

Abstract
Sickle cell disease (SCD) is characterized by reperfusion injury and chronic oxidative stress. Oxidative stress and hemolysis in SCD result in inactivation of nitric oxide (NO) and depleted arginine levels. We hypothesized that augmenting NO production by arginine supplementation will reduce oxidative stress in SCD. To this end, we measured the effect of arginine (5% in mouse chow) on NO metabolites (NOx), lipid peroxidation (LPO) and selected antioxidants in transgenic sickle mouse models. Untreated transgenic sickle (NY1DD) mice (expressing ~75% βS-globin of all β-globins; mild pathology) and knockout sickle (BERK) mice (expressing exclusively hemoglobin S; severe pathology) showed reduced NOx levels and significant increases in the liver LPO compared with C57BL mice, with BERK mice showing maximal LPO increase in accordance with the disease severity. This was accompanied by reduced activity of antioxidants (glutathione [GSH], total superoxide dismutase [SOD], catalase and glutathione peroxidase [GPx]). However, GSH levels in BERK were higher than in NY1DD mice indicating a protective response to greater oxidative stress. Importantly, dietary arginine significantly increased NOx levels, reduced LPO and increased antioxidants in both sickle mouse models. In contrast, L-NAME, a potent non-selective NOS inhibitor, worsened the oxidative stress in NY1DD mice. Thus, attenuating effect of arginine on oxidative stress in SCD mice suggests its potential application in the management of this disease.
Keywords: Sickle cell disease, oxidative stress, arginine, lipid peroxidation, antioxidants
INTRODUCTION
Increasing evidence accumulated over the last decade indicates that reactive oxygen species (ROS) play a crucial role in the pathophysiology of various ischemic diseases. The concept that sickle cell disease (SCD) is a state of inflammation has been substantiated by the reports of endothelial injury/activation and excessive generation of ROS in this disease, and enhanced leukocyte-endothelium interactions [3,13,15,31]. The oxidative stress in SCD is likely the result of intravascular sickling and transient vaso-occlusive events.
In SCD, growing evidence shows reduced bioavailability of nitric oxide (NO), probably due to consumption of NO by free oxygen radicals, and/or by cell free-plasma heme as a result of hemolysis [3,35]. In SCD and sickle mouse models, a preponderance of recent observations shows depleted levels of L-arginine, a substrate for NO, and NO metabolites (NOx) [24,25,36]. However, there is no definitive data on the status of antioxidant enzyme and lipid peroxidation according to the disease severity, as well as on the effect of NO replenishment. Among the likely sources of O2•− in SCD are plasma membrane NADPH oxidase [12,44], enzyme NO synthase due to depleted substrate arginine [40] and increased plasma xanthine oxidase (XO) activity [3,41].
NO has important salutary physiological properties like vascular tone regulation, quenching reactive oxygen intermediates and inhibiting leukocyte recruitment in the microcirculation [2,15,17,22,27,29]. Depleted NO production in a transgenic-knockout (BERK) model is associated with increased tyrosine nitration [16], suggesting that hemolysis in this model may trigger the reported hemin-mediated nitrotyrosine formation in the presence of nitrite and hydrogen peroxide (H2O2) [38]. L-arginine (a substrate for NO) and several NO donors can decrease the damage resulting from post-ischemic reperfusion [14]. Arginine blunts the inhibitory effect of XO on vascular function [42]. In renal ischemic-reperfusion injury, NO can protect the kidneys against leukocyte adhesion to vascular endothelium [29]. In contrast, inhibition of NO production by L-NAME, a potent nonselective inhibitor of nitric oxide synthase (NOS) causes in an inflammatory response, resulting in increased leukocyte-endothelium interaction [17] and oxidant generation [18].
Although the increased leukocyte counts in human SCD and transgenic sickle mouse models suggest a systemic effect of oxidative stress, a direct evidence of organ oxidative stress requires biochemical estimations of oxidative stress and cellular antioxidants. Among the many consequences of ischemia/reperfusion injury are increased oxidant generation, peroxidation of the membrane lipids and reduced antioxidants, both non-enzymatic (e.g., glutathione [GSH]) and enzymatic antioxidants (e.g., SOD, catalase and glutathione peroxidase [Gpx]) [28]. Lipid peroxidation is perhaps the most recognized biological effect of oxygen radicals that occurs when free radicals react directly with cellular lipids. Lipid peroxidation results in damage to the cell membrane, as well as to the membrane of cellular organelles (e.g. mitochondria). Among the best characterized enzymatic pathways are SOD, catalase and GPx. SOD dismutates toxic O2•− into less toxic H2O2. Because H2O2 is a potential source for hydroxyl (.OH) radical, two additional enzymatic antioxidants (catalase and GPx) transform it to water. GPx is specific for GSH (a non-enzymatic antioxidant) as a hydrogen donor, but accepts peroxides as well as H2O2. During reperfusion, these endogenous defenses are likely to be ineffective as a result of overproduction of oxygen radicals. Additionally, during ischemia/reperfusion, degradation/consumption of antioxidants by the release of oxygen radicals has been well-documented [28]. Thus, failure to replenish antioxidants is expected to exacerbate the reperfusion injury and organ damage in SCD.
We hypothesize that in SCD, increased oxidative stress will result in increased lipid peroxidation and a decreased activity of antioxidants. To test this aspect, we have used transgenic mouse models of mild (NY1DD mice) and severe pathology (BERK mice). Transgenic NY1DD mice show a mild phenotype but develop an inflammatory phenotype after hypoxia/reoxygenation as evident by increased lipid peroxidation and leukocyte recruitment [15,31]. Transgenic-knockout BERK mice express exclusively human hemoglobin S, and recapitulate many features of human sickle cell disease, i.e., hemolytic anemia, reticulocytosis, presence of irreversibly sickled cells (ISCs) and multiple organ damage [33,37]. In the current studies, we have investigated the level of oxidative stress in unperturbed (untreated) sickle mouse models and after arginine supplementation and L-NAME treatment. Our result show distinct effect of disease severity in these sickle mouse models on oxidative stress, and a protective effect of arginine supplementation.
MATERIALS AND METHODS
Chemicals
Butylated hydroxytoluene (BHT), desferrioxamine (Desferal), 5,5'-dithiobis-2-nitrobenzoic acid (DTNB), glutathione (GSH), oxidized glutathione (GSSG), pyrogallol, triton X-100, ethylenediamine tetracetic acid (EDTA), bovine serum albumin (BSA), thiobarbituric acid (TBA), reduced nicotinamide adenine dinucleotide (NADH) and reduced nicotinamide adenine dinucleotide phosphate (NADPH) were obtained from Sigma/Aldrich (St. Louis, MO).
