

Abstract
Spinocerebellar ataxia-1 (SCA1) is caused by the expansion of a polyglutamine repeats within the disease protein, ataxin-1. The mutant ataxin-1 precipitates as large intranuclear aggregates in the affected neurons. These aggregates may protect neurons from mutant protein and/or trigger neuronal degeneration by encouraging recruitment of other essential proteins. Our previous studies have shown that calcium binding protein calbindin-D28k (CaB) associated with SCA1 pathogenesis is recruited to ataxin-1 aggregates in Purkinje cells of SCA1 mice. Since our recent findings suggest that tissue transglutaminase 2 (TG2) may be involved in cross-linking and aggregation of ataxin-1, the present study was initiated to determine if TG2 has any role in CaB-ataxin-1 interaction. The guinea pig TG2 covalently cross-linked purified rat brain CaB. Time dependent progressive increase in aggregation produced large multimers, which stayed on top of the gel. CaB interaction with ataxin-1 was studied using HeLa cell lysates expressing GFP and GFP tagged ataxin-1 with normal and expanded polyglutamine repeats (Q2, Q30 and Q82). The reaction products were analyzed by Western blots using anti- polyglutamine, CaB or GFP antibodies. CaB interacted with ataxin-1 independent of TG2 as the protein-protein cross-linker DSS stabilized CaB-ataxin-1 complex. TG2 cross-linked CaB preferentially with Q82 ataxin-1. The cross-linking was inhibited with EGTA or TG2 inhibitor cystamine. The present data indicate that CaB may be a TG2 substrate. In addition, aggregates of mutant ataxin-1 may recruit CaB via TG2 mediated covalent cross-linking, further supporting the argument that ataxin-1 aggregates may be toxic to neurons.
Keywords: SCA1, Transglutaminase, Ataxin-1, Cerebellum
Introduction
Spinocerebellar ataxia type 1(SCA1), an autosomal dominant neurodegenerative disorder is caused by the expansion of unstable CAG repeats which encodes for the polyglutamine (polyQ) tract within the disease protein, ataxin-1 [1]. SCA1 is characterized by -progressive degeneration of selective neurons within the cerebellum, spinal tracts, and brainstem [1]. In normal neuronal tissues, ataxin-1 localizes to the nucleus in a diffuse fashion. Whereas, in the affected neurons of humans as well as the Purkinje cells of SCA1 transgenic mice, ataxin-1 precipitates as larger intranuclear aggregates [2]. These aggregates may protect neurons from mutant protein and/or trigger neuronal degeneration by encouraging recruitment of other essential proteins [3,4]. However, the given reasoning for the pathological sequence of events observed because of aggregation of these mutated proteins is still debated. Two widely believed mechanisms of aggregate formation are either via β-pleated sheets [4,5] or by transglutaminase (TG) dependent covalent incorporation of polyQ proteins into the aggregates [6,7].
Transglutaminases belong to a family of calcium dependent enzymes, which catalyze the post translational modification of proteins through the exchange of primary amines for ammonia at the γ-carboxamide group of glutamine residues [7]. In addition, mammalian TGs are known to stabilize biological structures via cross linking of proteins [8]. Tissue transglutaminase 2 (TG2) is expressed in mammalian nervous system and human brain, localizing mainly in the neurons[9,10] and is highly expressed in Purkinje cells of the cerebellum [11]. TG2 also plays a role in pathogenesis of polyglutamine neurodegenerative disorders [7,12]. Recently, we have demonstrated that SCA1 gene product ataxin-1 is a substrate of TG2 [13]. Further, our previous studies have shown that calcium binding protein calbindin D28k (CaB) associated with SCA1 pathogenesis is recruited to ataxin-1 aggregates in Purkinje cells of SCA1 mice [14,15]. Therefore, the present study was initiated to assess the role of TG2 in CaB-ataxin-1 interaction.
Materials and Methods
Materials
Mouse anti-GFP was obtained from Roche (Roche Diagnostics Corp). Mouse anti-calbindin D-28K and tissue transglutaminase type 2 were obtained from Sigma Chemical Co. (St. Louis, MO).
