

Abstract
Ribonucleotide reductase is a highly regulated cell cycle-controlled activity that is essential for DNA synthesis and repair. A retroviral vector for the R2 component of mammalian ribonucleotide reductase, the rate-limiting protein for enzyme activity and DNA synthesis in proliferating cells, was constructed and introduced into mammalian cells. Expression of Myc epitope-tagged R2 protein in benign BALB/c 3T3 and NIH 3T3 cells leads to a greatly increased frequency of focus formation in cooperation with H-ras transformation. Four lines of H-ras-transformed mouse 10T½ fibroblasts showed increased growth efficiency in soft agar after infection with the recombinant R2 expression virus vector. Furthermore, cells with altered R2 expression also exhibited significantly reduced subcutaneous tumor latency and increased tumor growth rates in syngeneic mice, and showed markedly elevated metastatic potential in lung metastasis assays. The results indicate that altered R2 gene expression cooperates with ras in mechanisms of malignant progression. A major Ras pathway involves the Raf-1 protein, which is recruited to the plasma membrane for activation. We show that recombinant R2 expression leads to significant increases in membrane-associated Raf-1 protein and mitogen-activating protein kinase-2 activity suggesting a mechanism for the observed Ras/R2 synergism. In support of this finding, we observed that activated Rac-1, which operates parallel to Raf-1 and cooperates with Raf-1 in Ras activated pathways, also cooperates with R2 in cellular transformation. These studies demonstrate that the R2 protein can participate in other critical cellular functions in addition to ribonucleotide reduction, and that deregulated R2 is a novel tumor progressor determinant that cooperates in oncogene-mediated mechanisms, which control malignant potential.
The first unique step leading to DNA synthesis is the conversion of ribonucleotides to their corresponding deoxyribonucleotides, a reaction that is catalyzed in a cell cycle-specific manner by ribonucleotide reductase (1, 2, 3). The enzyme is composed of two dissimilar components often called R1 and R2, which are differentially regulated during the cell cycle. Although the levels of the R1 protein do not appear to change substantially during the cell cycle, there is an S-phase correlated increase in the R2 protein resulting from its de novo synthesis (1, 4). Interestingly, the activity of ribonucleotide reductase, and therefore DNA synthesis and cell proliferation, is controlled during the cell cycle by the synthesis and degradation of the R2 component (5). The rate-limiting R2 component is a phosphoprotein capable of being phosphorylated by the CDC2 and CDK2 protein kinase mediators of cell cycle progression (6), and contains non-heme iron that stabilizes a unique tyrosyl-free radical required for enzyme activity (1, 2, 7). Chemotherapeutic compounds like hydroxyurea inhibit ribonucleotide reductase activity by destabilizing the iron center of the R2 protein causing the destruction of the tyrosyl-free radical (7), and preventing cells from progressing through S-phase of the cell cycle (8). In addition to cell cycle control, ribonucleotide reductase can be regulated by an S-phase independent mechanism that is important for DNA repair (9). Previous work has also shown that regulation of ribonucleotide reductase, and in particular the R2 component, is markedly altered in malignant cells exposed to tumor promoters or to the growth factor TGF-β (transforming growth factor β) (10, 11, 12, 13, 14). Interesting alterations in ribonucleotide reductase activity and in the levels of the R2 component have also been described in tumor cells obtained from rodent and human tissues (15, 16, 17, 18) and in cultured cells selected for resistance to anti-tumor agents such as hydroxyurea (1, 16). These latter observations, which are mainly correlative in nature, suggest that ribonucleotide reductase, and particularly the rate-limiting R2 component, may be critically involved in mechanisms controlling malignant progression. To directly test this hypothesis, we have constructed a retroviral expression vector for the R2 component, and have used it to investigate in vitro and in vivo malignancy-related properties of vector-infected cells.
MATERIALS AND METHODS
Expression Vectors.