Transgenic mice
Transgenic (NY1DD) and knockout (BERK), used in the present studies, were raised in a specific pathogen-free facility at the University of Minnesota Medical School. NY1DD mice were backcrossed an average of 8 generation into C57BL/6J background. In NY1DD mice, βS-globin forms symmetrical tetramers with human α-globin (42%) and with mouse α-globin (~30%). In these mice, the total βS -globin levels are ~ 75% βS of all β-globins [8]. NY1DD mice show mild pathology, but exhibit multiple organ damage [7]. NY1DD mice show no hemolysis and systemic hematocrit (%) in NY1DD mice is similar to that found for C57BL control mice (mean ± SD: NY1DD mice, 47.5 ± 3; controls, 46.5 ± 2.1) [7]. Transgenic-knockout BERK mice express 100% humanα, >99% human βS, and show severe pathology. BERK mice exhibit several features of human SCD, i.e., hemolysis, reticulocytosis, low hematocrit and extensive multiple organ damage [33,37]. Hemolytic anemia in BERK mice is associated with significantly low systemic hematocrit, i.e., 28.7 ± 4.0 (P<0.00001 vs. controls) [16]. Globin composition was determined by denaturing HPLC as described [9].
L-arginine supplementation
Mice of each genotypes (C57BL/6J, NY1DD and BERK mice, n = 4–6; 4–6 months of age) received dietary 5% arginine supplementation in mouse chow (Harlan-Teklad, Madison, WI) for a period of 15 days. In each case, controls (untreated) were maintained on normal mouse chow without added arginine.
L-NAME (nitro-L-arginine methylester) treatment
Control C57BL mice (n=9) and NY1DD mice (n=7) were given 1mg/ml L-NAME (Sigma/Aldrich) in drinking water for seven days. Controls were given plain drinking water ad libitum.
NO metabolites (NOx) determination
For the determination of NO metabolites, NOx, the blood was drawn from bifurcating abdominal aorta using EDTA as an anticoagulant. Total NOx concentration in plasma was determined using nitrate/nitrite colorimetric assay kit (Cayman Chemical, Ann Arbor, MI) using manufacturer’s instructions. In brief, the method involves a two-step process. The first step involves conversion of nitrate to nitrite utilizing nitrate reductase. The second step consists of adding Greiss Reagents that convert nitrite into deep purple azo compound. The photometric determination of the absorbance at 540 nm of this azo compound accurately determines nitrite concentration.
Preparation of homogenates, cytosol and microsome fractions
Mice were sacrificed by cervical dislocation and the entire liver was then perfused immediately with cold 0.9% NaCl and thereafter carefully removed, trimmed free of extraneous tissue and rinsed in chilled 0.15M Tris-KCl buffer (0.15M KCl + 10mM Tris-HCL, pH 7.4) . The liver was then blotted dry, weighed quickly and homogenized in ice cold 0.15 M Tris-KCl buffer (pH 7.4) to yield 10% (w/v) homogenate. An aliquot of this homogenate (0.5 ml) was used for assaying reduced glutathione levels while the remainder was centrifuged at 10,000 rpm for 30 min. The resultant supernatant was transferred into pre-cooled ultracentrifugation tubes and centrifuged at 105, 000 x g for 60 min in a Beckman ultracentrifuge (Model - L870M). The supernatant (cytosol fraction), after discarding any floating lipid layer and appropriate dilution, was used for the assay of antioxidant enzymes, whereas the pellet representing microsomes was suspended in homogenizing buffer and used for assaying lipid peroxidation. The kidney, and the muscle from the legs were carefully removed, along with the liver, trimmed free of extraneous tissue and rinsed in chilled 0.15 M Tris-KCl (pH 7.4). The organs were then blotted dry, weighed quickly and homogenized in ice cold 0.15 M Tris-KCl buffer (pH 7.4) to yield a 10% (w/v) homogenate. 0.5ml aliquot of this homogenate was used for assaying reduced glutathione. The rest of the homogenate was processed as described for the liver.
Assay methods
Lipid peroxidation (LPO)
LPO in the microsomes was estimated spectrophotometrically by thiobarbituric acid reactive substances (TBARS) method, as described by Ohkawa et al. [30] and is expressed in terms of malondialdehyde (MDA) formed per mg protein. To validate our TBARS results, in a series of experiments, we measured LPO (i) using an iron chelator and an antioxidant in the microsomal preparation and (ii) using an enzyme immunoassay (EIA) for the determination of 8-isoprostanes.
In the first approach (TBARS), liver and/or muscle microsomal fraction was suspended in Tris-KCl, so as to contain approximately 1 mg protein in 0.1 ml suspension. An aliquote of 0.25 ml was mixed with 1.75 ml LPO buffer containing 50 μM desferrioxamine (desferal) and 0.1 ml of 2% butylated hydroxytoluene (BHT) in ethanol. Thereafter, 0.5 ml of 30% trichloroacetic acid (TCA) and 0.5 ml of 52mM thiobarbituric acid (TBA) was added to the mixture. Desferal was added to prevent further iron-catalyzed TBARS formation [5] and BHT was used to prevent further lipid peroxidation [10]. The reaction mixture was processed for the measurement of TBARS as described [30].
In the second approach, we measured total (free and esterified) 8-isoprostanes in the liver using an EIA kit (Cayman Chemicals, Ann Arbor, MI). The liver tissue were weighed and homogenized in three volume of 0.1 M phosphate buffer, pH 7.4, containing 1 mM EDTA and 10 μM indomethacin under cold conditions (1 g tissue in 3 ml phosphate buffer). The homogenate was centrifuged at 1680 x g at 4°C for 10 min. Further, alkaline hydrolysis was performed adding an equal volume of 15% wt/vol KOH and was incubated for 60 min at 40°C. Thereafter, 2 volumes of ethanol containing 0.01% BHT was added to the sample and incubated for 5 min at 4°C. This was followed by centrifugation at 1,500 x g for 10 min to remove precipitated proteins. The supernatant was decanted into a clean test tube and ethanol was evaporated (to<10% v/v) under a gentle stream of nitrogen. Samples were finally rediluted in 500 μl of 0.1 M phosphate buffer (pH 7.4). The assay employs an Ellman’s reaction producing an absorbance at 412 nm proportional to the amount of 8-isoprostane tracer bound to the well and it is inversely proportional to 8-isoprostane in the well. To test for interference, initially few test samples (n=7) were diluted to obtain two different dilutions of each sample between ~5 and 200 pg/ml. Two different dilutions of the samples showed good correlation (i.e., differed by less than 8%). Since the difference was less than 20%, purification was not needed according to the manufacturer’s instructions.
Glutathione (GSH)
GSH was estimated as total non-protein sulphydryl group by the method as described by Moron et al. [23].
Superoxide dismutase (SOD)
Total SOD was assayed utilizing the technique of Marklund and Marklund [21], which involves inhibition of pyrogallol autoxidation at pH 8.0. A single unit of enzyme was defined as the quantity of superoxide dismutase required to produce 50% inhibition of autoxidation.
Catalase
Catalase was estimated at 240 nm by monitoring the disappearance of H2O2 as described by Aebi [1]. The reaction mixture (1 ml, vol) contained 0.02 ml of suitably diluted cytosol in phosphate buffer (50 mM, pH 7.0) and 0.1ml of 30 mM H2O2 in phosphate buffer. Catalase enzyme activity has been expressed as moles of H2O2 reduced / min / mg protein.
Glutathione peroxidase (GPx)
GPx activity was measured by the coupled assay method as described by Paglia and Valentine [32]. One unit of enzyme activity have been defined as nmoles of NADPH consumed /min /mg protein based on an extinction coefficient of 6.22 mM−1 cm−1.