Methods
HeLa cell culture, transfection and preparation of cell extracts
HeLa cells from the American Type Culture Collection (ATCC) were cultured, transfected with ataxin-1(Q2, 30 or 82)-GFP constructs and processed as described [13, 16]. Briefly, for obtaining whole cell lysates, transfected cells were lysed in 50 mM Tris, pH 7.5, 150 mM NaCl, 1% NP-40,1% sodium deoxycholate, 0.1% SDS, 2 mM EDTA plus 1 tablet of complete protease inhibitor cocktail (Roche, Mannheim, Germany) per 50 ml lysis solution. The lysates were then repeatedly passed through a 25-gauge needle to shear DNA.
In vitro crosslinking by TG2 or disuccinimidyl suberate (DSS)
The reaction was performed at 37°C for various time intervals in a buffer containing 125 mM Tris (pH 8.5), 2.5 mM CaCl2, 10 mM DTT, plus an appropriate amount of CaB protein or HeLa cell lysates. TG2 was added to a final concentration of 0.01U/mg protein. In some reactions 1mM EGTA or 10–40 mM cystamine was used to inhibit TG2 activity. The crosslinking of CaB with GFP-ataxin-1 using protein crosslinker DSS was performed as described previously [17]. In addition to CaB and HeLa cell lysates, the above reaction mixture contained 0.1mM DSS in the final volume of 50μl and no TG2.
Western blots
At different time intervals, the above reactions were subjected to SDS-PAGE (4–20% or 15% acrylamide gels) as desribed previously [13]. Equal amounts of proteins were used. Protein estimations were carried out in the HeLa cell extracts using protein assay kit (Bio-Rad). Proteins were transferred to PVDF membrane (Bio-Rad), blocked for 1 hr with blocking solution (Western Breeze, Invitrogen) and incubated for 1 hr or overnight with appropriate concentration of primary antibodies (monoclonal anti-GFP, 1:1000 and monoclonal calbindin D28k, 1:5000). Immunoreactive proteins were visualized by incubation (for 1 hr) in the goat anti- mouse alkaline phosphatase-labeled secondary antibody followed by reaction with the luminescent substrates (Western Breeze, Invitrogen). The blots were then exposed to hyperfilm-ECL (Amersham). Some blots were stripped and reprobed with appropriate antibodies. Images were processed using Adobe Photoshop CS (version 8.0; San Jose, CA).
Filter retardation assay
To isolate the aggregates of ataxin-1 from nuclear fractions the procedure described by Nagao et al [18] was used. Briefly, the aliquots of nuclear fraction were centrifuged at 10,000 x g for 10 min. The pellet was washed with PBS once and then suspended in suspension buffer (100mM NaCl, 10mM Tris-HCl pH 7.8, 1mM EDTA). This suspension was then sonicated thoroughly and centrifuged at 15,000 x g for 5 min. The pellet, which contained the insoluble material, was resuspended in 100 μl DNAse buffer (10 mM Tris-HCl, pH 7.5, and 10 mM MgCl2) and the protein concentration was determined using Bio-Rad protein assay kit. DNAse I was added to a final concentration of 1mg/ml followed by incubation at 37°C for 1h. After DNAse treatment, a sample containing 25μg protein was diluted into 100μl of 1% SDS and 8% beta-mercaptoethanol in PBS, boiled for 5 min and filtered through a cellulose acetate membrane (Schleicher and Schuell, 0.2 μm pore size) using a Bio-Rad dot-blot filtration unit. After filtration, two washes were performed with 200μl of 0.1% SDS. The cellulose acetate membrane was processed for immunodetection in the same way as regular immunoblotting as described above.
Results
To determine if calcium binding protein CaB is a substrate of TG2, we studied the effects of exogenous TG2 on purified rat brain CaB. The guinea pig TG2 covalently cross-linked CaB (Fig. 1). Time dependent progressive increase in aggregation produced large multimers, which stayed on top of the gel (Fig. 1). The cross-linking was inhibited by 1mM EGTA or 10 mM TG2 inhibitor cystamine (Fig. 1).
Figure 1.