The retroviral expression vector for the human Myc epitope-tagged mouse R2 component, SH/mR2, was constructed and packaged as recently described (19). The infectivity of the viral stock was ≥1 × 104 colony-forming units/ml. Plasmid pH06Ti, which expresses T-24 H-ras and a selective marker neo, has been used for malignant transformation (20, 21, 22). The activated Rac-1 plasmid (V12 Rac-1) was kindly provided by M. Symons (23).
Cells and Cell Culture.
The mouse cell lines, BALB/c 3T3, NIH 3T3, and four lines of T24 H-ras-transformed 10T½ cells named C1, NR4, r-2, and r-3 have been used as recipients of the R2 retroviral vector (19). Cells were routinely cultured in α-minimal essential medium (α-MEM) (GIBCO) supplemented with 10% calf serum (Fetalclone III, HyClone). Infection of cells with SH/mR2 or control virus LXSH in the presence of polybrene was carried out (24), and stable infectants (≥1 × 104 clones) were obtained with hygromycin selection and pooled (19, 24). Determinations of cell division times, plating efficiencies, and relative sensitivities to hydroxyurea cytotoxicity by estimating relative colony forming efficiencies, were carried out as described (1, 20, 25). Growth in soft agar was estimated in 10-cm tissue culture plates containing 15 ml of base agar (0.5% Bacto agar in α-MEM plus 10% calf serum) and 10 ml of growth agar (0.33% agar in α-MEM containing 10% calf serum). Cells were obtained from subconfluent cultures, and colonies were scored 10–15 days later (20, 21, 25). Transformation was also analyzed by determining focus formation after cells were infected with SH/mR2 or LXSH or transfected with T-24 Ras or V12 Rac-1 plasmids by calcium phosphate precipitation (22). At 40 hr after infection or transfection, cells were split into three 10-cm tissue culture plates, which were provided daily with 20 ml of fresh complete medium (α-MEM plus 10% calf serum) for 10–14 days, stained with methylene blue, after which the foci were scored (22). The transfection frequency in all the experiments were routinely determined by cotransfection of a mammalian expression plasmid for β-galactosidase from Esherichia coli, with the T-24 Ras or V-12 Rac-1 plasmids, followed by treatment of cells with the 5-bromo-4-chloro-3-indolyl β-d-galactoside, and counting the number of blue cells (26). In some cases, T-24 Ras plasmid transfected plates were selected with geneticin, and drug-resistant colonies were scored 14 days after staining with methylene blue.
Assays for Tumorigenicity and Metastasis.
Immunocompetent C3H/HeN syngeneic mice (Charles River Breeding Laboratories) were used in these assays as described earlier (20, 21, 22). Tumor latency was determined by injecting cells subcutaneously and recording the time required to form a tumor (2 × 2 mm) detectable by palpation. Tumor size was determined by multiplying the dimensions of the cross-section of the tumor. For experimental metastasis assays cells were injected into the tail veins of 6–8 week-old C3H/HeN syngeneic mice and an estimate of the number of lung tumors was made 21 days later.
Protein R2 Analysis.
The procedures for Western blot analysis have been described previously, for example, using either the anti-Myc mouse monoclonal 9E10 antibody (American Type Culture Collection) (19) or the anti-R2 rabbit polyclonal antibody (6, 27). To determine recombinant R2 protein expression during the cell cycle, flow cytometry analysis was performed following 9E10/fluorescein isothiocyanate-antibody labeling as described (28, 29).
Determination of Membrane-Associated Raf-1 Protein.
The membrane fraction was prepared as described by Qiu et al. (30) and used for Western blot analysis with a polyclonal antibody specific for Raf-1 protein (Santa Cruz Biotechnology), after the protein content was determined by the standard Bio-Rad assay. Densitometry analysis of the Raf-1 band was performed, and the amount of Raf-1 protein from each sample was corrected by densitometry analysis of a well-separated band on a parallel gel stained with Coomassie blue.
Ribonucleotide Reductase Assay.
