

Abstract
The molecular mechanisms of the defining event in fertilization, gamete fusion, remain poorly understood. The FUS1 gene in the unicellular, biflagellated green alga Chlamydomonas is one of the few sex-specific eukaryotic genes shown by genetic analysis to be essential for gamete fusion during fertilization. In Chlamydomonas, adhesion and fusion of the plasma membranes of activated mt+ and mt- gametes is accomplished via specialized fusion organelles called mating structures. Herein, we identify the endogenous Fus1 protein, test the idea that Fus1 is at the site of fusion, and identify the step in fusion that requires Fus1. Our results show that Fus1 is a ∼95-kDa protein present on the external surface of both unactivated and activated mt+ gametes. Bioassays indicate that adhesion between mating type plus and mating type minus fusion organelles requires Fus1 and that Fus1 is functional only after gamete activation. Finally, immunofluorescence demonstrates that the Fus1 protein is present as an apical patch on unactivated gametes and redistributes during gamete activation over the entire surface of the microvillous-like activated plus mating structure, the fertilization tubule. Thus, Fus1 is present on mt+ gametes at the site of cell-cell fusion and essential for an early step in the fusion process.
INTRODUCTION
Gamete fusion defines the beginning of a new organism in all sexual species. In most organisms, the events that precede fusion have been well characterized at the cellular level (Snell and White, 1996; Wassarman et al., 2001; Primakoff and Myles, 2002). Initial cell–cell interactions between sperm surface proteins and extracellular matrix molecules on the egg trigger activation of the sperm and exposure of previously cryptic regions of the plasma membrane proposed to be involved in gamete fusion. Once the sperm has made its way to the egg plasma membrane, the membranes of the two gametes interact more intimately, finally bringing about gamete fusion. Several sperm and egg molecules implicated in activation of the sperm have been identified, and we know much about the morphological events that accompany gamete fusion (Yanagimachi, 1994). On the other hand, little is known about the molecular mechanisms that underlie the late steps in fertilization, when the plasma membranes of the interacting gametes adhere and fuse (for review, see Primakoff and Myles, 2002). In the absence of any purely eukaryotic systems in which bona fide fusion proteins have been identified, the best models for fusion of the external plasma membrane of cells have come from studies of fusion of viruses with their eukaryotic target cells (White, 1996; Eckert and Kim, 2001). In these systems, a viral transmembrane surface protein interacts with a receptor in the host cell plasma membrane, leading to docking of the virus particle on the cell surface. Subsequent conformational changes in the interacting proteins finally lead to complete fusion (Dimitrov, 2000; Malashkevich et al., 2001; Mayer, 2001).
A small number of eukaryotic genes has been shown by gene disruption to be essential for zygote formation and likely to be required for the late step in fertilization during which the gamete plasma membranes undergo adhesion and fusion. Mouse CD9, a member of the tetraspanin family of proteins, is an egg protein that is essential for fertility. The protein is also found in several nonreproductive cell types in the mouse and is proposed to play a scaffold-like role during gamete fusion (Le Naour et al., 2000; Miller et al., 2000; Miyado et al., 2000). Another mouse protein essential for fertility is the endoplasmic reticulum resident chaperone calmegin, which likely is involved in proper folding of key sperm proteins (Watanabe et al., 1995; Ikawa et al., 2001; Yamagata et al., 2002). The best candidate for a fusion protein in yeast is PRM1p. PRM1p is present at sites of adhesion between a and α haploid Saccharomyces cerevisiae cells, and when it is disrupted in both cell types, fusion is inhibited by 50% (Heiman and Walter, 2000; White and Rose, 2001).
We have been studying fertilization and gamete fusion in the unicellular, biflagellated green alga Chlamydomonas reinhardtii. Fertilization in Chlamydomonas comprises many of the cellular events that typify fertilization in most organisms. During the Chlamydomonas life cycle, diploid zygotes undergo meiosis to yield mt+ and mt- vegetative cells, which undergo gametogenesis when transferred into nitrogen-free (N-free) medium. When wild-type mt+ and mt- gametes are mixed, they initially adhere to each other via sex-specific cell adhesion molecules, agglutinins, on their flagella (Adair, 1985). Interactions between the mating type plus and mating type minus agglutinin molecules on the flagellar membranes activate a gamete-specific flagellar adenylyl cyclase and the resultant increases in intracellular cAMP lead to gamete activation (Pasquale and Goodenough, 1987; Saito et al., 1993; Zhang and Snell, 1994). Both of the gametes release their extracellular matrices (cell walls), they recruit additional agglutinins and a protein kinase from the cell body to the flagella, and they activate sex-specific mating structures at their apical ends, which are the sites for cell-cell fusion (Detmers et al., 1983; Wilson et al., 1997; Wilson and Snell, 1998; Pan and Snell, 2000b). The activated minus mating structure is a small dome-like membrane protrusion ∼0.3 μm in diameter by 0.2 μm in height. The activated plus organelle, which is termed the fertilization tubule, is a more prominent, 0.5 × 3 μm microvillous-like organelle, filled with 60–80 actin filaments. The mating structures of both types of gametes display an extracellular coat of material called fringe (Goodenough et al., 1982).
Because the mating structures are located at the bases of the two flagella on each interacting gamete, flagellar adhesion brings the activated fusion organelles into intimate contact, allowing them to adhere to each other (Goodenough et al., 1982) and leading rapidly to fusion of their plasma membranes. Immediately after mating structure fusion, a cytoplasmic bridge representing the combined mating structures joins the two gametes to each other. Within seconds, the bridge shortens and expands and the formerly distinct, biflagellated gametes merge into a single cell with four flagella, the quadriflagellated zygote. Fertilization is a rapid process in this organism, and zygotes occur within minutes after mt+ and mt- gametes are mixed. Zygote formation is accompanied by inactivation and loss of flagellar agglutinins (the Chlamydomonas equivalent to a block to polyspermy) and activation of transcription of new genes as the zygote developmental pathway commences (Goodenough, 1991; Goodenough et al., 1995b; Pan and Snell, 2000b; Zhao et al., 2001).
In studies to delineate the cellular and molecular mechanisms of gamete fusion in Chlamydomonas, we developed methods for isolating and characterizing activated plus mating structures, the fertilization tubules (Wilson et al., 1997). We showed that the isolated organelles retained their ability to bind to mating structures on activated mt- gametes. An important next step in dissecting the molecular mechanisms for fusion in Chlamydomonas will be to identify proteins on the fertilization tubule responsible for the functions of the organelle. One candidate for the molecule responsible for mating structure adhesion and fusion is the protein encoded by the FUS1 gene. FUS1 is a sex-specific gene that is located in the mt+ locus, a chromosomal region that contains several genes involved in sex- and gamete-specific events (Ferris et al., 2002).
The fus1-1 mutant was generated in mt+ cells >20 years ago in a screen for mt+ gametes that were capable of flagellar adhesion but were unable to fuse when mixed with wild-type mt- gametes (Goodenough et al., 1976, 1995a). More extensive characterization of fus1-1 gametes has shown that they undergo normal flagellar adhesion and gamete activation, and produce a fertilization tubule as robust as those produced by wild-type mt+ gametes. On the other hand, the fus1-1 fertilization tubule fails to fuse with the activated minus mating structure and the cells in such mixtures continue to agglutinate for days. Ultrastructural analysis of the fertilization tubules on fus1 gametes indicated that the organelles do not contain fringe (Goodenough et al., 1982). More recently, the FUS1 gene was identified by its unique presence in the mt+ locus, the sex-restricted expression of its transcript and by its ability in transformation experiments to restore fusion competence to several mt+ mutant strains with lesions in the FUS1 gene (Ferris et al., 1996). Analysis of the predicted amino acid sequence suggested that the Fus1 protein would be an integral membrane protein with a single transmembrane region and a short cytoplasmic tail at the C termini. These properties, along with the reappearance of fringe on the mating structures of the FUS1-rescued mutants, led to the proposal that the protein encoded by the FUS1 gene is a component of fringe or required for production of fringe and is involved in mating structure interactions (Ferris et al., 1996).