Statistical analysis
Statistical analysis of the data (mean ± SD) was performed using student’s t-test. P<0.05 was considered significant. The statistical analysis was performed using Statgraphics plus 5.0 program for Windows (Manugistics, Inc. Rockville, MD). Isoprostane calculation was performed using the analysis spread sheet provided by Cayman chemical at http://www.caymanchem.com/app/template/Home.vm.
RESULTS
The effect of arginine supplementation
Plasma NOx levels
As shown in Figure 1, NO metabolites (NOx) levels in untreated NY1DD and BERK mice were 51.7% and 78.7% lower, respectively, than control C57BL values (P<0.000001 each), indicating the minimal NO production in more severe BERK mice. While arginine caused ~20% increase in NOx concentration in control C57BL mice (P<0.0004), it resulted in marked 86.2% and 107% increases in NOx levels, respectively, in NY1DD and BERK mice (P<0.0001 and P<0.001) compared with untreated mice, indicating that arginine caused significant increases in NO production in both transgenic sickle mice.
Figure 1.
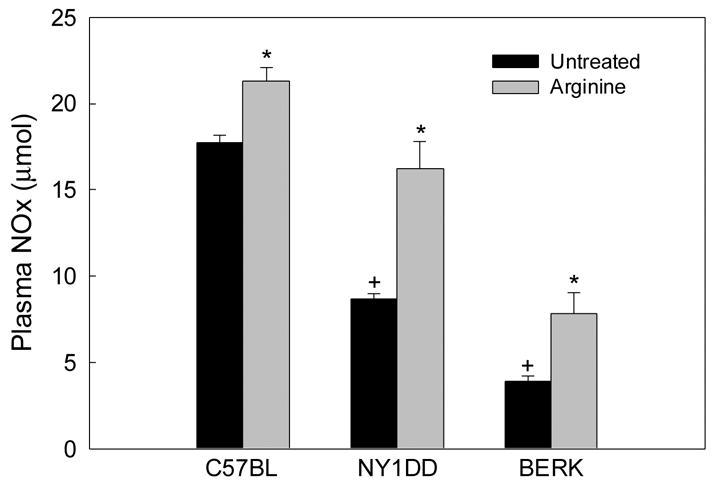
Plasma NOx levels in C57BL, NY1DD and BERK mice: the effect of arginine supplementation. Note the increased NOx levels after arginine treatment. *P<0.001–0.000001 vs. respective untreated controls; +P<0.00001 vs. untreated C57BL mice.
Lipid peroxidation (LPO)
Figure 2A depicts the results of LPO estimations in the liver of C57BL, NY1DD and BERK mice in the presence of desferal plus BHT. LPO values showed marked 67% and 150% increases, respectively, in untreated NY1DD and BERK mice as compared to C57BL controls (each P<0.001). Arginine supplementation (15 days in mouse chow) had no effect on LPO levels in C57BL mice. On the other hand, LPO levels decreased 11% in NY1DD mice (P<0.05 vs. untreated NY1DD), while maximal decrease (49%) was observed in BERK mice (P<0.0001 vs. untreated BERK).
Figure 2.
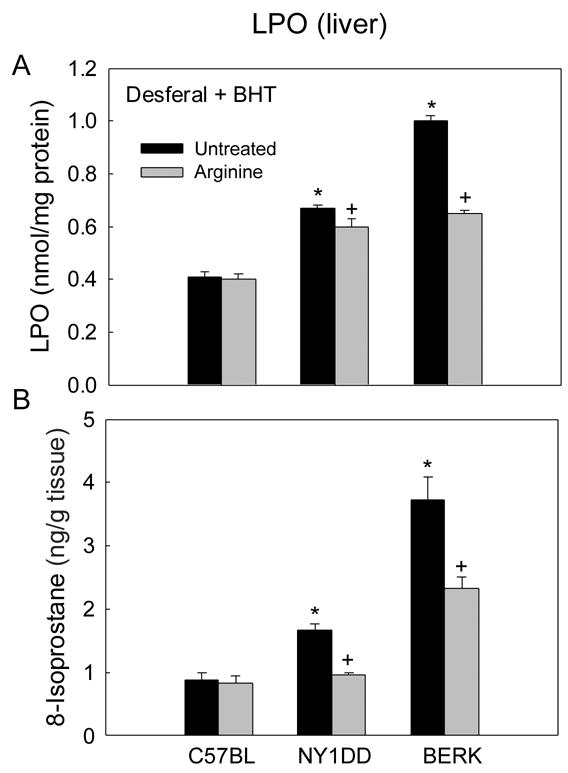
The effect of arginine supplementation on lipid peroxidation (LPO) in the liver of C57BL, NY1DD and BERK mice. (A) LPO estimation in the presence of desferal and BHT. (B) 8-isoprostane concentrations. *P<0.001 vs. untreated C57BL, +P<0.005–P<0.001 vs. untreated NY1DD or BERK mice
Measurement of total 8-isoprostanes (in the presence of BHT) in the liver validated the marked differences among untreated groups (Figure 2B). Isoprostanes showed 91% and 326% increases in NY1DD and BERK mice as compared with C57BL mice (each P<0.001), confirming the trend observed with TBARS method. Again, 8-isoprotane levels in these mice were directly proportional to the disease severity. In contrast to the above TBAR results, arginine had a pronounced ameliorating effect on isoprostane levels. While arginine had no effect in controls as also shown by TBARS method, it caused 43% and 37.7% decreases in isoprostane levels in NY1DD and BERK mice as compared with corresponding untreated groups (P<0.001 and P<0.005, respectively).
Antioxidants
Glutathione (GSH)
As shown in Figure 3, GSH levels in various tissues were significantly reduced in both NY1DD and BERK mice as compared with C57BL controls (P<0.05–0.001). Importantly, in both sickle mouse models, arginine supplementation caused recovery of GSH levels almost to the control baseline values. Interestingly, among untreated mice, GSH levels in untreated BERK mice were ~23%–27% lower in any given tissue than those in the control C57BL mice (P<0.05–0.01), but were ~35%–39% higher as compared with less severe NY1DD mice (P<0.01). The possible significance of this finding is discussed later.
Figure 3.
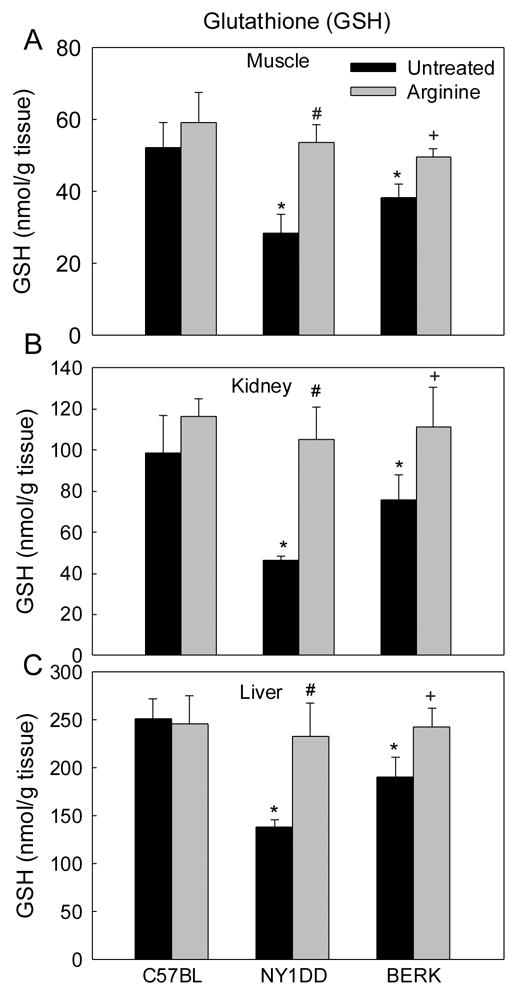
Glutathione (GSH) in C57BL, NY1DD and BERK mice: the effect of arginine supplementation. *P<0.05–<0.001 vs. C57BL controls; #P<0.01–0.001 vs untreated NY1DD mice; +P<0.01–0.001 vs. untreated BERK mice.