Enzyme preparations were obtained from logarithmically growing cells lysed in phosphate-buffered saline (pH 7.2), containing 1 mM dithiothreitol and 1 mM protease inhibitor, AEBSF (Calbiochem), by three cycles of freeze-thawing. Following centrifugation, the supernatant was used for enzyme activity assays with [14C]CDP (Moravek Biomedicals, Brea, CA), as detailed elsewhere (1, 9, 19, 27). In some experiments, enzyme assays were performed by combining purified recombinant R1 protein (31) with 9E10 antibody-precipitated R2 protein (9). In this case, 20 μg of the 9E10 antibody and 50 μl of staphylococcal protein A-agarose (Sigma) was added to 1 ml of the supernatant of centrifuged lysed cells, and placed on a rocker at 4°C for 2 hr. The staphylococcal protein A-agarose immunocomplex was washed three times with 1 ml of cold phosphate buffer containing 1 mg/ml bovine serum albumin, and then assayed for ribonucleotide reductase activity as described (1, 9, 19, 27).
Assay of MAPK Activity.
Cultures with ≥90% confluency were stressed in serum-free medium (23, 32) and extracted as described (33). Mitogen-activating protein kinase (MAPK)-2 protein was immunoprecipitated by agarose beads conjugated with non-neutralizing antibody specific for the protein (Santa Cruz Biotechnology), and the kinase activity of the immunocomplex was assayed by measuring its ability to phosphorylate myelin basic protein using a MAPK assay kit from Upstate Biotechnology (Lake Placid, NY).
RESULTS AND DISCUSSION
Expression of Biologically Active R2 Protein.
To determine the malignant potential of deregulated expression of the rate-limiting R2 component of ribonucleotide reductase, we investigated the properties of cells stably infected with a retroviral expression vector (SH/mR2) carrying the R2 component, which we have recently constructed (19). The use of this expression vector allowed us to achieve high infection efficiency and stable expression of the R2 protein after selecting the infectants with hygromycin (19). To distinguish the vector gene product from the endogenous R2, we added a human c-Myc epitope coding for 10 amino acids plus methionine to the 5′ end of the R2 cDNA. Fig. 1a shows that Western blots with the 9E10 antibody, which specifically recognizes the Myc-epitope sequence, detects the 45-kDa R2 protein in SH/mR2 stably infected BALB/c 3T3 and NIH 3T3 cells (named B3/mR2 and N3/mR2, respectively), but not in control vector (LXSH)-infected B3/SH or N3/SH cells, which were derived the same way as B3/mR2 and N3/mR2 except in B3/SH and N3/SH cells, where the vector expresses only the selective marker hyg. R2 specific antibodies detected the endogenous as well as the recombinant R2 protein in expression vector-infected cells, and as expected only the endogenous protein was observed in control vector-infected cells (Fig. 1b). Flow cytometry analysis following 9E10/fluorescein isothiocyanate antibody labeling demonstrated that the recombinant R2 protein was constitutively expressed throughout the cell cycle (Fig. 1c). Indirect fluorescence microscopic analysis using the 9E10 antibody indicated that essentially every cell in the B3/mR2 and N3/mR2 populations expressed the Myc-tagged R2 protein (data not shown). Several experiments were performed to show that the vector-expressed R2 is biologically active. First, we observed that B3/mR2 and N3/mR2 cells were resistant in colony-forming experiments to the cytotoxic effects of hydroxyurea, an inhibitor of the R2 protein (3, 16), when compared with B3/SH and N3/SH cells (ref. 19 and data not shown). Second, we assayed ribonucleotide reductase activity (9, 19), and found that the CDP reductase activities in B3/mR2 and N3/mR2 cells in three independent experiments were 1.96 ± 0.32 and 1.71 ± 0.11 nmols/mg protein/hr, respectively, which was 2.6 and 2.1 times higher than observed with B3/SH and N3/SH cells (0.74 ± 0.14 and 0.83 ± 0.08 nmols/mg/hr, respectively). Finally, enzyme assays were carried out by combining purified recombinant R1 protein (31) with 9E10 antibody precipitated R2 protein. Significant levels of activity (15–20 nmols/mg/hr) were detected when B3/mR2 and N3/mR2 cells were used as a source for Myc-tagged R2, and as expected no activity was found when B3/SH or N3/SH cells were used.
Figure 1.