Herein, we set out to test the idea that the FUS1 gene product is located at the site of gamete fusion and to examine its role in fusion. We wanted to reexamine the Fus1 protein sequence for possible clues about its function, to investigate the step in mating structure interactions that requires the FUS1 gene product, to identify the endogenous Fus1 protein, and to determine its cellular location in unactivated and activated gametes. Analysis of the sequence of the Fus1 protein revealed that it shows similarity to bacterial adhesion proteins, the invasins and intimins, and that it has five internal repeats of a 90 amino acid domain. Bioassays with imp12 mt- mutants that are fusion competent, but defective in flagellar adhesion, indicate that Fus1 is required for docking between mt- and mt+ gametes at their activated mating structures. Studies with an anti-Fus1 peptide antibody show that Fus1 is a 95-kDa polypeptide expressed only in mt+ gametes; that it is present in an inactive form on the outer surface of the unactivated plus mating structure; and that it becomes distributed over the surface of the entire fertilization tubule during gamete activation.
MATERIALS AND METHODS
Materials
Dibutyryl cAMP, papaverine, Plant Protease Inhibitor Cocktail, trypsin, Nα-tosyl-l-lysine chloromethyl ketone HCl (TLCK), cold water fish gelatin, and glutathione-agarose beads were from Sigma-Aldrich (St. Louis, MO). Electron microscopy-grade paraformaldehyde and electron microscopy-grade glutaraldehyde were from Electron Microscopy Sciences (Ft. Washington, PA). Alexa 488-phalloidin, Alexa 546-phalloidin, SYTOX-green, and Alexa 488-conjugated goat anti-rabbit antibody were from Molecular Probes (Eugene, OR). Kaleidoscope and Precision Prestained molecular weight markers and horseradish peroxidase-conjugated anti-rabbit antibody were from Bio-Rad (Hercules, CA). Glutathione S-transferase (GST)-antibody was from Santa Cruz Biotechnology (Santa Cruz, CA). All other chemicals were of reagent grade.
Cells and Cell Culture
Chlamydomonas reinhardtii strains 21gr (wild-type mt+) (CC-1690), fus1-1 (mt+) (CC-1158), fus1-2 (mt+) (CC-2062), fus1-3 (mt+) (CC-2392), 6145C (wild-type mt-) (CC-1691), and imp12 (mt-) (CC-1149) (available from the Chlamydomonas Genetic Center, Duke University, Durham, NC) were cultured vegetatively with aeration at 23°C in medium I of Sager and Granick on a 13:11-h light/dark cycle. Gametogenesis was induced by transferring vegetatively growing cells into N-free medium as described previously (Snell, 1976a; Pan and Snell, 2000a). Gametes were activated by incubation with 15 mM dibutyryl cAMP and freshly prepared 0.15 mM papaverine in N-free medium for 45–60 min with vigorous aeration (Pasquale and Goodenough, 1987). Gamete activation was confirmed by measuring release of cell walls as described previously (Wilson et al., 1997). Where indicated, cell walls were removed from unactivated gametes by incubation with Chlamydomonas g-lysin for 30 min before use (Buchanan et al., 1989).
Isolation of Fertilization Tubules
Fertilization tubules were isolated from activated wild-type mt+ gametes by differential centrifugation and fractionation on sucrose and Percoll gradients as described previously (Wilson et al., 1997). This method yields an overall purification of fertilization tubules of 160- to 300-fold. Protein concentrations were determined by use of a Bio-Rad protein assay kit with bovine serum albumin (Albumin Standard; Pierce Chemical, Rockford, IL) as a standard.
Gamete Docking and Fusion Assays
To prepare the fixed gametes for the docking assay, activated gametes were incubated in 2.5% glutaraldehyde in N-free medium for 10 min, washed twice in 1% glycine in 10 mM phosphate buffered N-free (bN-free) medium for 5 min, incubated with the live-cell impermeant, nucleic acid fluorochrome SYTOX-green (1 μM in bN-free medium) for 10 min, and washed twice in N-free medium. To carry out the docking assay, fixed, SYTOX-labeled mt+ gametes were mixed with live, activated imp12 mt- gametes and allowed to interact for 30 min. The mixed samples were placed on a microscope slide, fixed in 2% paraformaldehyde, covered with a coverslip supported by petroleum jelly posts, and viewed by fluorescence and differential interference contrast (DIC) microscopy. Using a fluorescein isothiocyanate long-pass barrier filter (filter set 09; Carl Zeiss, Thornwood, NY), the fixed mt+ cells (bright green SYTOX fluorescence) were easily distinguished from the imp12 mt- gametes (low red background autofluorescence). Percentage of docking was defined as (number of fixed cells docked to a live gamete)/(total number of fixed cells counted) × 100. At least 100 randomly chosen cells were counted.
To assay gamete fusion in the absence of flagellar adhesion, activated mt+ and imp12 mt- gametes were mixed at 1:1 ratio, centrifuged at 20,000 × g for 14 s, and resuspended after 20 min. Samples were fixed in 2.5% glutaraldehyde for 1 h after resuspension and the numbers of biflagellated, unfused gametes and quadriflagellated, fused cells were determined by phase-contrast microscopy. Percentage of zygote formation was defined as (number of zygotes × 2)/(number of zygotes × 2 + number of gametes) × 100. At least 100 randomly chosen cells were counted.
Treatment of Cells with Trypsin
Activated or unactivated mt+ gametes were mixed with a freshly prepared stock solution of trypsin (50 mg/ml in 1 mM HCl) at a concentration of 5 × 107 cells/ml to yield the final concentrations of trypsin indicated in the figure legends. Control cells were incubated with equivalent dilutions of the stock solution buffer. After 20 min at room temperature, by which time cells had lost their ability to undergo flagellar agglutination with tester mt- gametes, the cells were diluted 10-fold with N-free medium, centrifuged, and resuspended in fresh N-free medium containing 1 mM TLCK (diluted from freshly prepared 10 mM stock in 1 mM HCl). After a 10-min incubation with TLCK, an aliquot of cells was fixed as described above for immunofluorescence. For immunoblotting, the remaining cells were washed twice more with N-free media containing 1 mM TLCK before analysis by SDS-PAGE and immunoblotting. Both 0.05% and 0.5% trypsin were used for the protease treatment and yielded similar results for loss of agglutinin activity, Fus1 immunofluorescence, and Fus1 immunoblotting.
Production of Recombinant Protein
To prepare a GST-tagged, truncated, recombinant Fus1 protein, a polymerase chain reaction (PCR) product consisting of a FUS1 cDNA fragment corresponding to amino acids 17–741 (which excludes the putative signal peptide and transmembrane domain) and 5′ and 3′ multiple cloning sites was generated using a full-length FUS1 cDNA plasmid (kindly provided by Drs. Patrick Ferris and Ursula Goodenough, Washington University, St. Louis, MO) as the template. The originally obtained, full-length FUS1 cDNA was modified and sequenced before the PCR by standard methods to remove an intron and to ensure that the sequence was correct. The truncated FUS1 PCR product was cloned into the pGEX-2T expression vector (Amersham Biosciences, Piscataway, NJ) and subsequently transfected into M15 bacteria (QIAGEN, Valenica, CA) for expression. Protein expression was induced by adding 1 mM isopropyl β-d-thiogalactoside at 30°C for 1–3 h. GST-Fus1 protein was purified using glutathione-agarose beads according to the manufacturer's instructions (Sigma-Aldrich).