Superoxide dismutase (SOD)
As shown in Figure 4, both NY1DD and BERK mice showed significant decreases (~34% to 60%) in total SOD activity in the organs examined (P<0.02–0.001 vs. C57BL). In arginine-treated C57BL mice, SOD activity showed somewhat variable tissue responses: no change was noted in the muscle; a ~36% increase in kidney (P<0.001); and a 38% decrease in the liver (P<0.001). In contrast, arginine significantly augmented SOD activity in both NY1DD and BERK (Figure 4) as compared to the respective untreated controls (muscle- P<0.05 and P<0.001, respectively; kidney- each P<0.01; liver- P<0.01 and P<0.001), with BERK mice showing a greater recovery in the muscle and kidney SOD activity (not different from C57BL baseline values).
Figure 4.
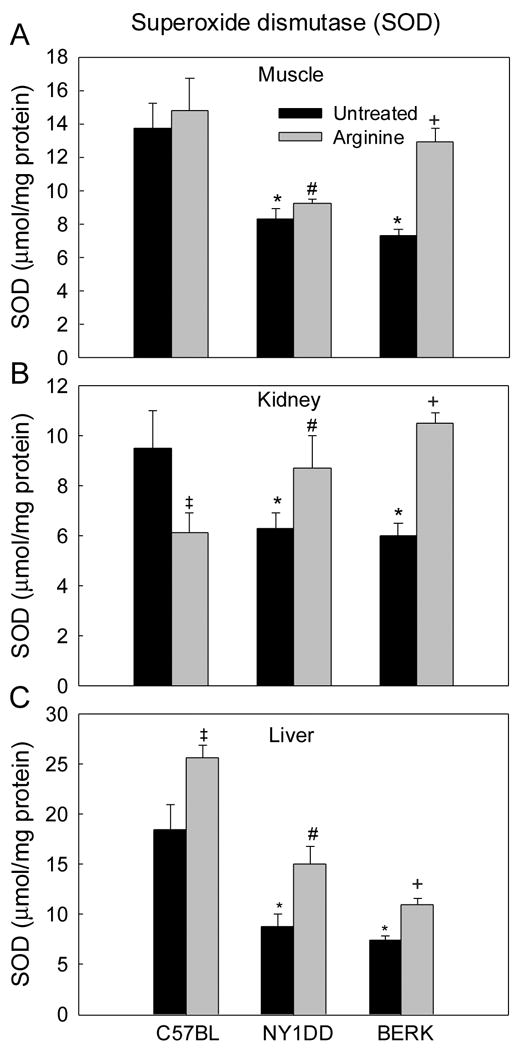
Superoxide dismutase (SOD) activity in C57BL, NY1DD and BERK mice: the effect of arginine supplementation. *P<0.02–0.001 vs. C57BL controls, #P<0.05–0.001 vs. untreated NY1DD mice, +P<0.01–0.001 vs. untreated BERK mice.
Catalase
Untreated NY1DD and BERK mice showed marked decreases in catalase activity (Figure 5). The muscle catalase showed maximal decreases (57.4% in NY1DD and ~50% in BERK mice, each P<0.001 vs. C57BL), while the kidney and liver of both transgenic models showed ~36% to 43% decreases (P<0.02–0.01 vs. C57BL). In C57BL mice, arginine resulted in no significant changes in catalase activity except a 23% increase in the kidney (P<0.01). In contrast, NY1DD mice treated with arginine showed ~98% and 25% increases in catalase activity in the muscle and liver tissues as compared with the untreated NY1DD mice; the resulting activity was not significantly different from the baseline values in untreated C57BL mice. In BERK mice arginine caused marked 52%–86% increases in catalase activity in all the tissues (P<0.003–0.001 vs. untreated BERK) and the values in the kidney and liver were not significantly different from the C57BL values.
Figure 5.
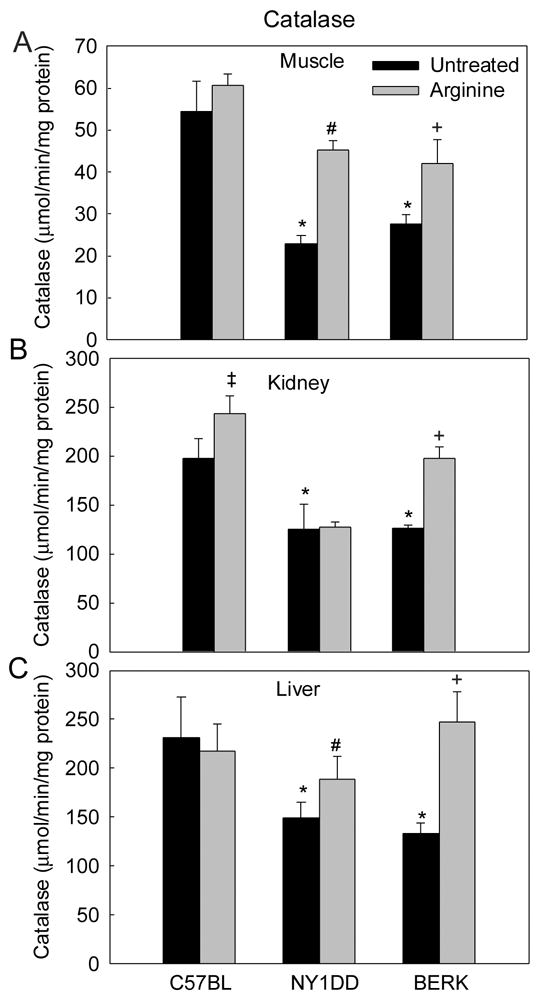
Catalase activity in C57BL, NY1DD and BERK mice: the effect of arginine supplementation. *P<0.01–0.001 vs. C57BL controls, #P<0.034–0.001 vs. untreated NY1DD mice, +P<0.003–0.001 vs. untreated BERK mice.
Glutathione peroxidase (GPx)
In the tissues examined, GPx activity showed significant 38% to 45% decreases in NY1DD (P<0.01–0.001) and even more marked 56% to 69% decreases in BERK mice (P<0.02–0.001) as compared with C57BL controls (Figure 6). Thus, GPx activity was directly proportionate to the severity of SCD mice. In C57BL mice, arginine supplementation had no significant effect on GPx activity in the muscle and kidney, but resulted in a significance increase in the liver (P<0.001). In contrast, arginine supplementation in NY1DD mice increased GPx activity in all the tissues by 29% to 48% (P<0.03–0.01 vs. untreated NY1DD), and resulted even in more marked increase in the BERK tissues, i.e., ~1.5, to ~3.0 fold increases (P<0.02–0.001 vs. untreated BERK) (Figure 6). GPx activity in the kidney and liver of arginine-treated BERK mice was not significantly different from the C57BL values.