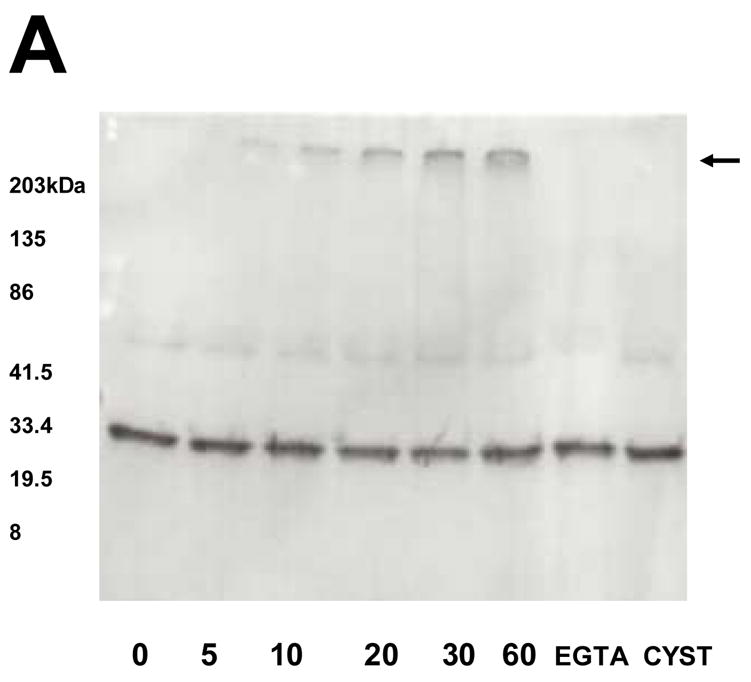
Western blots showing time course cross-linking of rat brain calbindin-D 28k by TG2. When indicated, 10μl aliquots were removed and electrophoresed on a reducing 4–20% polyacrylamide gel. The reaction produced dimers and larger multimers, which stayed on top of the gel (arrow). Multimer formation was inhibited by addition of the 10 mM TG2 inhibitor, cystamine, or by addition of 1 mM calcium chelator EGTA.
To evaluate if ataxin-1 nuclear aggregates co-localize CaB in vivo a filter retardation assay was performed. Figure 2 shows a strong CaB immunoreactivity in insoluble aggregates prepared from 6 wks old SCA1 homozygous mice. In contrast, a weak immunoreaction was detected in age-matched wildtype animals (Fig. 2).
Figure 2.
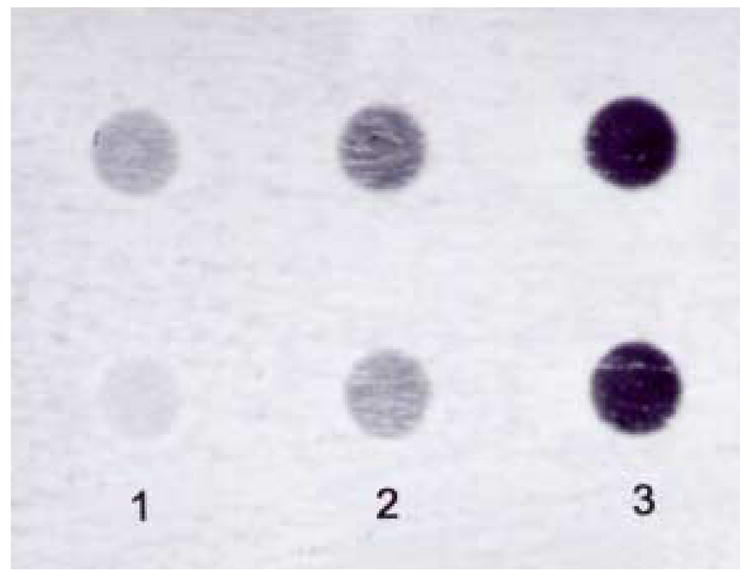
Dot blot analysis showing calbindin-D28k immunoreactivity in insoluble aggregates isolated from crude cerebellar nuclear fractions of 6 wks old wildtype and SCA1 homozygous mice. Four cerebella in each group were pooled to prepare the samples. Equal amounts of proteins were used.
Column 1: Wildtype (incubated with calbindin primary antibody); Column 2: SCA1 homozygous (incubated without calbindin antibody); Column 3: SCA1 homozygous (incubated with calbindin antibody).
CaB interaction with ataxin-1 was studied (Fig. 3) using HeLa cell lysates expressing GFP or GFP tagged ataxin-1 with expanded polyglutamine repeats (Q82). Reactions containing HeLa cell lysates did not show any insoluble CaB aggregates. In contrast, lysates of Q82 expressing cells showed insoluble CaB deposits on top of the stacking gel. The aggregate formation was inhibited by 10 mM cystamine (Fig. 3).