Antibody Production
Anti-Fus1 peptide antibodies were prepared by Biosource International, Quality Controlled Biochemicals (Hopkinton, MA). The peptide SDRFTNWIREKSIATQLRVC was synthesized, verified by mass spectrometry, and used to generate polyclonal antibodies. Antibodies were affinity purified on a peptide affinity column. For immunoblotting, the affinity-purified antibody was absorbed against methanol extracted, lyophilized wild-type mt- gametes to remove nonspecific background staining. To do this, ∼1010 gametes were extracted twice with ice cold 100% methanol, resuspended in 1 ml of methanol, and 250-μl aliquots were lyophilized. For absorption of the antibody, the lyophilized methanol extracts were resuspended in a 1:10 dilution of the antibody in phosphate-buffered saline and incubated with gentle agitation at room temperature for 1–3 h. The sample was cleared by centrifugation at 20, 000 × g at 4°C for 10 min. Antibodies were stored at 4°C with 0.05% NaN3. For additional absorption for immunofluorescence, the antibody sample was mixed with wild-type mt- gametes that had been fixed in 2.5% glutaraldehyde as described above. After an overnight incubation at 4°C the sample was cleared by centrifugation. Absorptions with vegetative mt+ cells yielded similar results.
SDS-PAGE and Immunoblot Analysis
Cells harvested by centrifugation were resuspended in buffer containing 10 mM NaCl, 10 mM HEPES, pH 7.2, and 1 × Plant Protease Inhibitor Cocktail; sonicated on ice three times for 10 s each; mixed with an equal volume of 2 × SDS sample buffer (0.125 M Tris, pH 6.8, 20% glycerol, 4% SDS, 0.2 M dithiothreitol, and 0.05% bromphenol blue); and boiled for 5 min. In some experiments, detergent extracts of cells were used for immunoblotting. To prepare the detergent extracts, cells were resuspended in 10 mM HEPES, pH 7.2, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and 1 × Plant Protease Inhibitor Cocktail at a concentration of 8 × 108 cells/ml and sonicated as described above. After 30 min on ice samples were centrifuged at 10,000 rpm for 10 min at 4°C and the supernatant was used for immunoblotting. Control experiments indicated that essentially all of Fus1 protein was extracted into the supernatant with the detergent solution.
The samples were subjected to electrophoresis on 6% polyacrylamide mini-slab gels at 100 V in buffer containing 25 mM Tris, 192 mM glycine, and 0.1% SDS. Typically, each well was loaded with 50 μl of sample containing ∼2 × 107 cell equivalents. After SDS-PAGE, proteins were transferred to a nitrocellulose membrane (Protran; Schleicher & Schuell, Keene, NH) for anti-Fus1 immunoblotting. For the GST and Chlamydomonas aurora-like protein kinase (CALK) immunoblots a polyvinylidene difluoride membrane (Immobilon P; Millipore, Bedford, MA) was used (Pan and Snell, 2000). Transfer was carried out overnight at 36 V at 4°C in buffer containing 25 mM Tris, 192 mM glycine, and 20% methanol. For detection of Fus1, the membrane was rinsed several times with 25 mM KPO4 buffer, pH 7.0 and fixed with 0.2% glutaraldehyde for 45 min, followed by rinsing twice with TBST (20 mM Tris, pH 7.6, 137 mM NaCl, and 0.05% Tween 20) (Hulen et al., 1991; Wilson et al., 1997). The membrane was blocked with 5% Carnation dry milk (Nestle) in TBST for 2 h and incubated with the primary antibody at a final dilution of 1:1000 in 3% Carnation dry milk in TBST. After 1 h, the membrane was washed 3 × for 7 min each with TBST, followed by incubation for 30 min with a horseradish peroxidase-conjugated goat anti-rabbit IgG antibody diluted 1:10,000 in TBST containing 3% Carnation dry milk. The membrane was washed as before and incubated in enhanced chemiluminescence immunoblotting reagents (Pierce Chemcial) for 1 min as described by the manufacturer, exposed to Hyperfilm ECL (Amersham Biosciences), and the film was developed in an automatic film processor. For GST and CALK immunoblots, the procedure was similar except the fixation step was omitted and the primary antibody was used at a dilution of 1:1,000 and 1:5,000, respectively.
Microscopy
Samples for microscopy were fixed with either 2.5% glutaraldehyde in bN-free medium for visible light microscopy or 2% paraformaldehyde in bN-free medium for fluorescence microscopy. Cells were affixed to eight-well glass slides coated with 0.1% polyethylenimine (Sanders and Salisbury, 1994). Fertilization tubules were visualized by fluorescence microscopy by using the actin-specific fluorochromes Alexa 488-phalloidin or Alexa 546-phalloidin. Samples were incubated in 5 U of fluorochrome/ml in bN-free medium for 15 min and processed as described by Wilson et al., 1997. Samples for immunofluorescence were prepared as follows: paraformaldehyde-fixed cells were permeabilized in a cold acetone series (80%, 100%, 6 min each), blocked for 30 min at 37°C in blocking buffer (1% cold water fish gelatin, 0.1% bovine serum albumin, 5% glycerol, 30 mM NaCl, and 2 mM sodium phosphate buffer, pH 7.3) (Sanders and Salisbury, 1994), incubated in absorbed primary antibody (1:200 dilution in blocking buffer) at 37°C for 1 h, washed three times in phosphate-buffered saline and incubated in absorbed secondary antibody (Alexa 488-conjugated goat anti-rabbit, 1:2000) for 30 min at 37°C.
Microscopy was performed using either an Axioplan2 or a III RS microscope (Carl Zeiss) equipped with epifluorescence and DIC or phase-contrast optics. Images were acquired using Hammamatsu Orca digital cameras and Openlab (Improvision) image acquisition software. Final composite images were constructed using Adobe Photoshop (Adobe Systems, San Jose, CA).
RESULTS
Fus1 Displays Similarity to Bacterial Adhesion Proteins and Contains Five Internal Repeats of ∼90 Amino Acids
At the time the cloning of the FUS1 gene was originally reported no similarities to known proteins were detected (Ferris et al., 1996). More recent analysis, however, indicates that Fus1 protein has sequence similarity to members of the invasin/intimin family of bacterial proteins. The BLAST-link feature on the National Center for Biotechnology Information Entrez PubMed Web site indicates 22% identity and 35% similarity between Fus1 and enteropathogenic Escherichia coli invasin (accession number BAA15799) protein >405 amino acids. This region of invasin contains contiguous repeats of an ∼90 amino acid domain found in all members of the invasin/intimin family of proteins (Oelschlaeger, 2001), which are used for bacterial adhesion to their mammalian host cells (Kalman et al., 1999; Vallance and Finlay, 2000).
Further BLAST analysis and visual inspection of the Fus1 sequence indicated that it contained five internal repeats of ∼90 amino acids. Interestingly, these regions bore some resemblance to a repeating domain in invasins/intimins. Figure 1 shows an alignment of an invasin/intimin domain consensus sequence and the five repeats of Fus1. These similarities between Fus1 and the bacterial adhesion proteins were consistent with the possibility that Fus1 plays a role in adhesion of Chlamydomonas mt+ and mt- gametes during gamete fusion.
Figure 1.

Alignment of Fus1 domains and the bacterial invasin/intimin Ig-like consensus sequence. The sequence in the first line (INV) is a consensus derived from 25 diverse, invasin-related BID 1, bacterial Ig-like (group 1) domains in proteins with the following accession numbers: 1F00_I, 1CWV_A, gi12513096, gi12516151, gi12519346, gi1706558, gi2125981, gi124714, gi3257750, gi11044949, and gi7462086. The remaining five sequences, whose positions are indicated by the numbers at the left, are from Fus1. The alignment was optimized by visual inspection. Identical and similar amino acids are indicated in bold.
Gamete Activation and a Functional FUS1 Gene Are Essential for Gamete Docking
To learn more about the role of the FUS1 gene product in gamete fusion, we developed methods for identifying and characterizing the properties of mating structures at discrete phases of fertilization. To do this required a system in which flagellar adhesion did not interfere with our ability to examine mating structure interactions. We bypassed flagellar adhesion and focused directly on the interactions between mating structures by exploiting two features of the Chlamydomonas system: the availability of mutant mt- gametes (imp12) that do not express functional flagellar agglutinins (Pasquale and Goodenough, 1987; Goodenough, 1991; Goodenough et al., 1995a) and the ability to activate gametes of a single mating type experimentally by incubating them in dibutyryl cAMP (Pjist et al., 1984; Pasquale and Goodenough, 1987; Wilson et al., 1997). As shown in Figure 2A, flagellar adhesion between wild-type mt+ and mt- gametes is characterized by close interactions between the flagella and accompanied by intimate contacts between the cell bodies at the sites of the activated mating structures. On the other hand, and confirming previous reports (Pasquale and Goodenough, 1987), no cellular interactions were observed when imp12 mt- gametes and wild-type mt+ gametes were mixed (our unpublished data), unless both types of gametes were activated by incubation in dibutyryl cAMP and papaverine. The activated mt- and mt+ gametes still did not interact with each other via their flagella (Figure 2B). Rather, the random collisions that occurred as a consequence of the high motility of these cells brought their activated mating structures into contact (Figure 2B), an interaction that was followed by fusion to form a quadriflagellated zygote (Figure 2C).