Figure 6.
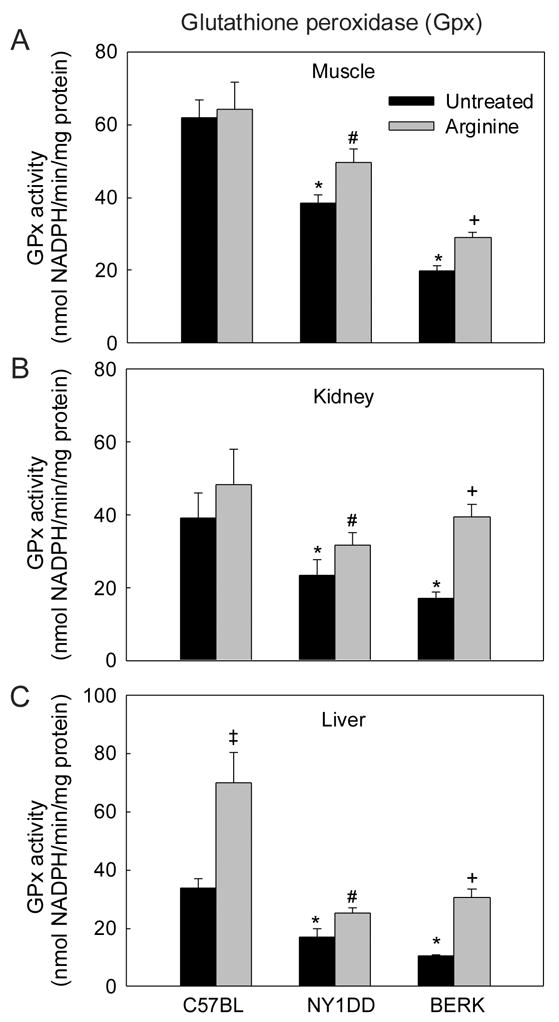
Glutathione peroxidase (GPX) activity in C57BL, NY1DD and BERK mice: the effect of arginine supplementation. *P<0.02–0.001 vs. C57BL controls, #P<0.03–0.01 vs. untreated NY1DD mice, +P<0.02–0.001 vs. untreated BERK mice.
The effect of L-NAME
We investigated the effect of L-NAME, a potent non-selective inhibitor of NO synthase (NOS), (1mg/ml in drinking water for 7 days) in control C57BL and transgenic NY1DD mice. The objective of these experiments was to ascertain if NO-depletion in NY1DD mice with mild pathology would induce an inflammatory phenotype.
Lipid peroxidation (LPO)
TBARS (with desferal and BHT) method was performed in both muscle and liver tissue whereas 8-isoprostane was estimated in the liver. As shown in Table 1, L-NAME pretreatment (1 mg/ml in drinking water for 1 wk) resulted in marked 4- and 2.2-fold increases in the muscle LPO levels (TBARS) in C57BL and NY1DD mice as compared with respective controls (each P<0.01). Also, L-NAME treatment resulted ~2.0- and 2.2-fold increases in the liver of C57BL and NY1DD mice (P<0.01 vs. respective controls).
Table 1.
Lipid peroxidation (LPO) and antioxidants in L-NAME treated control and NY1DD mice
| Tissue | C57BL mice | NY1DD mice | ||
|---|---|---|---|---|
| Untreated | L-NAME | Untreated | L-NAME | |
|
1. Lipid peroxidation
A. Desferal + BHT (nmol/mg protein) i. Muscle ii. Liver |
n=3
0.59 ± 0.01 0.32 ± 0.02 |
n=3
2.4 ± 0.1* 0.63 ± 0.01* |
n=3
1.19 ± 0.02 0.69 ± 0.04 |
n=3
2.61 ±0.1* 1.52 ± 0.28* |
| B. 8-Isoprostane (ng/g tissue), liver | 0.87 ±0.12 | 1.93 ±0.06* | 1.67 ±0.1 | 2.2 ±0.06* |
|
2. GSH (nmol/g tissue)
i. Muscle ii. Kidney iii. Liver |
n=6
52.3 ± 6.9 98.0 ± 18.2 251.3 ± 20.5 |
n=4
51.5 ± 6.1 96.8 ± 3.1 235.4 ± 33.1 |
n=6
28.5 ± 5.3* 46.5 ± 2.1* 138.1 ± 7.7* |
n=4
18.3 ± 1.8+ 28.2 ± 5.1+ 104.4 ± 5.7* |
|
3. SOD (μmol/mg protein)
i. Muscle ii. Kidney iii. Liver |
n=6
13.7 ± 1.4 9.5 ± 1.4 18.5 ± 2.4 |
n = 4
9.6 ± 1.3* 3.6 ± 0.8* 11.6 ± 0.3* |
n = 6
8.3 ± 0.5* 6.3 ± 0.6* 8.8 ± 1.2* |
n= 4
6.7 ± 0.2+ 4.4 ± 0.4+ 6.6 ± 1.1+ |
|
4. Catalase (μmol H2O2/min/ mg protein)
i. Muscle ii. Kidney iii. Liver |
n=6
54.5 ± 7.1 197.7 ± 19.7 231.2 ± 41.3 |
n= 4
27.1 ± 5.3* 118.8 ± 5.8* 124.9 ± 17.6* |
n=6
22.9 ± 2.0* 125.6 ± 24.7* 149.8 ± 15.4* |
n= 4
17.7 ± 3.7+ 69.6 ± 18.0+ 93.6 ± 8.5+ |
|
5. GPx (nmol NADPH/min/ mg protein)
i. Muscle ii. Kidney iii. Liver |
N=6
61.9 ± 5.0 39.2 ± 6.8 33.7 ± 3.4 |
n= 4
28.4 ± 2.7* 21.5 ± 1.1* 17.±1.5* |
n=6
38.4 ± 2.4* 23.3 ± 4.4* 17.0 ± 2.7* |
n= 4
14.7 ± 2.3+ 18.5 ± 2.8+ 9.8 ± 2.4+ |
Values are expressed as mean ± SD
P<0.01 vs untreated C57BL mice;
P<0.01 vs. untreated NY1DD mice.
Similarly, 8-isoprostanes showed significant increases in the liver tissue with L-NAME treatment in C57BL and NY1DD mice. L-NAME resulted in 2.2 and 1.3-fold increases in 8-isoprostanes in C57BL and NY1DD mice, respectively (each P<0.01 vs. respective controls). Notably, in untreated BERK mice, liver isoprostane levels were ~1.7-fold higher (P<0.002) than L-NAME-treated NY1DD mice (see Figure 2B), suggesting that chronic depletion of NO substrate arginine [37] in BERK mice resulted in a greater lipid peroxidation.
Antioxidants
Glutathione (GSH)
In control C57BL mice, L-NAME pretreatment caused only slight insignificant decrease in GSH in any given tissue (Table 1). In contrast, untreated mice NY1DD mice showed a distinct 24% to 39% reduction in GSH in these tissues after L-NAME treatment (P<0.01–0.001).