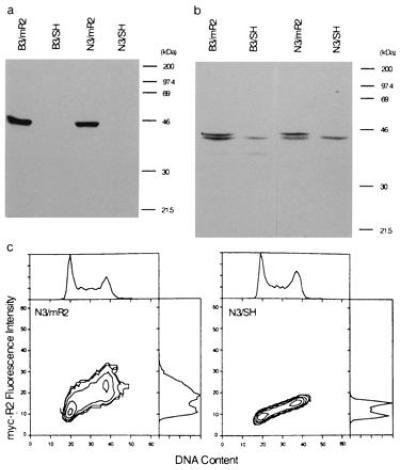
Analysis of Myc-tagged R2 expression from stable infectants by Western blot analysis using monoclonal anti-Myc epitope antibody 9E10 (a), polyclonal rabbit anti-R2 serum (b), and during the cell cycle by flow cytometry, using antibody 9E10 (c).
Ras Transformation Potential Determined by Aberrant R2 Gene Expression.
The above results indicate that we succeeded in obtaining cells altered in the regulation of biologically active R2 protein. Therefore, we determined whether or not altered R2 expression further transformed cells like BALB/c 3T3 or NIH 3T3. We observed that similar to control B3/SH and N3/SH cells, as well as the parental non-infected lines, B3/mR2 and N3/mR2 cultures remained in a flat, non-transformed morphology on tissue culture plates and exhibited contact and density inhibited growth (data not shown). No transformed foci were observed with BALB/c 3T3 or NIH 3T3 cells after infection with the retroviral SH/mR2 vector (Fig. 2A, a and b). The results suggest that deregulation of R2 gene expression does not on its own transform BALB/c 3T3 or NIH 3T3 fibroblasts. However, previous studies have shown alterations in the regulation of R2 gene expression in H-ras-transformed cells (13, 14, 34), suggesting that R2 deregulation may be important in H-ras-mediated malignancies. To test this hypothesis, we transfected an expression plasmid containing T24 H-ras into established recombinant R2-expressing cell populations derived from BALB/c 3T3 or NIH 3T3. Interestingly, we observed a consistent and significant increase (3.4-fold) in the number of foci formed with H-ras-transfected N3/mR2 cells when compared with N3/SH control cells (Fig. 2 B, c and d and C), and an even more marked increase of about 70-fold was observed when H-ras transfected B3/mR2 cells were compared with B3/SH cells (Fig. 2 B, a and b and C). This occurred even though the transfection efficiency with N3/mR2 and B3/mR2 cells as determined by scoring G418 selected colonies, and/or counting blue cells following cotransfection of H-ras with an expression plasmid for E. coli β-galactosidase (26), were actually lower (by about 50%) than with N3/SH and B3/SH cells.
Figure 2.
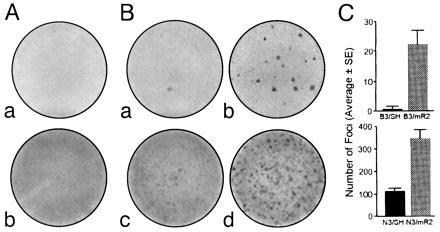
(A) Infection of BALB/c 3T3 (a) and NIH 3T3 (b) cells with SH/mR2 did not lead to focus formation. (B) There was an increase in focus formation with B3/mR2 (b) and N3/mR2 (d) compared with B3/SH (a) and N3/SH (c) after transfection with the T24 H-ras plasmid. (C) The number of foci formed in three independent ras transfection experiments was plotted.
Ras Malignancy Potential Determined by Aberrant R2 Gene Expression.