Figure 2.

Use of imp12 gametes to study gamete docking. (A) Phase-contrast micrograph of a pair of interacting wild-type mt+ and mt- gametes showing their adhering, intertwined flagella (arrows). (B) Phase-contrast micrograph of a gamete pair composed of an activated wild-type mt+ gamete and an activated imp12 mt- gamete. The flagella (arrows) are not interacting, and the cells are adherent only via their apical ends at the sites of their mating structures. (C) Phase-contrast micrograph of a quadriflagellated zygote formed from fusion of a live, activated wild-type mt+ gamete and a live, activated imp12 mt- gamete. (D) Low-magnification, fluorescent image of several pairs of docked gametes. The arrows indicate the activated, fixed, SYTOX-labeled wild-type mt+ gametes in the pairs. (E and E′) corresponding DIC (E) and fluorescent micrographs (E′) of an activated, live imp 12 mt- gamete (arrowheads) docked to an activated, fixed, SYTOX-labeled wild-type mt+ gamete (asterisks). (F and F′) Corresponding DIC (F) and fluorescent images (F′) of an activated, fixed, SYTOX-labeled imp12 mt- gamete (asterisks) docked with an activated, live wild-type mt+ (arrowheads) gamete showing a phalloidin-stained fertilization tubule joining them (F′). Gametes are ∼10 μm in diameter.
This system made it possible to establish a gamete docking assay, in which gametes of opposite mating type would adhere to each other via their mating structures without fusing. To do this, we activated wild-type mt+ gametes, fixed them briefly with glutaraldehyde, and mixed them with live, activated imp12 mt- gametes. As expected, we could find no evidence for fusion, nor did we observe large aggregates of cells. Rather, we detected numerous pairs of gametes (Figure 2D). Within a pair, no flagellar adhesion was detected; each cell was oriented with its apical end directed toward the apical end of the other cell and the sites of their activated mating structures were juxtaposed (Figure 2, E and F). Similar results were obtained when activated, fixed imp12 mt- gametes were mixed with live activated wild-type mt+ gametes. In both circumstances, the interactions were stable when prepared for examination by phase contrast microscopy, and the docked gametes remained as pairs even as the live member of the pair propelled its fixed partner rapidly through the medium. On the other hand, if the samples were vigorously pipetted or otherwise subjected to strong agitation, the pairs came apart, indicating that the gamete mating structure interactions were easily disrupted.
In addition to determining that the cells interacted at their apical ends, we documented that the pairs were composed exclusively of one mt+ gamete and one imp12 mt- gamete adherent to each other at their mating structures. No pairs were observed in suspensions of activated cells of a single mating type, only one cell in a pair was fixed (Figure 2, D and E′), and only one cell in a pair contained a fertilization tubule, which was localized at the site of cell adhesion (Figure 2F′).
With these tools in hand it became possible to examine whether gamete activation was required for docking and to identify the particular step in fertilization that was abrogated in fus1 mutants. To examine the requirement for gamete activation, we mixed unactivated, SYTOX-labeled, glutaraldehyde-fixed, wild-type mt+ gametes with activated, live imp12 mt- gametes and assessed pair formation by using the docking assay. Before mixing, we removed the extracellular matrix (cell wall) that encloses unactivated gametes by use of the wall-degrading Chlamydomonas collagenase, g-lysin (Buchanan and Snell, 1988; Kinoshita et al., 1992). We found that only activated gametes were capable of docking (Figure 3). Thus, although 70% of the control, activated wild-type mt+ gametes formed pairs with activated imp12 mt- gametes (Figure 3A), unactivated, lysin-treated wild-type mt+ gametes were unable to form pairs with the activated imp12 mt- gametes (UnA-L). The lysin treatment did not interfere with docking, as wild-type mt+ gametes that were activated after lysin treatment still formed pairs in the assay (L-A), and activated gametes subsequently treated with the lysin preparation (A–L) also retained functional mating structure adhesion molecules. Thus, the results indicated that gamete activation was required for gamete docking and suggested that molecules involved in docking either were stored intracellularly or were present at the cell surface in an inactive form.
Figure 3.

Activation is required for gamete docking. Live, activated imp12 mt- mutants were mixed with fixed, SYTOX-labeled wild-type mt+ gametes and the percentage of docked cells was determined as described in MATERIALS AND METHODS. Unactivated mt+ gametes were treated under the following conditions before fixation: A, activated with dibutyryl cAMP and papaverine; UnA→L, cell walls were removed with lysin; L→A, cells treated with lysin followed by activation; and A→L, activation followed by lysin treatment. Triplicate samples were counted for each experiment (bar, SEM).
Although it was known that the FUS1 gene product was essential for gamete fusion, we could now ask whether the gene was also required for gamete docking. We activated fus1-1 gametes by incubating them in dibutyryl cAMP and papaverine and assayed for their docking ability as described above. As shown in Figure 4, not only were the activated fus1-1 gametes incapable of gamete fusion (Figure 4B), but also docking was abrogated (Figure 4A). Thus, the FUS1 gene product is essential for a key membrane adhesion event during gamete fusion.
Figure 4.

fus1-1 mt+ gametes are defective in mating structure docking and fusion. (A) Activated, live imp12 mt- gametes were mixed with activated, fixed, SYTOX-labeled mt+ wild-type or fus1-1 gametes and percent docking was determined as described in MATERIALS AND METHODS (n = 5). (B) Activated wild-type or fus1-1 mt+ gametes were mixed with activated imp12 mt- gametes and scored for quadriflagellated zygote formation (n = 4). (bar, SEM).
The Endogenous Fus1 Protein is ∼95 kDa and Is Enriched in Preparations of Isolated Fertilization Tubules
Although the genetic data and the docking experiments were consistent with the idea that Fus1 plays a role in adhesion and fusion of mating structures, the endogenous protein had not been identified or characterized. To study endogenous Fus1, we used a polyclonal antibody raised against a 19 amino acid peptide located near the N terminus of the polypeptide (Figure 5A). The immunoblots in Figure 5B show that the antibody recognized a bacterially expressed GST-Fus1 fusion protein (left, anti-Fus1-peptide antibody; right, anti-GST antibody). The arrow indicates the full-length GST-Fus1, whose identity was confirmed by mass spectrometry (our unpublished data). The lower molecular mass bands presumably were GST-Fus1 fragments, some of which contained only the N-terminally positioned GST.
Figure 5.

A polyclonal antibody directed against a synthetic Fus1 peptide recognizes recombinant Fus1 protein. (A) Amino acid sequence of the Fus1 protein with the antigenic peptide underlined. (B) Immunoblot of a recombinant Fus1-GST fusion protein probed with the anti-Fus1 polyclonal antibody or an anti-GST polyclonal antibody. Migration of prestained molecular weight markers is indicated on the left. The arrow indicates the full-length GST-Fus1 fusion protein.