Figure 3.
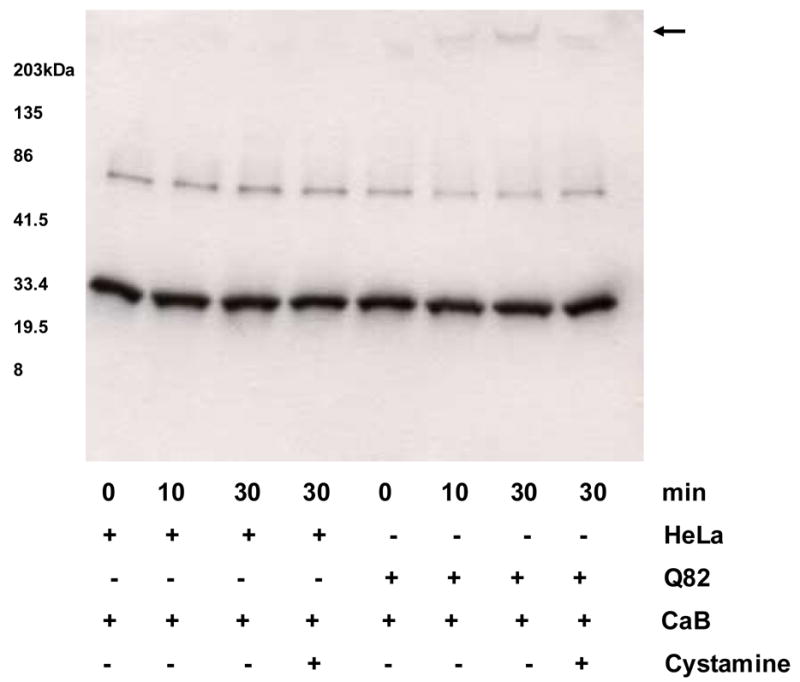
Calbindin-D 28k (CaB) Western blot showing TG2 catalyzed time-dependent crosslinking of CaB with ataxin-1. HeLa cell lysates expressing GFP and GFP-Q82 ataxin-1 were used. No insoluble CaB aggregates are seen in lanes containing GFP expressing HeLa cell lysates. In contrast, lysates of Q82 expressing cells showed insoluble deposits containing CaB on top of the stacking gel (arrow). The aggregate formation was inhibited by 10 mM cystamine.
Further, to investigate if CaB interacted with ataxin-1 independent of TG2, we incubated CaB with lysates of nontransfected HeLa cells as controls and cells expressing GFP-ataxin-1 with Q2, Q30 or Q82 repeats. These interactions were stabilized with protein-protein cross-linker DSS. Figure 4A shows cross-linking of Q2, Q30 or Q82 ataxin-1-GFP to form large multimers. CaB preferentially interacted with Q30 and Q82 ataxin-1 as indicated by decreased intensity of CaB immunoreactive bands in Q30 and Q82 lanes (Fig. 4B). In contrast, another calcium binding protein S100B did not cross-link to Q2-Q82 ataxin-1-GFP (not shown).
Figure 4.
