMBO J. 1995;14:2133–2143. doi: 10.1002/j.1460-2075.1995.tb07207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dirr H., Reinemer P., Huber R. X-ray crystal structures of cytosolic glutathione S-transferases. Implications for protein architecture, substrate recognition and catalytic function. Eur. J. Biochem. 1994;220:645–661. doi: 10.1111/j.1432-1033.1994.tb18666.x. [DOI] [PubMed] [Google Scholar]
- 9.Rossjohn J., Feil S. C., Wilce M. C. J., Sexton J., Spithill T. W., Parker M. W. Crystallization, structural determination and analysis of a novel parasite vaccine candidate: Fasciola hepatica glutathione S-transferase. J. Mol. Biol. 1997;273:857–872. doi: 10.1006/jmbi.1997.1338. [DOI] [PubMed] [Google Scholar]
- 10.Stenberg G., Board P. G., Carlberg I., Mannervik B. Effects of directed mutagenesis on conserved arginine residues in a human class Alpha glutathione transferase. Biochem. J. 1991;274:549–555. doi: 10.1042/bj2740549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu S., Zhang P., Ji X., Johnson W. W., Gilliland G. L., Armstrong R. N. Contribution of tyrosine 6 to the catalytic mechanism of isoenzyme 3-3 of glutathione S-transferase. J. Biol. Chem. 1992;267:4296–4299. [PubMed] [Google Scholar]
- 12.Kolm R. H., Sroga G. E., Mannervik B. Participation of the phenolic hydroxyl group of Tyr-8 in the catalytic mechanism of human glutathione transferase P1-1. Biochem. J. 1992;285:537–540. doi: 10.1042/bj2850537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caccuri A. M., Antonini G., Nicotra M., Battistoni A., Lo Bello M., Board P. G., Parker M. W., Ricci G. Catalytic mechanism and role of hydroxyl residues in the active site of theta class glutathione S-transferases. Investigation of Ser-9 and Tyr-113 in a glutathione S-transferase from the Australian sheep blowfly, Lucilia cuprina. J. Biol. Chem. 1997;272:29681–29686. doi: 10.1074/jbc.272.47.29681. [DOI] [PubMed] [Google Scholar]
- 14.Tan K.-L., Chelvanayagam G., Parker M. W., Board P. G. Mutagenesis of the active site of the human Theta-class glutathione transferase GSTT2-2: catalysis with different substrates involves different residues. Biochem. J. 1996;319:315–321. doi: 10.1042/bj3190315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Winayanuwattikun P., Ketterman A. J. Catalytic and structural contributions for glutathione binding residues in a Delta class glutathione S-transferase. Biochem. J. 2004;382:751–757. doi: 10.1042/BJ20040697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Winayanuwattikun P., Ketterman A. J. An electron-sharing network involved in the catalytic mechanism is functionally conserved in different glutathione transferase classes. J. Biol. Chem. 2005;280:31776–31782. doi: 10.1074/jbc.M502612200. [DOI] [PubMed] [Google Scholar]
- 17.Winayanuwattikun P., Ketterman A. J. Glutamate 64, a newly identified residue of the functionally conserved electron-sharing network contributes to catalysis and structural integrity of glutathione transferases. Biochem. J. 2007;402:339–348. doi: 10.1042/BJ20061253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. UCSF chimera – a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 19.Vararattanavech A., Ketterman A. Multiple roles of glutathione binding-site residues of glutathione S-transferase. Protein Peptide Lett. 2003;10:441–448. doi: 10.2174/0929866033478654. [DOI] [PubMed] [Google Scholar]
- 20.Vararattanavech A., Prommeenate P., Ketterman A. J. The structural roles of a conserved small hydrophobic core in the active site and an ionic bridge in domain I of Delta class glutathione S-transferase. Biochem. J. 2006;393:89–95. doi: 10.1042/BJ20050555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Habig W. H., Pabst M. J., Jakoby W. B. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J. Biol. Chem. 1974;249:7130–7139. [PubMed] [Google Scholar]