Because combinations of altered R2 gene expression and activated H-ras were synergistic in focus-forming experiments in which ras was transfected into altered R2-expressing cells, we tested this gene combination further by infecting four independent H-ras-transformed 10T½ cell lines, C1, NR4, r-2, and r-3, which we previously characterized (20, 21, 22, 23), with the retroviral vector SH/mR2. Stable infectants were selected with hygromycin, and Western blot analyses and enzyme activity assays confirmed that these infectants expressed biologically active Myc-tagged R2 protein (data not shown). Soft agar growth experiments revealed that H-ras-transformed cells containing the recombinant R2 sequence were much more efficient at producing colonies in semi-solid growth agar than the uninfected parental populations (e.g., r-3) or control vector infected cells (C1, NR4, r-2) (Table 1). In addition, many of the colonies formed by cells infected with recombinant R2 were larger in size (Fig. 3A). Because each pair of recombinant R2-expressing and control cell populations have almost identical growth rates (12.9 hr for C1/SH and 12.2 hr for C1/mR2, 13.5 hr for r-2/SH and 13.9 hr for r-2/mR2, 11.6 hr for r-3 and 11.9 hr for r-3/mR2, 14.1 hr for NR4/SH and 14.3 hr for NR4/mR2), plating efficiencies (58% for C1/SH and 55% for C1/mR2, 59% for r-2/SH and 63% for r-2/mR2, 91% for r-3 and 88% for r-3/mR2, 73% for NR4/SH and 75% for NR4/mR2), and cell cycle-phase distributions (data not shown) when grown on solid surfaces, the alterations observed in soft agar and in focus-forming experiments suggest that a combination of deregulated R2 expression and activated H-ras may lead to greater malignant potential in vivo. Therefore, we compared the tumorigenic and metastatic potential of C1/mR2 and C1/SH cells in syngeneic C3H/HeN mice. Marked differences in malignant potential were observed. C1/mR2 cells exhibited shorter tumor latency and greater tumor growth when compared with C1/SH cells (Fig. 3B). Furthermore, metastasis assays clearly indicated that C1/mR2 cells were more malignant than C1/SH cells and produced significantly more lung tumors (Fig. 3C).
Table 1.
Increased colony formation in soft agar by ras-transformed cells containing the recombinant R2 vector
| Cell line | No. of soft
agar colonies with varying cell inoculum
|
||
|---|---|---|---|
| 103 | 104 | 105 | |
| C1/SH | 0 | 4 ± 3 | 66 ± 9 |
| C1/mR2 | 3 ± 3 | 28 ± 7 | 347 ± 45 |
| r-2/SH | ND | 9 ± 2 | 105 ± 7 |
| r-2/mR2 | ND | 24 ± 1 | 298 ± 11 |
| NR4/SH | 0 | 3 ± 1 | 32 ± 4 |
| NR4/mR2 | 2 ± 1 | 14 ± 2 | 127 ± 10 |
| r-3 | 7 ± 1 | 100 ± 11 | ND |
| r-3/mR2 | 31 ± 4 | 309 ± 17 | ND |
The number of colonies presented (means ± SE) were the results obtained in three independent experiments, except those obtained for r-2/SH and r-2/mR2 cells, which were the results from single experiments with triplicate dishes. ND, not determined.
Figure 3.
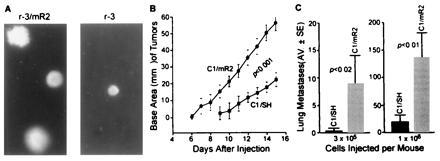
(A) Expression of Myc-R2 in ras-transformed cells resulted in an increased growth efficiency in soft agar. Examples shown are r-3/mR2 and uninfected r-3 cells. (See Table 1 for further information.) (B) C1/mR2 cells showed reduced tumor latency and increased growth rate when compared with C1/SH control cells. Three ×105 cells from logarithmically growing cultures were collected and subcutaneously injected into five syngeneic C3H/HeN mice per cell line per experiment. Results presented were from two independent experiments. The P value of t test analysis of tumor growth rates is shown, and indicates that the growth rates for the two cell lines are significantly different. (C) C1/mR2 cells exhibited elevated metastatic potential.
R2 Gene Expression and Oncogene Cooperativity.