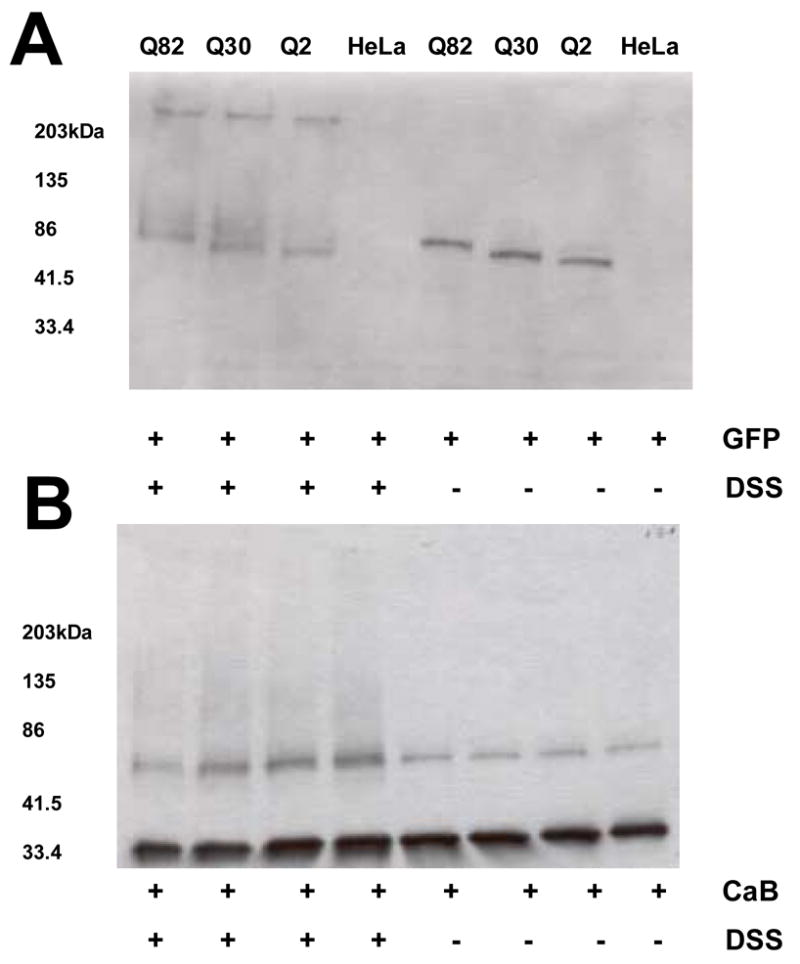
Western blots of GFP (A) and CaB (B). Reactions containing lysates of non-transfected HeLa cells and cells expressing ataxin-1 (Q2, 30 or 82) were incubated for 30 min at room temperature with rat brain CaB followed by the addition of 0.1 mM protein-protein cross-linker DSS. The reactions were then incubated for additional 15 min. In the presence of DSS ataxin-1 formed insoluble aggregates due to self-association (A). A decrease in the intensity of CaB bands in Q30 and Q82 lanes (in the presence of DSS) indicate that CaB may preferably interact with Q30 and Q82 ataxin-1 (B).
Incubation of Q82 ataxin-1-GFP with increasing concentrations of CaB in the presence of DSS resulted in higher molecular weight CaB immunoreactive bands, which were otherwise absent in reactions without Q82 lysates (Fig. 5), further suggesting that ataxin-1 interacts with CaB.
Figure 5.
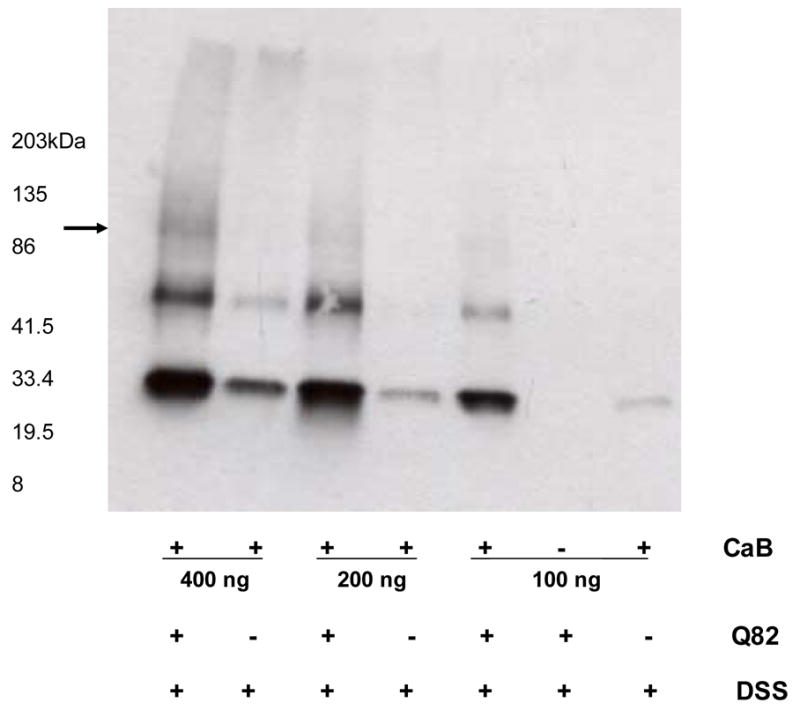
Western blot of CaB showing DSS crosslinked Q82 ataxin-1 with various concentrations of CaB. With increasing concentrations a high molecular weight bands of CaB (arrow), probably crosslinked with Q82 ataxin-1 are visible.