Immunoblot analysis of wild-type mt+ Chlamydomonas gametes showed reactivity with a protein of ∼95 kDa, which was not present in wild-type mt- gametes (Figure 6A). (An unidentified, weakly staining, cross-reactive band of higher molecular mass was present in both samples.) Consistent with the localization of the FUS1 gene exclusively at the mt+ locus and the previous report that the FUS1 transcript was not detected in vegetative mt+ cells (Ferris et al., 1996), immunoblot analysis of mt+ and mt- gametes and vegetative cells showed that Fus1 was expressed only after gametogenesis and only in wild-type mt+ cells (Figure 6B). Moreover, as expected, gametes of three fus1 mutant strains, each with unique lesions in the FUS1 gene, also failed to express the protein (Figure 6C). The observation that the observed 95-kDa molecular mass of endogenous Fus1 was close to the ∼88-kDa mass predicted by its peptide sequence suggested that the expressed protein may not be heavily glycosylated and is mostly polypeptide.
Figure 6.
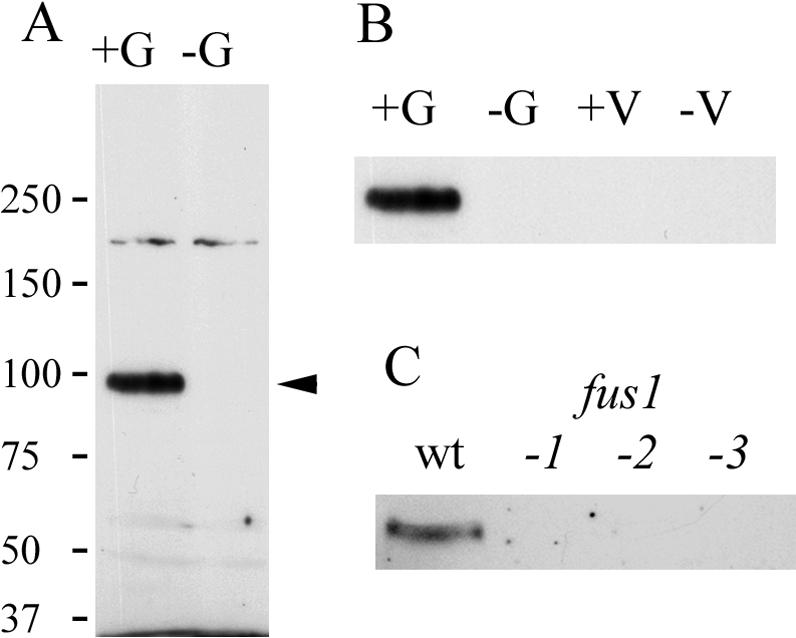
Endogenous Fus1 protein migrates as a ∼95-kDa protein and is expressed only in wild-type mt+ gametes. (A) Wild-type mt+ (+G) and mt- (-G) gametes were analyzed by SDS-PAGE and immunoblotting. The arrow indicates the endogenous Fus1 protein. (B) Anti-Fus1 immunoblot analysis of detergent extracts of 2 × 107 cell equivalents of wild-type mt+ gametes (+G), mt- gametes (-G), mt+ vegetative cells (+V), and mt- vegetative cells (-V). (C) Anti-Fus1 immunoblot analysis of gametes of wild-type mt+ (wt) and three fus1 mutant strains.
We used cell fractionation to determine if endogenous Fus1 was enriched in isolated fertilization tubules. Fertilization tubules were isolated from activated wild-type mt+ gametes as described previously (Wilson et al., 1997) and the following three fractions obtained during purification were analyzed by immunoblotting: the starting cells, partially purified fertilization tubules from the sucrose gradient, and more highly purified fertilization tubules harvested from the final Percoll gradient. Figure 7A shows activated wild-type mt+ gametes stained for actin with fluorescent phalloidin. Figure 7B shows similarly stained, isolated fertilization tubules from the Percoll gradient. Immunoblot analysis of equal amounts of protein from each of the three samples from the purification procedure showed that Fus1 became highly enriched during the purification of fertilization tubules (Figure 7C).
Figure 7.

Fus1 is enriched in isolated fertilization tubules. (A) Alexa 488-phalloidin staining of activated mt+ gametes. The activated gametes display a prominent, actin-rich fertilization tubule. (B) Percoll gradient fraction of isolated fertilization tubules were stained with Alexa 488-phalloidin. (C) Samples (5 μg each) of homogenized cells (HC), and sucrose fractions (SP), and Percoll gradient fractions were analyzed by immunoblotting with the anti-Fus1 antibody. Bar, 5 μm.
Fus1 Is on the External Surface of the Activated plus Mating Structure
Having shown by cell fractionation and immunoblotting that Fus1 was present in fertilization tubules isolated from activated mt+ gametes, we also used immunolocalization methods to determine the cellular distribution of Fus1. To do this, wild-type mt+ gametes were activated as described above and Fus1 location was assessed by indirect immunofluorescence. As shown in Figure 8, the protein was distributed over the entire surface of the fertilization tubules. Figure 8A shows a low-magnification view of a field of activated wild-type mt+ gametes stained only with the anti-Fus1 peptide antibody. Almost every cell displayed an antibody-reactive structure with the morphology of a fertilization tubule. Figure 8B shows a higher magnification view of several activated wild-type mt+ gametes with their fertilization tubules brightly stained by the antibody. Figure 8B′ is a corresponding image of the same cells, which were also stained for actin with fluorescent phalloidin. In each case, the structures that stained with the anti-Fus1 antibody were also stained with phalloidin. Thus, the Fus1 protein is present at fertilization tubules and distributed along their length. As expected, although activated fus1-1 mutant mt+ gametes erected prominent fertilization tubules that could be visualized with fluorescent phalloidin, their fertilization tubules did not stain with the Fus1 antibody (Figure 8, C and C′) because the cells do not express the Fus1 protein (Figure 6).
Figure 8.

Indirect immunofluorescence localization of Fus1 to the fertilization tubule of activated mt+ gametes. (A) Anti-Fus1 indirect immunofluorescence of wild-type mt+ gametes. (B and B′) Corresponding micrographs of wild-type mt+ gametes dual-labeled with the Fus1 polyclonal antibody (B, green) and the filamentous actin-specific fluorochrome Alexa 546-phalloidin (B′, red). (C and C′) Corresponding micrographs of fus1-1 mt+ gametes dual-labeled with the Fus1 polyclonal antibody (C) and the actin-specific fluorochrome Alexa 546-phalloidin (C′). The arrowheads point to the position of fertilization tubules.
We tested the idea that Fus1 is a cell surface protein by using three independent methods. In one approach, we carried out immunolocalization studies in which the antibody would have access only to surface-exposed molecules by using cells that had been fixed, but not permeabilized. The indirect immunofluorescence images shown in Figure 8 were obtained from samples of cells that had been fixed with paraformaldehyde and permeabilized with acetone or methanol before incubation with the antibody. Consistent with the predicted topology, Fus1 was accessible to the antibody in the nonpermeabilized sample (Figure 9A).
Figure 9.

Fus1 is on the external surface of fertilization tubules in activated wild-type mt+ gametes and in a discrete patch on the external surface of unactivated wild-type mt+ gametes. (A) Fluorescence micrograph of activated gametes not permeabilized before immunolocalization with the Fus1 antibody. (B and C) Activated wild-type mt+ gametes incubated with 0% (B) or 0.5% trypsin (C). The insets for B and C each show control (B) and trypsin-treated (C) samples dual labeled with anti-Fus1 antibody (green) and Alexa 546-phalloidin (red). (D) Activated wild-type mt+ gametes incubated with 0.05% trypsin were analyzed for Fus1 by immunoblotting (top). Identical samples also were immunoblotted for CALK, an intracellular protein (bottom). In each lane, 2 × 107 cells were loaded. (E and E′) Corresponding micrographs of unactivated wild-type mt+ gametes dual labeled with the anti-Fus1 antibody (E, green) and the actin-specific fluorochrome, Alexa 546-phalloidin (E′, red). (F) Unactivated fus1 mt+ gametes incubated with the anti-Fus1 antibody. (G) Indirect immunofluorescence image of unactivated nonpermeabilized wild-type mt+ gametes stained with the anti-Fus1 antibody. (H and I) Anti-Fus1 indirect immunofluorescence images of control (H) and 0.5% trypsin-treated (I) unactivated wild-type mt+ gametes. (J) Anti-Fus1 immunoblot of unactivated wild-type mt+ gametes treated with 0.05% trypsin (top). The lower panel shows an anti-CALK immunoblot of identical samples.