The above results indicate that altered R2 expression can cooperate with activated H-ras in in vitro transformation and in in vivo malignancy assays. Because no obvious differences in growth rates or cell cycle-phase distributions were found that may account for this cooperation, by for example changes in cell cycle regulation, we wondered if deregulated R2 expression might synergize with ras by elevating the activity of a Ras signal pathway. This would be consistent with studies showing a direct correlation between ras expression and malignant potential (20, 21, 34, 35). A major Ras pathway for regulating gene expression involves the Raf-1 protein kinase. Activated Ras recruits Raf to the plasma membrane where Raf and downstream signaling molecules like MAPKs become activated (23, 32, 36). Using a Raf-1 specific antibody, we compared the levels of membrane-associated Raf-1 in six BALB/c 3T3-, NIH 3T3-, and 10T½-derived cell lines containing deregulated R2 expression, with control cells containing only endogenous R2 protein (Fig. 4A). Interestingly, in all six cases, cell lines containing deregulated R2 showed increased membrane-associated Raf-1, with an average increase of about 30%, which was highly significant (P < 0.001). In agreement with the above observation, we also found that cell lines with deregulated R2 expression exhibited a consistent and significant increase of about 70% (P < 0.001) in MAPK-2 activity (Fig. 4B). Oncogenic Ras also activates the Rac pathway, which is parallel to the Raf pathway, and therefore constitutively active Rac-1 cooperates with membrane-targeted Raf-1 in malignant transformation (30). If MAPK activation mediated by Raf-1 translocation and activation is important in the R2/ras synergism described in this study, then aberrant R2 expression should cooperate with activated Rac-1 in cellular transformation, because it has been shown previously that activated Raf-1 and Rac-1 cooperate in mechanisms of transformation (30). Fig. 4C indicates that this prediction is correct, since we observed positive cooperation in transformation between activated Rac-1 and R2 in a manner similar to Ras and R2, as measured by focus formation with N3/mR2 and N3/SH cells transfected with activated V12 Rac-1 (30). These observations are consistent with the view that deregulated R2 gene expression cooperates with oncogenes like ras and rac by upregulating Raf translocation and MAPK pathway activity, but they do not rule out the possibility that other transduction pathways involving activated Raf may also be involved, since there is evidence that Raf can regulate some cellular activities through a MAPK-independent pathway(s) (37, 38, 39).
Figure 4.
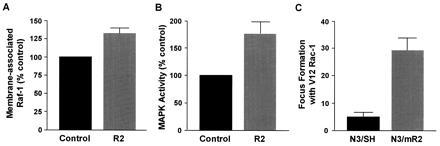
(A) There was an increased amount of Raf-1 protein associated with the membrane in R2 over expressing cells. The recombinant R2-expressing cell lines B3/mR2, N3/mR2, C1/mR2, r-2/mR2, r-3/mR2, and NR4/mR2 (R2) were compared with their respective control lines, B3/SH, N3/SH, C1/SH, r-2/SH, r-3, and NR4 (control). In all cases, cells expressing recombinant R2 exhibited increased membrane-associated Raf-1 protein, and when the two groups of cell lines were compared, they were found to be significantly different by t test analysis (P < 0.001). (B) There was also an increase in the activity of MAPK-2 in R2 overexpressing cells. The recombinant R2-expressing lines B3/mR2, N3/mR2, 10T/mR2, C1/mR2, r-2/mR2, and NR4/mR2 (R2) were compared with their respective control lines infected with LXSH (controls). In all cases tested, cells expressing recombinant R2 showed increased enzyme activity, and the difference between two groups was highly significant (P < 0.001). (C) Increased focus formation with N3/mR2 cells compared with N3/SH cells after transfection with the activated V12 Rac-1 plasmid. The difference was highly significant (P < 0.001). The number of foci shown represents the average ± SE from two independent experiments.
Summary.
The results obtained in this study indicate for the first time, to the best of our knowledge, that the R2 component of mammalian ribonucleotide reductase is a novel malignancy determinant that can synergize with activated oncogenes to modify malignant potential, and supports a model in which these effects are mediated through alterations in major Ras pathways, which are brought about by deregulated R2 gene expression. It is important to note that the only role described for R2 in the cell prior to this report is as a rate-limiting component of ribonucleotide reductase (2, 3). Here we demonstrate that R2 can also participate in other critical cellular functions, and can play a direct role in determining malignant potential through oncogene cooperativity.