At this time we do not know if calbindin interacts with aggregated and/or non–aggregated form of ataxin-1. One could argue that in Fig. 4 the decrease in intensity of CaB bands is an indirect approach to show interaction between the ataxin-1 aggregates and CaB. However, the absence of CaB in the high molecular weight ataxin-1 aggregates may indicate that in the absence of transglutaminase, CaB interacts with non-aggregated ataxin-1. HeLa cell lysates were prepared 24 hr after transfection and as such do not contain aggregated form of ataxin-1 [13]. Here we show that CaB either colocalizes to TG2 mediated ataxin-1aggregates or interacts with non-aggregated Q30 or Q82 ataxin-1. Further, DSS crosslinked ataxin-1 does not colocalize CaB. This could be due to crosslinking of CaB to native ataxin-1, which may not form aggregates. This argument is further supported by the data in Fig. 5, which shows low molecular weight (about 100–120 kDa) CaB-ataxin-1 crosslinks.
Another intriguing observation was that CaB band intensity was stronger in lanes containing HeLa lysates plus CaB as compared to lanes with CaB alone (Fig. 4B and Fig. 5). These changes were not dependent on components of lysis buffer or free calcium, but were sensitive to heat-treatment. Heating HeLa cell lysates at 95°C for 5 min. prior to the addition of CaB eliminated these effects (not shown).
Discussion
In this report we show that the calcium binding protein CaB may be a substrate of TG2. In addition, TG2 crosslinks CaB to ataxin-1 in vitro, supporting our previous findings, which demonstrated a colocalization of CaB and ataxin-1 to Purkinje cell nuclear inclusions in SCA1 mice [15].
Data on different transgenic lines of mice suggest that nucleus is the primary site of the pathogenesis of ataxin-1 in Purkinje cells of the cerebellum [2,19]. Recently, we observed elevated levels of TG2 in the SCA1 mouse cerebellar nuclei [13]. Transglutaminases have a nuclear translocalization signal (NLS) [20], and nuclear TG has been described in neuroblastoma cells in vitro [21]. However, their function in the nucleus is not fully understood. Using a live-cell nucleocytoplasmic transport assay, it was shown that wildtype, but not polyglutamine expanded mutant ataxin-1, has the ability to export from the nucleus [22, 23]. Therefore, it is possible that in SCA1 mice TG2 is translocated to the nucleus [13] due to retention of mutant ataxin-1 within Purkinje cell nuclei.
Calbindin-D28k, a widely expressed calcium binding protein that belongs to the EF- hand calmodulin superfamily also localizes to the nucleus in cerebellar Purkinje cells [24,25]. Although CaB does not possess a classical NLS, it may use an alternative mechanism to enter the nucleus due to its small size [24]. The functions of CaB in the nucleus are unknown. However, CaB has a higher affinity for Ca2+ and would be operative at lower free Ca2+ concentrations than required by other calcium binding proteins like calmodulin[26,27]. Also, calmodulin is ubiquitously expresses in cells, whereas CaB exhibits tissue-specific expression [27]. Therefore, CaB may be involved in tissue-specific activation of Ca2+ -mediated processes or may act as a Ca2+ -dependent regulator of target proteins in Purkinje cells [24].
CaB interacts with a variety of target proteins [29]. Most importantly, CaB regulates myo-inositol monophosphatase (IMPase), a key enzyme of the inositol-1,4,5-trisphosphate (IP3) signaling cascade [30]. CaB targets IMPase in Purkinje cell spines and dendrites [31]. This interaction and CaB-dependent activation of IMPase accelerates breakdown/recycling of IP3 and finally, allow the cell to maintain its responsiveness to inositiol messengers [31]. Thus, a disturbance of CaB/IMPase interaction may cause substantial deterioration of IP3-mediated signaling [31]. Moreover, using a PCR-based cDNA subtractive-hybridization strategy Lin et al [32] have shown that the expression of genes involved in IP3 signaling are downregulated in SCA1 transgenic mice. In addition, earlier we have demonstrated that CaB levels were altered in Purkinje cells in SCA1 mice, and CaB colocalized to ataxin-1 nuclear inclusions [15]. Therefore, we believe that sequestration of CaB into ataxin-1 aggregates may deplete CaB levels, thus affecting IP3 signaling pathway.