In the second approach, the surface localization of Fus1 was assessed by use of the protease trypsin, which, based on the amino acid sequence of Fus1, would cleave the protein at multiple sites. To do this, live, activated wild-type mt+ gametes were incubated with trypsin for 20 min and then prepared for indirect immunofluorescence. Consistent with previous reports (Snell, 1976b; Hunnicutt et al., 1990), the trypsin-treatment did not have any effects on the morphology or motility of the cells, although flagellar adhesion and gamete fusion were blocked (our unpublished data). On the other hand, although the control samples showed typical Fus1 staining (Figure 9B), Fus1 staining was eliminated by the trypsin treatment (Figure 9C). The higher magnification views in the insets show that the fertilization tubules on control samples were stained for Fus1 and actin (Figure 9B, inset), whereas only actin staining remained in the trypsin-treated samples (Figure 9C, inset).
Finally, these indirect immunofluorescence experiments demonstrating that Fus1 was on the external surface of the fertilization tubules were also confirmed by immunoblotting; Figure 9D). Control, activated wild-type mt+ gametes exhibited typical levels of Fus1, but the protein was almost completely absent from the trypsin-treated cells (Figure 9D). Immunoblot analysis with an antibody against the CALK (Pan and Snell, 2000a) showed that this cytoplasmic protein was not accessible to the trypsin in this experiment (Figure 9D), although CALK was sensitive to trypsin if cells were sonicated before the trypsin treatment (our unpublished data). Finally, and consistent with our previous studies, assays for docking (our unpublished data) documented that the trypsin treatment eliminated mating structure adhesion.
Fus1 Is on the External Cell Surface of Unactivated Gametes in a Patch at the Mating Structure
Having shown that Fus1 was localized on the surface of activated plus mating structures, we also determined its location on unactivated gametes. One interpretation of the failure of unactivated wild-type mt+ gametes to adhere to activated imp12 mt- gametes in the docking assay (Figure 3) was that Fus1 might not be on the cell surface before activation. Analysis of unactivated wild-type mt+ gametes by indirect immunofluorescence, however, showed that Fus1 was present as an apical patch at the site of the unactivated mating structure (Figure 9E). Figure 9E′, which shows the corresponding cells stained for actin, documents the expected absence of actin in the unactivated mating structures (Goodenough et al., 1982; Detmers et al., 1983). Indirect immunofluorescence analysis of unactivated fus1 gametes again documented the specificity of the antibody, as these cells do not contain Fus1 protein and did not stain with the antibody (Figure 9F).
Surface localization experiments similar to those carried out with activated gametes demonstrated that, like the Fus1 on activated gametes, Fus1 on unactivated gametes also was on the external surface of the cell. The protein was accessible to the antibody on nonpermeabilized, unactivated gametes (Figure 9G), and trypsin treatment of live gametes eliminated Fus1 as assessed both by indirect immunofluorescence (Figure 9H, control cells; 9I, trypsin-treated cells) and by immunoblotting (Figure 9J). Thus, the results indicated that the Fus1 protein is present on the surface of unactivated mating structures in an inactive form. Furthermore, the results strongly suggest that all of the Fus1 that covers the fully formed fertilization tubule is derived from the Fus1 present on the surface of the inactive mating structure.
DISCUSSION
This work demonstrates that the Fus1 protein is localized on the external surface of the specialized fusion organelle, the fertilization tubule, of activated mt+ gametes where it plays an essential role in the initial membrane adhesion event that precedes membrane fusion during zygote formation. At the end of gametogenesis, but before gamete activation, the FUS1 gene product is present in an inactive form in a highly localized apical patch at the site of the inactive plus mating structure. The gamete activation that is induced when mt+ gametes and mt- gametes are mixed brings about a dramatic redistribution of Fus1. It becomes displayed along the entire length of the newly formed fusion organelle that assembles at the site of the erstwhile apical patch.
Gamete Docking Requires the Fus1 Protein
Even though it is likely that fusion proceeds through a mating structure adhesion step as originally proposed by Friedman et al. (1968), fusion is such a rapid process in Chlamydomonas that it has been difficult experimentally to identify membrane adhesion, in large part because of the inability to determine whether cell pairs are adherent via their mating structures or via their flagella. Rare images of adherent mating structures were obtained in mixtures of wild-type cells in which the mt+ gametes had been treated with cytochalasin to disrupt the actin filaments in the fertilization tubules (Goodenough et al., 1982; Detmers et al., 1983). Presumably, the absence of actin filaments within the fertilization tubules slowed the process of fusion. Related studies with the pseudo-plus fertilization mutant, imp11, which is genotypically mt- and has a lesion in the master sex-determining gene mid, also provided suggestive evidence for docking (Ferris and Goodenough, 1997). After being mixed with wild-type mt- gametes, imp11 cells transformed with the FUS1 gene underwent flagellar adhesion and formed pairs of cells that seemed to be adherent also via their mating structures. Attempts to determine whether the pairs were adherent via their mating structures or their flagella by deflagellating the interacting gametes with a pH shock led to fusion of the gametes. Although intriguing, such a system did not lend itself to a good method for assaying mating structure adhesion. In other studies, fusion-defective mt- mutants, including the gam-1 mutant, were reported to bind to mt+ gametes via their mating structures (Forest, 1987). Those experiments are difficult to interpret, though, because the genes disrupted in the mt- mutants are unknown and the mt- mutants exhibited normal flagellar adhesion, making it impossible to determine whether the cells were adherent via their mating structures or via their flagella. Moreover, the gam-1 mutant is reported to be defective in gamete activation (Forest et al., 1978) and, therefore, the mating structures of the gam-1 cells would not have been activated in those experiments.
More direct observation of mating structure adhesion came from previous studies with isolated fertilization tubules. We showed that fertilization tubules isolated from activated, wild-type mt+ gametes bound to the mating structures of wild-type mt- gametes. Only a single fertilization tubule bound to each mt- gamete and organelles isolated from trypsin-treated wild-type mt+ gametes did not bind (Wilson et al., 1997). In the experiments reported herein, we examined docking by use of activated imp12 mt- gametes. These cells contained activated mating structures, as evidenced by their ability to fuse with activated wild-type mt- gametes, but they did not express functional flagellar agglutinins. Therefore, we were able experimentally to detect mating structure adhesion without the interference of flagellar adhesion. Our results that activated fus1-1 gametes failed to form pairs with activated imp12 gametes (Figure 4) directly demonstrated that the FUS1 gene product is required for adhesion between the plasma membranes of plus and minus mating structures during gamete fusion.
The observation that Fus1 is required for membrane adhesion does not address the question of whether it is also required for the next stage in cell fusion, the actual merging of the lipid bilayers of the two adherent membranes. During fusion of intracellular membranes in the secretory pathway, the proteins involved in vesicle adhesion are also strongly implicated in the subsequent fusion event. In this case, transmembrane proteins on both interacting membranes are proposed to participate directly in bilayer fusion (Jahn and Sudhof, 1999). In several viral systems, the virus adheres to its target cell via interactions between a viral transmembrane protein and a receptor protein on the target cell. After undergoing a conformational change, which exposes a socalled fusion peptide that inserts into the target cell membrane, the viral protein participates directly in bilayer fusion (Doms and Moore, 2000; Eckert and Kim, 2001).
The analysis of the Fus1 sequence offers only limited insights into its role in fertilization, especially because no cell-cell fusion proteins have been identified in eukaryotes. The resemblance of Fus1 to bacterial adhesion proteins described above and the absence of an obvious “fusion peptide” (Ferris et al., 1996) or other domains found in viral or vesicle fusion proteins, suggest that Fus1 might be involved only in adhesion. On the other hand, because we do not yet have an even rudimentary understanding of the molecular mechanisms of fusion initiated at the external surfaces of plasma membranes in any eukaryotic system, it is too early to establish whether Fus1 has more than one role in bilayer fusion. Future studies, in which intact FUS1 constructs and FUS1 constructs with selected domains deleted are used to transform fus1 mutants, should provide new insights about whether Fus1 also participates directly in membrane fusion.