Superoxide dismutase (SOD)
In C57BL mice, L-NAME treatment resulted in a marked 30% to 62% reduction in SOD activity in the tissues (P<0.01–0.001 vs. untreated C57BL) (Table 1). In contrast, L-NAME treatment in NY1DD mice caused further decrease in SOD activity in all the tissues (19.3 to 30.2%, P<0.027–0.001 vs. untreated NY1DD).
Catalase
Catalase activity in untreated NY1DD mice was decreased 35.6% to 57.4% in all the tissues as compared with C57BL mice (P<0.01) (Table 1). In NY1DD mice, L-NAME treatment caused a further decrease in catalase activity in the organs examined (P<0.05–0.001 vs. untreated NY1DD).
Glutathione peroxidase (GPx)
L-NAME treatment caused significant reduction in GPx activity in both C57BL and NY1DD mice in all the investigated organs (p<0.001 vs. respective untreated controls), with NY1DD mice showing the minimal values (Table 1).
DISCUSSION
The primary event of the SCD is polymerization of deoxygenated sickle hemoglobin that can disrupt blood flow to the affected tissue. Oxidative damage resulting from transient occlusive events can be attributed to oxygen free radicals after restitution of blood flow, (“reperfusion injury”) [13,31,43]. The present studies show that extent of oxidative stress (e.g. lipid peroxidation [LPO] level and activity of certain endogenous antioxidants) in sickle mouse models is reflective of the disease severity. The disease severity in the context of this study refers to the sickling ability of red cells in NY1DD (mild pathology) and BERK mice (severe pathology) which is proportional to the percent of βS-globin synthesis in these models (see Methods). BERK mice, expressing exclusively human α- and βS-globins, show maximal intravascular sickling, hence frequent transient vaso-occlusive events [16,33,39]. Importantly, we show that the replenishment of NO by dietary arginine results in significant decrease in oxidative stress in these transgenic sickle models.
Effect of disease severity
The results show that decreased NO bioavailability, as indicated reduced NO metabolites (NOx) levels, is proportional to the disease severity in sickle mouse models, with BERK mice showing minimal NOx levels. The reduced NOx levels in sickle mice are in agreement with findings in human sickle cell patients and are associated with with arginine deficiency [24,25].
Furthermore, depleted NO metabolites in transgenic (NY1DD) and knockout (BERK) sickle mice are associated with increased oxidative stress. We show that LPO, a marker of peroxidation of membrane lipids by peroxides, is significantly increased in both transgenic NY1DD mice and knockout BERK mice as compared with C57BL mice. Notably, BERK mice showed higher LPO levels compared with NY1DD mice (see Figure 2). This pattern was confirmed whether using TBARS method in the presence of desferal and BHT (Figure 2A), or using using 8-isoprostane determination in the liver tissue (see Figure 2B). In BERK mice as in human SCD, chronic depletion of arginine levels, reduced NO production and NO consumption by the plasma heme as reported before [11,24,26,35] may all contribute to a pro-inflammatory milieu and in greater LPO levels.
Distinct increases in LPO among NY1DD and BERK mice were accompanied by marked decreases in antioxidant enzymes (i.e, total SOD, catalase and GPx) in the tissues compared with control C57BL values. The decreases in antioxidant enzymes in sickle mouse models suggest consumption/inactivation of antioxidant enzymes by excessive reactive oxygen species in these models. This is in conformity with the observations that ischemia-reperfusion injury and increased oxidative stress (e.g., superoxide [O2•−]) and hemolysis (e.g., BERK model) lead to consumption and depletion of endogenous NO and antioxidants [26].
Non-enzymatic GSH also showed significant decreases in the same tissues in both untreated NY1DD and BERK mice, but the decrease was not proportionate with the βS-globin level and the disease severity of these mice. For example, the GSH level was higher in BERK mice as compared with NY1DD mice (P<0.05–0.001) (see Figure 4). GSH is initially depleted by ischemia/reperfusion and by inflammatory cytokines (e.g., TNF-α) but shows a rebound as an adaptive response to oxidative stress [28,34]. The higher GSH activity in BERK mice as compared with less severe NY1DD mice may be a protective response to a greater oxidative stress in this model caused by recurring ischemic-reperfusion events, increased levels of inflammatory cytokines and higher levels of protein nitration [16] that are known to enhance GSH synthesis [34]. GSH effectively scavenges reactive oxygen species directly and indirectly via enzymatic reactions [45]. Furthermore, GPx catalyzes GSH-dependent reduction of H2O2 and other peroxides [20]. Induction of mitochondrial peroxynitrite by sodium nitroprusside, an NO donor, can also induce GSH synthesis [19]. In fact, BERK mice show several-fold increase in nitrotyrosine [16], probably because initial higher production of NO in this model and in human SCD is followed by consumption of NO by oxygen radicals and plasma heme that could lead to increased metabolism and depletion of arginine substrate [25,35].
Protective effect of arginine
Both human SCD and sickle mouse models are characterized by reduced NO bioavailability due to enhanced consumption of NO as noted above. In BERK mice, the destruction of NO and concomitant arginine deficiency results in induction of other vasodilatory mechanisms (e.g., induction of cyclooxygenase-2 and enhanced prostacyclin production) to compensate for NO deficiency and to maintain optimal blood flow in the face of chronic hemolytic anemia in this model [16].
Arginine supplementation resulted in significant increases in plasma NOx levels in C57BL control, NY1DD and BERK mice. In contrast to C57BL mice wherein arginine resulted in a smaller increase in NOx levels (perhaps due to already higher NOx levels), both NY1DD and BERK mice showed marked increases in NOx levels over the untreated control groups. Although arginine treatment in NY1DD mice raised the NOx values almost to the level seen in C57BL mice, the increase in BERK mice was not sufficient to normalize the values to C57BL baseline values (see Figure 1).
While arginine had minimal effect on the liver LPO in C57BL mice, NY1DD mice with moderate oxidative stress exhibited significant decrease in LPO (e.g., isoprostanes) levels in the liver; the resulting values were not significantly different from control C57BL baseline values. Also, arginine resulted in an overall reduction in LPO in BERK mice, though it did not normalize the values. However, the marked overall efficacy of dietary arginine on LPO reduction in BERK mice is a likely due to the replenishment of endogenous arginine levels, as noted before.
Antioxidative effect of arginine supplementation was further evident by a marked recovery of GSH in both NY1DD and BERK mice almost to the C57BL baseline levels in any given tissue. Also, following arginine supplementation, antioxidant enzymes showed a marked increase: SOD activity showed almost complete normalization in the kidney and liver of NY1DD mice and in the muscle and kidney of BERK mice, accompanied by marked increase in catalase activity in all the tissues examined except for the kidney in NY1DD mice; GPx which along with catalase scavenges H2O2 showed significant increases in all the tissues in both NY1DD and BERK mice.
These results strongly suggest that increased NO production after arginine treatment exerts anti-inflammatory effect not only by reducing LPO levels, but also by enhancing protective antioxidant mechanisms in both transgenic NY1DD and knockout BERK models of SCD. Although NY1DD mice do not exhibit hemolysis, BERK mice show more than 2-fold increase in cell-free plasma heme compared with controls [16] that may result in consumption/inactivation of NO by ferrous hemoglobin as described [35]. In addition, transient vaso-occlusive events in BERK mice, caused by intravascular sickling, will result in reperfusion injury and oxidant generation contributing to NO consumption by O2•− [3,16]. Thus, arginine supplementation is likely to enhance NO production and reduce oxidative stress in models of hemolysis and reperfusion injury [4,6,24].