- 22.Segel I. H. New York: John Wiley and Sons; 1993. Enzyme Kinetics, Behavior and Analysis of Rapid Equilibrium and Steady-state Enzyme Systems. [Google Scholar]
- 23.Jirajaroenrat K., Pongjaroenkit S., Krittanai C., Prapanthadara L., Ketterman A. J. Heterologous expression and characterization of alternatively spliced glutathione S-transferases from a single Anopheles gene. Insect Biochem. Mol. Biol. 2001;31:867–875. doi: 10.1016/s0965-1748(01)00032-7. [DOI] [PubMed] [Google Scholar]
- 24.Caccuri A. M., Ascenzi P., Lo Bello M., Federici G., Battistoni A., Mazzetti P., Ricci G. Are the steady state kinetics of glutathione transferase always dependent on the deprotonation of the bound glutathione? New insights in the kinetic mechanism of GST P1-1. Biochem. Biophys. Res. Commun. 1994;200:1428–1434. doi: 10.1006/bbrc.1994.1610. [DOI] [PubMed] [Google Scholar]
- 25.Wolf A. V., Brown M. G., Prentiss P. G. Boca Raton, FL: CRC Press; 1985. Handbook of Chemistry and Physics. [Google Scholar]
- 26.Armstrong R. N., Rife C., Wang Z. Structure, mechanism and evolution of thiol transferases. Chem. Biol. Interact. 2001;133:167–169. [Google Scholar]
- 27.Caccuri A. M., Ascenzi P., Antonini G., Parker M. W., Oakley A. J., Chiessi E., Nuccetelli M., Battistoni A., Bellizia A., Ricci G. Structural flexibility modulates the activity of human glutathione transferase P1-1. Influence of a poor co-substrate on dynamics and kinetics of human glutathione transferase. J. Biol. Chem. 1996;271:16193–16198. doi: 10.1074/jbc.271.27.16193. [DOI] [PubMed] [Google Scholar]
- 28.Caccuri A. M., Lo Bello M., Nuccetelli M., Rossi P., Antonini G., Federici G., Ricci G. Proton release upon glutathione binding to glutathione transferase P1-1: kinetic analysis of a multistep glutathione binding process. Biochemistry. 1998;37:3028–3034. doi: 10.1021/bi971903g. [DOI] [PubMed] [Google Scholar]
- 29.Armstrong R. N. Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem. Res. Toxicol. 1997;10:2–18. doi: 10.1021/tx960072x. [DOI] [PubMed] [Google Scholar]
- 30.Gustafsson A., Pettersson P. L., Grehn L., Jemth P., Mannervik B. Role of the glutamyl α-carboxylate of the substrate glutathione in the catalytic mechanism of human glutathione transferase A1-1. Biochemistry. 2001;40:15835–15845. doi: 10.1021/bi010429i. [DOI] [PubMed] [Google Scholar]
- 31.Widersten M., Björnestedt R., Mannervik B. Involvement of the carboxyl groups of glutathione in the catalytic mechanism of human glutathione transferase A1-1. Biochemistry. 1996;35:7731–7742. doi: 10.1021/bi9601619. [DOI] [PubMed] [Google Scholar]
- 32.Johnson W. W., Liu S., Ji X., Gilliland G. L., Armstrong R. N. Tyrosine 115 participates both in chemical and physical steps of the catalytic mechanism of a glutathione S-transferase. J. Biol. Chem. 1993;268:11508–11511. [PubMed] [Google Scholar]
- 33.Ricci G., Caccuri A. M., Lo Bello M., Rosato N., Mei G., Nicotra M., Chiessi E., Mazzetti A. P., Federici G. Structural flexibility modulates the activity of human glutathione transferase P1-1. Role of helix 2 flexibility in the catalytic mechanism. J. Biol. Chem. 1996;271:16187–16192. doi: 10.1074/jbc.271.27.16187. [DOI] [PubMed] [Google Scholar]
- 34.Dirr H. W., Little T., Kuhnert D. C., Sayed Y. A conserved N-capping motif contributes significantly to the stabilization and dynamics of the C-terminal region of class alpha glutathione transferases. J. Biol. Chem. 2005;280:19480–19487. doi: 10.1074/jbc.M413608200. [DOI] [PubMed] [Google Scholar]