The Fus1 Protein Is in the Right Place at the Right Time for a Direct Role in Docking and Fusion
The identification and characterization of the endogenous Fus1 protein document that Fus1 is in the right place at the right time to play a direct role in mating structure interactions. The results of our adhesion bioassays and results from previous studies (Ferris et al., 1996) were consistent with the idea that Fus1 is on the surface of the fertilization tubule. On the other hand, Fus1 could have been an intracellular membrane protein with only an indirect role in gamete fusion. For example, the protein calmegin is essential for normal fertility in mouse, but calmegin is an endoplasmic resident chaperone and not expressed at the cell surface (Watanabe et al., 1995; Ikawa et al., 2001; Yamagata et al., 2002). The result that Fus1 was localized to the external surface of wild-type plus mating structures was exciting because it placed the protein in the proper cellular compartment to be directly involved in interactions between the membranes of the mating structures. Moreover, the surface localization experiments revealed that not just a portion of total cellular Fus1 was on the surface of unactivated gametes; essentially all detectable Fus1 was surface localized (Figure 9).
It will be interesting to learn the molecular mechanisms that underlie this striking localization to such a restricted area of the gamete surface. It is likely that the molecular mechanisms that target Fus1 to the specialized microvillus in Chlamydomonas will be similar to microvillus targeting mechanisms in the gametes of multicellular organisms. Adhesion and fusion in mouse eggs occurs in the region of the egg surface that is replete with microvilli; and the microvillous-like acrosomal extension in the sperm of many invertebrates is specialized for membrane fusion (for review, see Wilson and Snell, 1998). The sequence analysis of Fus1 predicts that <10 amino acids are in the cytoplasm, and this short region does not contain obvious features, such as protein interaction domains, that might provide clues about how it is localized. The presence of the protein on the unactivated organelles was also surprising, because docking assays showed that unactivated mating structures are incapable of adhering (Figure 3). We should note that there is a precedent for the existence of inactive forms of cell surface adhesion molecules in Chlamydomonas. Flagellar agglutinins are present in an inactive form on the external surface of the cell body plasma membrane and become active only after delivery to the flagella (Hunnicutt et al., 1990). Cell surface integrins in mammalian cells also exist in active and inactive forms under the regulation of signal transduction pathways (Hughes and Pfaff, 1998). One explanation of our results is that that signals generated during gamete activation render Fus1 active for docking and possibly for fusion. For example, Fus1 alone could undergo posttranslational, activating modifications. Or, gamete activation might release inhibitory Fus1-associated proteins, or a second protein could become available or competent to interact with Fus1 to form an active complex.
Another facet of Fus1 demonstrated by the localization studies is that the mechanisms that constrain its location to the mating structure before gamete activation likely persist in some form after activation. Thus, the protein did not spread over the cell body plasma membrane after activation. On the other hand, it did not remain at the base of the mating structure after activation, nor did it all occur as a patch at the tip of the fertilization tubule. Thus, although its location still is restricted, it can be mobilized. Given its distribution along the length of the organelle, it will be important to learn whether the sides of the fertilization tubules are competent for adhesion and fusion. In addition, future studies that identify putative plus gamete proteins that interact with Fus1 should provide insights into the mechanisms that underlie the remarkable preactivation localization and subsequent activation-induced redistribution over the entire surface of the fertilization tubule.
Acknowledgments
We thank Drs. Patrick Ferris and Ursula Goodenough (Washington University, St. Louis, MO) for providing FUS1 plasmids and for helpful discussions. We also thank Dr. Fred Grinnell and Nick Grishin (University of Texas Southwestern Medical Center, Dallas, TX) for insightful discussions and Dr. Fred Grinnell for comments on the manuscript. This work was supported by grants from the National Institutes of Health (National Research Service Award GM-20329 to M.J.M. and GM-56778 to W.J.S.).
Abbreviations used: bN-free medium, 10 mM phosphate-buffered nitrogen-free medium; CALK, Chlamydomonas aurora-like protein kinase; GST, glutathione S-transferase; mt, mating type; N-free, nitrogen-free; TLCK, Nα-tosyl-l-lysine chloromethyl ketone HCl.
References
- Adair, W.S. (1985). Characterization of Chlamydomonas sexual agglutinins. J. Cell Sci. Suppl. 2, 233-260. [DOI] [PubMed] [Google Scholar]
- Buchanan, M.J., Imam, S.H., Eskue, W.A., and Snell, W.J. (1989). Activation of the cell wall degrading protease, lysin, during sexual signalling in Chlamydomonas: the enzyme is stored as an inactive, higher relative molecular mass precursor in the periplasm. J. Cell Biol. 108, 199-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan, M.J., and Snell, W.J. (1988). Biochemical studies on lysin, a cell wall degrading enzyme released during fertilization in Chlamydomonas. Exp. Cell Res. 179, 181-193. [DOI] [PubMed] [Google Scholar]
- Detmers, P.A., Goodenough, U.W., and Condeelis, J. (1983). Elongation of the fertilization tubule in Chlamydomonas: new observations on the core microfilaments and the effect of transient intracellular signals on their structural integrity. J. Cell Biol. 97, 522-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrov, D.S. (2000). Cell biology of virus entry. Cell 101, 697-702. [DOI] [PubMed] [Google Scholar]
- Doms, R.W., and Moore, J.P. (2000). HIV-1 membrane fusion: targets of opportunity. J. Cell Biol. 151, F9-F14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert, D.M., and Kim, P.S. (2001). Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 70, 777-810. [DOI] [PubMed] [Google Scholar]
- Ferris, P.J., Armbrust, E.V., and Goodenough, U.W. (2002). Genetic structure of the mating type locus of Chlamydomonas reinhardtii. Genetics 160, 181-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris, P.J., and Goodenough, U.W. (1997). Mating type in Chlamydomonas is specified by mid, the minus-dominance gene. Genetics 146, 859-869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris, P.J., Woessner, J.P., and Goodenough, U.W. (1996). A sex recognition glycoprotein is encoded by the plus mating-type gene fus1 of Chlamydomonas reinhardtii. Mol. Biol. Cell 7, 1235-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forest, C.L. (1987). Genetic control of plasma membrane adhesion and fusion in Chlamydomonas gametes. J. Cell Sci. 88, 613-621. [DOI] [PubMed] [Google Scholar]
- Forest, C.L., Goodenough, D.A., and Goodenough, U.W. (1978). Flagellar membrane agglutination and sexual signaling in the conditional GAM-1 mutant of Chlamydomonas. J. Cell Biol. 79, 74-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann, I., Colwin, A.L., and Colwin, L.H. (1968). Fine-structural aspects of fertilization in Chlamydomonas reinhardi. J. Cell Sci. 3, 115-128. [DOI] [PubMed] [Google Scholar]
- Goodenough, U.W. (1991). Chlamydomonas mating interactions. In: Microbial Cell-Cell Interactions, ed. M. Dworkin, New York: American Society of Microbiology, 71-112.