Acknowledgments
We thank A. Chan for technical assistance, D. Litchfield for the 9E10 antibody, A. D. Miller for pLXSH, M. Symons for V12 Rac-1, and H. Rubin and B. Cooperman for the R1 protein. J.A.W. is a Terry Fox Senior Scientist of the National Cancer Institute (Canada). We thank the National Cancer Institute (Canada), National Sciences and Engineering Research Council, and the Cancer Research Society, Inc. for financial support (to J.A.W.). H.F. has been the recipient of Medical Research Council of Canada and Medical Research Council of Canada–Pfizer Postdoctoral Fellowships.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviation: MAPK, mitogen-activating protein kinase.
References
- 1.Lewis W H, Kuzik B A, Wright J A. J Cell Physiol. 1978;94:287–298. doi: 10.1002/jcp.1040940306. [DOI] [PubMed] [Google Scholar]
- 2.Reichard P. Science. 1993;260:1773–1777. doi: 10.1126/science.8511586. [DOI] [PubMed] [Google Scholar]
- 3.Wright J A. Int Encycl Pharmacol Ther. 1989;128:89–111. [Google Scholar]
- 4.Mann G J, Musgrove E A, Fox R M, Thelander L. J Cell Biol. 1987;48:5151–5156. [PubMed] [Google Scholar]
- 5.Eriksson S, Graslund A, Skog A, Thelander L, Tribukait B. J Biol Chem. 1984;257:5711–5715. [PubMed] [Google Scholar]
- 6.Chan A K, Litchfield D W, Wright J A. Biochemistry. 1993;32:12835–12840. doi: 10.1021/bi00210a036. [DOI] [PubMed] [Google Scholar]
- 7.McClarty G A, Chan A K, Choy B K, Wright J A. J Biol Chem. 1990;265:7539–7547. [PubMed] [Google Scholar]
- 8.Ashihara T, Baserga R. Methods Enzymol. 1979;58:248–262. doi: 10.1016/s0076-6879(79)58141-5. [DOI] [PubMed] [Google Scholar]
- 9.Hurta R A R, Wright J A. J Biol Chem. 1992;267:7066–7071. [PubMed] [Google Scholar]
- 10.Amara F M, Chen F Y, Wright J A. J Biol Chem. 1994;269:6709–6715. [PubMed] [Google Scholar]
- 11.Chen F Y, Amara F M, Wright J A. EMBO J. 1993;12:3977–3986. doi: 10.1002/j.1460-2075.1993.tb06075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amara F M, Chen F Y, Wright J A. Nucleic Acids Res. 1995;23:1461–1467. doi: 10.1093/nar/23.9.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hurta R A R, Wright J A. J Cell Biochem. 1995;57:543–556. doi: 10.1002/jcb.240570319. [DOI] [PubMed] [Google Scholar]
- 14.Hurta R A R, Samuel S K, Greenberg A H, Wright J A. J Biol Chem. 1991;266:24097–24100. [PubMed] [Google Scholar]
- 15.Weber G. Cancer Res. 1983;43:3466–3492. [PubMed] [Google Scholar]
- 16.Wright J A, McClarty G A, Lewis W H, Srinivasan P R. In: Drug Resistance in Mammalian Cells. Gupta R S, editor. Vol. 1. Boca Raton, FL: CRC; 1989. pp. 15–27. [Google Scholar]
- 17.Saeki T, Takashashi T, Okabe M, Furuya A, Hanai N, Yamagami K, Mandai K, Moriwaki S, Doihara H, Takashima S, Salomon D S. Int J Oncol. 1995;6:523–533. doi: 10.3892/ijo.6.3.523. [DOI] [PubMed] [Google Scholar]
- 18.Jensen R A, Page D L, Holt J T. Proc Natl Acad Sci USA. 1994;91:9257–9261. doi: 10.1073/pnas.91.20.9257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan H, Villegas C, Wright J A. FEBS Lett. 1996;382:145–148. doi: 10.1016/0014-5793(96)00143-3. [DOI] [PubMed] [Google Scholar]