Inflammatory effect of L-NAME
Since L-NAME competes with endogenous arginine to reduce NO production, the marked increase in oxidative stress in both C57BL and NY1DD mice following L-NAME treatment is in accord with the inflammatory effects of reduced NO availability that characterizes SCD [16,35]. This was evident by not only increased LPO levels, but also a marked decrease in various antioxidants, i.e., GSH, SOD, catalase and GPx (see Table 1). We did not treat BERK mice with L-NAME as our previous studies have shown that L-NAME has no effect on blood pressure or vessel (arteriolar) diameters in these mice [16], suggesting drastically depleted endogenous arginine levels. Using transgenic NY1DD mice, these results suggest that reduced NO bioavailability would play a major role in the inflammatory state in SCD.
In conclusion, the present studies demonstrate that reperfusion injury in transgenic (NY1DD) and knockout (BERK) mice caused by intravascular sickling and vaso-occlusive events results in increased LPO and decreased antioxidants as compared with C57BL mice. Furthermore, there is a distinct relationship between disease severity and LPO levels, with pathologically more severe BERK mice showing maximal LPO. While antioxidant enzymes showed decreases in both models, GSH decrease in BERK mice was not as pronounced as in NY1DD mice, suggesting in severe BERK mice, recurring ischemia/reperfusion events, inflammatory cytokines and protein nitration might induce GSH synthesis as a protective response. Importantly, arginine supplementation in both the mouse models reduced oxidative stress and enhanced antioxidant activity, suggesting that increased NO bioavailability after arginine treatment (as indicated by increased NOx levels) had anti-inflammatory effect on the organ oxidative stress. In contrast, a decrease in NO bioavailability after L-NAME treatment resulted in a marked increase in oxidative stress in transgenic NY1DD mice. These results demonstrate that replenishment of NO by arginine supplementation significantly decreases oxidative stress in SCD mice, suggesting a potential therapeutic use of arginine in the management of this disease.
Acknowledgments
This work was supported by National Heart, Lung and Blood Institute grants, PO1 HL-55552 and RO1 HL-070047.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aebi H. Catalase in vitro. Methods Enzymol. 1984;105:121–126. doi: 10.1016/s0076-6879(84)05016-3. [DOI] [PubMed] [Google Scholar]
- 2.Ahluwalia A, Foster P, Scotland RS, McLean PG, Mathur A, Perretti M, Moncada S, Hobbs AJ. Antiinflammatory activity of soluble guanylate cyclase: cGMP-dependent down-regulation of P-selectin expression and leukocyte recruitment. Proc Natl Acad Sci U S A. 2004;101:1386–1391. doi: 10.1073/pnas.0304264101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aslan M, Ryan TM, Adler B, Townes TM, Parks DA, Thompson JA, Tousson A, Gladwin MT, Patel RP, Tarpey MM, Batinic-Haberle I, White CR, Freeman BA. Oxygen radical inhibition of nitric oxide-dependent vascular function in sickle cell disease. Proc Natl Acad Sci U S A. 2001;98:15215–15220. doi: 10.1073/pnas.221292098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Au A, Louch WE, Ferrier GR, Howlett SE. L-Arginine ameliorates effects of ischemia and reperfusion in isolated cardiac myocytes. Eur J Pharmacol. 2003;476:45–54. doi: 10.1016/s0014-2999(03)02175-7. [DOI] [PubMed] [Google Scholar]
- 5.Braughler JM, Duncan LA, Chase RL. The involvement of iron in lipid peroxidation. Importance of ferric to ferrous ratios in initiation. J Biol Chem. 1986;261:10282–10289. [PubMed] [Google Scholar]
- 6.Cao B, Li N, Wang Y, Li JS. Protective effect of L-arginine preconditioning on ischemia and reperfusion injury associated with rat small bowel transplantation. World J Gastroenterol. 2005;11:2994–2997. doi: 10.3748/wjg.v11.i19.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fabry ME, Costantini F, Pachnis A, Suzuka SM, Bank N, Aynedjian HS, Factor SM, Nagel RL. High expression of human beta S- and alpha-globins in transgenic mice: erythrocyte abnormalities, organ damage, and the effect of hypoxia. Proc Natl Acad Sci U S A. 1992;89:12155–12159. doi: 10.1073/pnas.89.24.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fabry ME, Nagel RL, Pachnis A, Suzuka SM, Costantini F. High expression of human beta S- and alpha-globins in transgenic mice: hemoglobin composition and hematological consequences. Proc Natl Acad Sci U S A. 1992;89:12150–12154. doi: 10.1073/pnas.89.24.12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fabry ME, Suzuka SM, Weinberg RS, Lawrence C, Factor SM, Gilman JG, Costantini F, Nagel RL. Second generation knockout sickle mice: the effect of HbF. Blood. 2001;97:410–418. doi: 10.1182/blood.v97.2.410. [DOI] [PubMed] [Google Scholar]
- 10.Garcia YJ, Rodriguez-Malaver AJ, Penaloza N. Lipid peroxidation measurement by thiobarbituric acid assay in rat cerebellar slices. J Neurosci Methods. 2005;144:127–135. doi: 10.1016/j.jneumeth.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 11.Gladwin MT, Lancaster JR, Jr, Freeman BA, Schechter AN. Nitric oxide's reactions with hemoglobin: a view through the SNO-storm. Nat Med. 2003;9:496–500. doi: 10.1038/nm0503-496. [DOI] [PubMed] [Google Scholar]
- 12.Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 13.Hebbel RP, Osarogiagbon R, Kaul D. The endothelial biology of sickle cell disease: inflammation and a chronic vasculopathy. Microcirculation. 2004;11:129–151. [PubMed] [Google Scholar]
- 14.Hoffman A, Goldstein S, Samuni A, Borman JB, Schwalb H. Effect of nitric oxide and nitroxide SOD-mimic on the recovery of isolated rat heart following ischemia and reperfusion. Biochem Pharmacol. 2003;66:1279–1286. doi: 10.1016/s0006-2952(03)00441-6. [DOI] [PubMed] [Google Scholar]
- 15.Kaul DK, Hebbel RP. Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice [see comments] J Clin Invest. 2000;106:411–420. doi: 10.1172/JCI9225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaul DK, Liu XD, Chang HY, Nagel RL, Fabry ME. Effect of fetal hemoglobin on microvascular regulation in sickle transgenic-knockout mice. J Clin Invest. 2004;114:1136–1145. doi: 10.1172/JCI21633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kubes P, Suzuki M, Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci U S A. 1991;88:4651–4655. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurose I, Wolf R, Grisham MB, Aw TY, Specian RD, Granger DN. Microvascular responses to inhibition of nitric oxide production. Role of active oxidants. Circ Res. 1995;76:30–39. doi: 10.1161/01.res.76.1.30. [DOI] [PubMed] [Google Scholar]