- Goodenough, U.W., Armbrust, E.V., Campbell, A.M., and Ferris, P.J. (1995a). Molecular genetics of sexuality in Chlamydomonas. Annu. Rev. Plant Physiol. Plant Mol. Biol. 46, 21-44. [Google Scholar]
- Goodenough, U.W., Armbrust, E.V., Campbell, A.M., and Ferris, P.J. (1995b). Molecular genetics of sexuality in Chlamydomonas. Annu. Rev. Plant Physiol. Plant Mol. Biol. 46, 21-44. [Google Scholar]
- Goodenough, U.W., Detmers, P.A., and Hwang, C. (1982). Activation for cell fusion in Chlamydomonas: analysis of wild type gametes and nonfusing mutants. J. Cell Biol. 92, 378-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodenough, U.W., Hwang, C., and Martin, H. (1976). Isolation and genetic analysis of mutant strains of Chlamydomonas reinhardi defective in gametic differentiation. Genetics 82, 169-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman, M.G., and Walter, P. (2000). Prm1p, a pheromone-regulated multispanning membrane protein, facilitates plasma membrane fusion during yeast mating. J. Cell Biol. 151, 719-730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, P.E., and Pfaff, M. (1998). Integrin affinity modulation. Trends Cell Biol. 8, 359-364. [DOI] [PubMed] [Google Scholar]
- Hulen, D., Baron, A., Salisbury, J., and Clarke, M. (1991). Production and specificity of monoclonal antibodies against calmodulin from Dictyostelium discoideum. Cell Motil. Cytoskeleton 18, 113-122. [DOI] [PubMed] [Google Scholar]
- Hunnicutt, G.R., Kosfiszer, M.G., and Snell, W.J. (1990). Cell body and flagellar agglutinins in Chlamydomonas reinhardtii: the cell body plasma membrane is a reservoir for agglutinins whose migration to the flagella is regulated by a functional barrier. J. Cell Biol. 111, 1605-1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikawa, M., Nakanishi, T., Yamada, S., Wada, I., Kominami, K., Tanaka, H., Nozaki, M., Nishimune, Y., and Okabe, M. (2001). Calmegin is required for fertilin α/β heterodimerization and sperm fertility. Dev. Biol. 240, 254-261. [DOI] [PubMed] [Google Scholar]
- Jahn, R., and Sudhof, T.C. (1999). Membrane fusion and exocytosis. Annu. Rev. Biochem. 68, 863-911. [DOI] [PubMed] [Google Scholar]
- Kalman, D., Weiner, O.D., Goosney, D.L., Sedat, J.W., Finlay, B.B., Abo, A., and Bishop, J.M. (1999). Enteropathogenic E. coli acts through WASP and Arp2/3 complex to form actin pedestals. Nat. Cell Biol. 1, 389-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita, T., Fukuzawa, H., Shimada, T., Saito, T., and Matsuda, Y. (1992). Primary structure and expression of a gamete lytic enzyme in Chlamydomonas reinhardtii: similarity of functional domains to matrix metalloproteases. Proc. Natl. Acad. Sci. USA 89, 4693-4697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Naour, F., Rubinstein, E., Jasmin, C., Prenant, M., and Boucheix, C. (2000). Severely reduced female fertility in CD9-deficient mice. Science 287, 319-321. [DOI] [PubMed] [Google Scholar]
- Malashkevich, V.N., Singh, M., and Kim, P.S. (2001). The trimer-of-hairpins motif in membrane fusion: Visna virus. Proc. Natl. Acad. Sci. USA 98, 8502-8506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer, A. (2001). What drives membrane fusion in eukaryotes? Trends Biochem Sci. 26, 717-723. [DOI] [PubMed] [Google Scholar]
- Miller, B.J., Georges-Labouesse, E., Primakoff, P., and Myles, D.G. (2000). Normal fertilization occurs with eggs lacking the integrin alpha6beta1 and is CD9-dependent. J. Cell Biol. 149, 1289-1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyado, K., et al. (2000). Requirement of CD9 on the egg plasma membrane for fertilization. Science 287, 321-324. [DOI] [PubMed] [Google Scholar]
- Oelschlaeger, T.A. (2001). Adhesins as invasins. Int. J. Med. Microbiol. 291, 7-14. [DOI] [PubMed] [Google Scholar]
- Pan, J., and Snell, W.J. (2000a). Regulated targeting of a protein kinase into an intact flagellum. An aurora/Ipl1p-like protein kinase translocates from the cell body into the flagella during gamete activation in Chlamydomonas. J. Biol. Chem. 275, 24106-24114. [DOI] [PubMed] [Google Scholar]
- Pan, J., and Snell, W.J. (2000b). Signal transduction during fertilization in the unicellular green alga, Chlamydomonas. Curr. Opin. Microbiol. 3, 596-602. [DOI] [PubMed] [Google Scholar]
- Pasquale, S.M., and Goodenough, U.W. (1987). Cyclic AMP functions as a primary sexual signal in gametes of Chlamydomonas reinhardtii. J. Cell Biol. 105, 2279-2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pjist, H.L.A., van Driel, R., Janssens, P.M.W., Musgrave, A., and van den Ende, H. (1984). Cyclic AMP is involved in sexual reproduction of Chlamydomonas eugametos. FEBS Lett. 174, 132-136. [Google Scholar]
- Primakoff, P., and Myles, D.G. (2002). Penetration, adhesion, and fusion in mammalian sperm-egg interaction. Science 296, 2183-2185. [DOI] [PubMed] [Google Scholar]
- Saito, T., Small, L., and Goodenough, U.W. (1993). Activation of adenylyl cyclase in Chlamydomonas reinhardtii by adhesion and by heat. J. Cell Biol. 122, 137-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders, M.A., and Salisbury, J.L. (1994). Centrin plays an essential role in microtubule severing during flagellar excision in Chlamydomonas reinhardtii. J. Cell Biol. 124, 795-805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell, W.J. (1976a). Mating in Chlamydomonas: a system for the study of specific cell adhesion. I. Ultrastructural and electrophoretic analyses of flagellar surface components involved in adhesion. J. Cell Biol. 68, 48-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell, W.J. (1976b). Mating in Chlamydomonas: a system for the study of specific cell adhesion. II. A radioactive flagella-binding assay for quantitation of adhesion. J. Cell Biol. 68, 70-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell, W.J., and White, J.M. (1996). The molecules of mammalian fertilization. Cell 85, 629-637. [DOI] [PubMed] [Google Scholar]
- Vallance, B.A., and Finlay, B.B. (2000). Exploitation of host cells by enteropathogenic Escherichia coli. Proc. Natl. Acad. Sci. USA 97, 8799-8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassarman, P.M., Jovine, L., and Litscher, E.S. (2001). A profile of fertilization in mammals. Nat. Cell Biol. 3, E59-E64. [DOI] [PubMed] [Google Scholar]
- Watanabe, D., Okabe, M., Hamajima, N., Morita, T., Nishina, Y., and Nishimune, Y. (1995). Characterization of the testis-specific gene `calmegin' promoter sequence and its activity defined by transgenic mouse experiments. FEBS Lett. 368, 509-512. [DOI] [PubMed] [Google Scholar]
- White, J.M. (1996). Membrane fusion: the influenza paradigm. Cold Spring Harbor Symp. Quant. Biol. 60. [DOI] [PubMed]
- White, J.M., and Rose, M.D. (2001). Yeast mating: getting close to membrane merger. Curr. Biol. 11, R16-R20. [DOI] [PubMed] [Google Scholar]
- Wilson, N.F., Foglesong, M.J., and Snell, W.J. (1997). The Chlamydomonas mating type plus fertilization tubule, a prototypic cell fusion organelle: isolation, characterization, and in vitro adhesion to mating type minus gametes. J. Cell Biol. 137, 1537-1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, N.F., and Snell, W.J. (1998). Microvilli and cell-cell fusion during fertilization. Trends Cell Biol. 8, 93-96. [DOI] [PubMed] [Google Scholar]
- Yamagata, K., Nakanishi, T., Ikawa, M., Yamaguchi, R., Moss, S.B., and Okabe, M. (2002). Sperm from the calmegin-deficient mouse have normal abilities for binding and fusion to the egg plasma membrane. Dev. Biol. 250, 348-357. [PubMed] [Google Scholar]
- Yanagimachi, R. (1994). Mammalian reproduction. In: The Physiology of Reproduction, vol. 1, eds. E. Knobil and J. Neill, New York: Raven Press, 189-317. [Google Scholar]
- Zhang, Y., and Snell, W.J. (1994). Flagellar adhesion-dependent regulation of Chlamydomonas adenylyl cyclase in vitro: a possible role for protein kinases in sexual signaling. J. Cell Biol. 125, 617-624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, H., Lu, M., Singh, R., and Snell, W.J. (2001). Ectopic expression of a Chlamydomonas mt+-specific homeodomain protein in mt- gametes initiates zygote development without gamete fusion. Genes Dev. 15, 2767-2777. [DOI] [PMC free article] [PubMed] [Google Scholar]