- 35.Lo Bello M., Nuccetelli M., Chiessi E., Lahm A., Mazzetti A. P., Parker M. W., Tramontano A., Federici G., Ricci G. Mutations of Gly to Ala in human glutathione transferase P1-1 affect helix 2 (G-site) and induce positive cooperativity in the binding of glutathione. J. Mol. Biol. 1998;284:1717–1725. doi: 10.1006/jmbi.1998.2270. [DOI] [PubMed] [Google Scholar]
- 36.Labrou N. E., Mello L. V., Clonis Y. D. Functional and structural roles of the glutathione-binding residues in maize (Zea mays) glutathione S-transferase I. Biochem. J. 2001;358:101–110. doi: 10.1042/0264-6021:3580101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Labrou N. E., Mello L. V., Clonis Y. D. The conserved Asn49 of maize glutathione S-transferase I modulates substrate binding, catalysis and intersubunit communication. Eur. J. Biochem. 2001;268:3950–3957. doi: 10.1046/j.1432-1327.2001.02307.x. [DOI] [PubMed] [Google Scholar]
- 38.Principato G. B., Danielson U. H., Mannervik B. Relaxed thiol substrate specificity of glutathione transferase effected by a non-substrate glutathione derivative. FEBS Lett. 1988;231:155–158. doi: 10.1016/0014-5793(88)80722-1. [DOI] [PubMed] [Google Scholar]
- 39.Jemth P., Mannervik B. Fast product formation and slow product release are important features in a hysteretic reaction mechanism of glutathione transferase T2-2. Biochemistry. 1999;38:9982–9991. doi: 10.1021/bi983065b. [DOI] [PubMed] [Google Scholar]
- 40.Wang J., Barycki J. J., Colman R. F. Tyrosine 8 contributes to catalysis but is not required for activity of rat liver glutathione S-transferase, 1-1. Protein Sci. 1996;5:1032–1042. doi: 10.1002/pro.5560050606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gustafsson A., Etahadieh M., Jemth P., Mannervik B. The C-terminal region of human glutathione transferase A1-1 affects the rate of glutathione binding and the ionization of the active-site Tyr9. Biochemistry. 1999;38:16268–16275. doi: 10.1021/bi991482y. [DOI] [PubMed] [Google Scholar]
- 42.Labrou N. E., Rigden D. J., Clonis Y. D. Engineering the pH-dependence of kinetic parameters of maize glutathione S-transferase I by site-directed mutagenesis. Biomol. Eng. 2003;21:61–66. doi: 10.1016/j.bioeng.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 43.Patskovsky Y. V., Patskovska L. N., Listowsky I. The enhanced affinity for thiolate anion and activation of enzyme-bound glutathione is governed by an arginine residue of human Mu class glutathione S-transferases. J. Biol. Chem. 2000;275:3296–3304. doi: 10.1074/jbc.275.5.3296. [DOI] [PubMed] [Google Scholar]
- 44.Björnestedt R., Tardioli S., Mannervik B. The high activity of rat glutathione transferase 8-8 with alkene substrates is dependent on a glycine residue in the active site. J. Biol. Chem. 1995;270:29705–29709. doi: 10.1074/jbc.270.50.29705. [DOI] [PubMed] [Google Scholar]
- 45.Garcia-Viloca M., Gao J., Karplus M., Truhlar D. G. How enzymes work: analysis by modern rate theory and computer simulations. Science. 2004;303:186–195. doi: 10.1126/science.1088172. [DOI] [PubMed] [Google Scholar]
- 46.Caccuri A. M., Antonini G., Board P. G., Parker M. W., Nicotra M., Lo Bello M., Federici G., Ricci G. Proton release on binding of glutathione to Alpha, Mu and Delta class glutathione transferases. Biochem. J. 1999;344:419–425. [PMC free article] [PubMed] [Google Scholar]
- 47.Caccuri A. M., Antonini G., Board P. G., Flanagan J., Parker M. W., Paolesse R., Turella P., Federici G., Lo Bello M., Ricci G. Human glutathione transferase T2-2 discloses some evolutionary strategies for optimization of substrate binding to the active site of glutathione transferases. J. Biol. Chem. 2001;276:5427–5431. doi: 10.1074/jbc.M002819200. [DOI] [PubMed] [Google Scholar]