- 20.Egan S E, McClarty G A, Jarolim L, Wright J A, Spiro I, Hager G, Greenberg A H. Mol Cell Biol. 1987;7:830–837. doi: 10.1128/mcb.7.2.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Egan S E, Wright J A, Jarolim L, Yanagihara K, Bassin R H, Greenberg A H. Science. 1987;238:202–205. doi: 10.1126/science.3659911. [DOI] [PubMed] [Google Scholar]
- 22.Taylor W R, Egan S E, Mowat M, Greenberg A H, Wright J A. Oncogene. 1992;7:1383–1390. [PubMed] [Google Scholar]
- 23.Stokoe D, Macdonald S G, Cadwaller K, Symons M, Hancock J F. Science. 1994;264:1463–1467. doi: 10.1126/science.7811320. [DOI] [PubMed] [Google Scholar]
- 24.Miller A D, Miller D G, Garcia J V, Lynch C M. Methods Enzymol. 1993;217:581–599. doi: 10.1016/0076-6879(93)17090-r. [DOI] [PubMed] [Google Scholar]
- 25.Hards R G, Wright J A. J Cell Physiol. 1981;106:309–319. doi: 10.1002/jcp.1041060218. [DOI] [PubMed] [Google Scholar]
- 26.Price J, Turner D, Cepko C. Proc Natl Acad Sci USA. 1987;84:156–160. doi: 10.1073/pnas.84.1.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choy B K, McClarty G A, Chan A K, Thelander L, Wright J A. Cancer Res. 1988;48:2029–2035. [PubMed] [Google Scholar]
- 28.Blosmanis R, Wright J A, Goldenberg G J. Cancer Res. 1987;47:1273–1277. [PubMed] [Google Scholar]
- 29.Chadee D N, Taylor W R, Hurta R A R, Allis C D, Wright J A, Davie J R. J Biol Chem. 1995;270:20098–20105. doi: 10.1074/jbc.270.34.20098. [DOI] [PubMed] [Google Scholar]
- 30.Qiu R-G, Chen J, Kirn D, McCormick F, Symons M. Nature (London) 1995;374:457–459. doi: 10.1038/374457a0. [DOI] [PubMed] [Google Scholar]
- 31.Salem J S, Scott C P, Li L-S, Cooperman B S, Rubin H. FEBS Lett. 1993;323:93–95. doi: 10.1016/0014-5793(93)81455-9. [DOI] [PubMed] [Google Scholar]
- 32.Jelinek T, Catling A D, Reuter C W M, Moodie S A, Wolfman A, Weber M J. Mol Cell Biol. 1994;14:8212–8218. doi: 10.1128/mcb.14.12.8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alessi D R, Cohen P, Ashworth A, Cowley S, Leevers S J, Marshall C J. Methods Enzymol. 1995;255:279–290. doi: 10.1016/s0076-6879(95)55031-3. [DOI] [PubMed] [Google Scholar]
- 34.Wright J A, Turley E A, Greenberg A H. Crit Rev Oncog. 1993;4:473–492. [PubMed] [Google Scholar]
- 35.Bradley M O, Kraynak A R, Storer R D, Gibbs J B. Proc Natl Acad Sci USA. 1986;83:5277–5281. doi: 10.1073/pnas.83.14.5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leevers S, Paterson H F, Marshall C J. Nature (London) 1994;369:411–414. doi: 10.1038/369411a0. [DOI] [PubMed] [Google Scholar]
- 37.Lenormand P, McMahon M, Pouysségur J. J Biol Chem. 1996;271:15762–15768. doi: 10.1074/jbc.271.26.15762. [DOI] [PubMed] [Google Scholar]
- 38.Koong A C, Chen E Y, Mirechi N F, Denko N C, Stambrook P, Giaccia A J. Cancer Res. 1994;54:5273–5279. [PubMed] [Google Scholar]
- 39.Agarwal S, Corbley M J, Roberts T M. Oncogene. 1995;11:427–438. [PubMed] [Google Scholar]