- 19.Kurozumi R, Takahashi M, Kojima S. Involvement of mitochondrial peroxynitrite in nitric oxide-induced glutathione synthesis. Biol Pharm Bull. 2005;28:779–785. doi: 10.1248/bpb.28.779. [DOI] [PubMed] [Google Scholar]
- 20.Lei XG. In vivo antioxidant role of glutathione peroxidase: evidence from knockout mice. Methods Enzymol. 2002;347:213–225. doi: 10.1016/s0076-6879(02)47021-8. [DOI] [PubMed] [Google Scholar]
- 21.Marklund S, Marklund G. Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur J Biochem. 1974;47:469–474. doi: 10.1111/j.1432-1033.1974.tb03714.x. [DOI] [PubMed] [Google Scholar]
- 22.Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. [Review] [404 refs] Pharmacological Reviews. 1991;43:109–142. [PubMed] [Google Scholar]
- 23.Moron MS, DePierre JW, Mannervik B. Levels of glutathione, glutathione reductase and glutathione S-transferase activities in rat lung and liver. Biochim Biophys Acta. 1979;582:67–78. doi: 10.1016/0304-4165(79)90289-7. [DOI] [PubMed] [Google Scholar]
- 24.Morris CR, Kuypers FA, Larkin S, Sweeters N, Simon J, Vichinsky EP, Styles LA. Arginine therapy: a novel strategy to induce nitric oxide production in sickle cell disease. Br J Haematol. 2000;111:498–500. doi: 10.1046/j.1365-2141.2000.02403.x. [DOI] [PubMed] [Google Scholar]
- 25.Morris CR, Morris SM, Jr, Hagar W, van Warmerdam J, Claster S, Kepka-Lenhart D, Machado L, Kuypers FA, Vichinsky EP. Arginine therapy: a new treatment for pulmonary hypertension in sickle cell disease? Am J Respir Crit Care Med. 2003;168:63–69. doi: 10.1164/rccm.200208-967OC. [DOI] [PubMed] [Google Scholar]
- 26.Nanobashvili J, Neumayer C, Fuegl A, Punz A, Blumer R, Mittlbock M, Prager M, Polterauer P, Dobrucki LW, Huk I, Malinski T. Combined L-arginine and antioxidative vitamin treatment mollifies ischemia-reperfusion injury of skeletal muscle. J Vasc Surg. 2004;39:868–877. doi: 10.1016/j.jvs.2003.10.060. [DOI] [PubMed] [Google Scholar]
- 27.Nath KA, Katusic ZS, Gladwin MT. The perfusion paradox and vascular instability in sickle cell disease. Microcirculation. 2004;11:179–193. doi: 10.1080/10739680490278592. [DOI] [PubMed] [Google Scholar]
- 28.Nita DA, Nita V, Spulber S, Moldovan M, Popa DP, Zagrean AM, Zagrean L. Oxidative damage following cerebral ischemia depends on reperfusion - a biochemical study in rat. J Cell Mol Med. 2001;5:163–170. doi: 10.1111/j.1582-4934.2001.tb00149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ogawa T, Nussler AK, Tuzuner E, Neuhaus P, Kaminishi M, Mimura Y, Beger HG. Contribution of nitric oxide to the protective effects of ischemic preconditioning in ischemia-reperfused rat kidneys. J Lab Clin Med. 2001;138:50–58. doi: 10.1067/mlc.2001.115648. [DOI] [PubMed] [Google Scholar]
- 30.Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;95:351–358. doi: 10.1016/0003-2697(79)90738-3. [DOI] [PubMed] [Google Scholar]
- 31.Osarogiagbon UR, Choong S, Belcher JD, Vercellotti GM, Paller MS, Hebbel RP. Reperfusion injury pathophysiology in sickle transgenic mice. Blood. 2000;96:314–320. [PubMed] [Google Scholar]
- 32.Paglia DE, Valentine WN. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J Lab Clin Med. 1967;70:158–169. [PubMed] [Google Scholar]
- 33.Paszty C, Brion CM, Manci E, Witkowska HE, Stevens ME, Mohandas N, Rubin EM. Transgenic knockout mice with exclusively human sickle hemoglobin and sickle cell disease [see comments] Science. 1997;278:876–878. doi: 10.1126/science.278.5339.876. [DOI] [PubMed] [Google Scholar]
- 34.Rahman I. Regulation of glutathione in inflammation and chronic lung diseases. Mutat Res. 2005 doi: 10.1016/j.mrfmmm.2005.02.025. [DOI] [PubMed] [Google Scholar]
- 35.Reiter CD, Wang X, Tanus-Santos JE, Hogg N, Cannon RO, III, Schechter AN, Gladwin MT. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002;8:1383–1389. doi: 10.1038/nm1202-799. [DOI] [PubMed] [Google Scholar]
- 36.Romero JR, Suzuka SM, Nagel RL, Fabry ME. Arginine supplementation of sickle transgenic mice reduces red cell density and Gardos channel activity. Blood. 2002;99:1103–1108. doi: 10.1182/blood.v99.4.1103. [DOI] [PubMed] [Google Scholar]
- 37.Ryan TM, Ciavatta DJ, Townes TM. Knockout-transgenic mouse model of sickle cell disease. Science. 1997;278:873–876. doi: 10.1126/science.278.5339.873. [DOI] [PubMed] [Google Scholar]
- 38.Thomas DD, Espey MG, Vitek MP, Miranda KM, Wink DA. Protein nitration is mediated by heme and free metals through Fenton-type chemistry: an alternative to the NO/O2- reaction. Proc Natl Acad Sci U S A. 2002;99:12691–12696. doi: 10.1073/pnas.202312699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Turhan A, Weiss LA, Mohandas N, Coller BS, Frenette PS. Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. Proc Natl Acad Sci U S A. 2002;99:3047–3051. doi: 10.1073/pnas.052522799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS, Karoui H, Tordo P, Pritchard KA., Jr Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci U S A. 1998;95:9220–9225. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.White CR, Darley-Usmar V, Berrington WR, McAdams M, Gore JZ, Thompson JA, Parks DA, Tarpey MM, Freeman BA. Circulating plasma xanthine oxidase contributes to vascular dysfunction in hypercholesterolemic rabbits. Proc Natl Acad Sci U S A. 1996;93:8745–8749. doi: 10.1073/pnas.93.16.8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.White CR, Parks DA, Patel RP, Shelton J, Tarpey MM, Freeman BA, Darley-Usmar VM. L-Arginine inhibits xanthine oxidase-dependent endothelial dysfunction in hypercholesterolemia. FEBS Lett. 2004;561:94–98. doi: 10.1016/S0014-5793(04)00137-1. [DOI] [PubMed] [Google Scholar]
- 43.Wood KC, Hebbel RP, Granger DN. Endothelial cell P-selectin mediates a proinflammatory and prothrombogenic phenotype in cerebral venules of sickle cell transgenic mice. Am J Physiol Heart Circ Physiol. 2004;286:H1608–H1614. doi: 10.1152/ajpheart.01056.2003. [DOI] [PubMed] [Google Scholar]
- 44.Wood KC, Hebbel RP, Granger DN. Endothelial cell NADPH oxidase mediates the cerebral microvascular dysfunction in sickle cell transgenic mice. FASEB J. 2005;19:989–991. doi: 10.1096/fj.04-3218fje. [DOI] [PubMed] [Google Scholar]
- 45.Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J Nutr. 2004;134:489–492. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]