Besides CaB, TG2 cross-links other calcium binding proteins like S100A11 [33]. Since TG2 catalyze the formation of covalent cross-links between the side groups of peptide-bound glutamine and lysine residues, it’s possible that TG2 targets at least one reactive glutamine and one reactive lysine in CaB [34] similar to that observed in S100A11 [33]. In sum, these data indicate that CaB may be a TG2 substrate. Further, mutant ataxin-1 interacts with CaB and TG2 mediated recruitment of CaB to ataxin-1 aggregates support the argument that ataxin-1 inclusions may be toxic to neurons.
Acknowledgments
This work was partly supported by grants from the National Institute of Neurological Disorders and Stroke and Lucky Day Foundation, USA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genet. 2005;6:743–755. doi: 10.1038/nrg1691. [DOI] [PubMed] [Google Scholar]
- 2.Skinner PJ, Koshy BT, Cummings CJ, Klement IA, Helin L, Servadio A, Zoghbi HY, Orr HT. Ataxin-1 with an expanded glutamine tract alters nuclear matrix-associated structures. Nature. 1997;389:971–974. doi: 10.1038/40153. [DOI] [PubMed] [Google Scholar]
- 3.Perez MK, Paulson HL, Pendse SJ, Saionz SJ, Bonini NM, Pittman RN. Recruitment and the role of nuclear localization in polyglutamine-mediated aggregation. J Cell Biol. 1998;143:1457–1470. doi: 10.1083/jcb.143.6.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perutz MF, Johnson T, Suzuki M, Finch JT. Glutamine repeats as polar zippers: Their possible role in inherited neurodegenerative diseases. PNAS. 1994;91:5355–5358. doi: 10.1073/pnas.91.12.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iuchi S, Hoffner G, Verbeke P, Djian P, Green N. Oligomeric and polymeric aggregates formed by proteins containing expanded polyglutamine. PNAS. 2003;100:2409–2414. doi: 10.1073/pnas.0437660100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gentile V, Sepe C, Calvani M, Melone MA, Cotrufo R, Cooper AJ, Blass JP, Peluso G. Tissue transglutaminase-catalyzed formation of high-molecular-weight aggregates in vitro is favored with long polyglutamine domains: a possible mechanism contributing to CAG-triplet diseases. Arch Biochem Biophys. 1998;352:314–321. doi: 10.1006/abbi.1998.0592. [DOI] [PubMed] [Google Scholar]
- 7.Bailey CD, Tucholski J, Johnson GV. Transglutaminases in neurodegenerative disorders. Prog Exp Tumor Res. 2005;38:139–157. doi: 10.1159/000084238. [DOI] [PubMed] [Google Scholar]
- 8.Aeschlimann D, Tomazy V. Protein crosslinking in assembly and remodeling of extracellular matrices: the role of transglutaminases. Connec Tissue Res. 2000;41:1–27. doi: 10.3109/03008200009005638. [DOI] [PubMed] [Google Scholar]
- 9.Kim SY, Grant P, Lee JH, Pant HC, Steinert PM. Differential expression of multiple transglutaminases in human brain. Increased expression and cross-linking by transglutaminases 1 and 2 in Alzheimer’s disease. J Biol Chem. 1999;274:30715–30721. doi: 10.1074/jbc.274.43.30715. [DOI] [PubMed] [Google Scholar]
- 10.Lesort M, Attanavanich K, Zhang J, Johnson GV. Distinct nuclear localization and activity of tissue transglutaminase. J Biol Chem. 1998;273:11991–11994. doi: 10.1074/jbc.273.20.11991. [DOI] [PubMed] [Google Scholar]
- 11.Maggio N, Sellitti S, Capano CP, Papa M. Tissue-transglutaminase in rat and human brain: Light and electron immunocytochemical analysis and in situ hybridization study. Brain Res Bull. 2001;56:173–182. doi: 10.1016/s0361-9230(01)00649-9. [DOI] [PubMed] [Google Scholar]
- 12.Kim S, Nollen EA, Kitagawa K, Bindokas VP, Morimoto RI. Polyglutamine protein aggregates are dynamic. Nat Cell Biol. 2002;4:826–831. doi: 10.1038/ncb863. [DOI] [PubMed] [Google Scholar]
- 13.D’Souza DR, Wei J, Shao Q, Hebert MD, Subramony SH, Vig PJS. Tissue transglutaminase crosslinks ataxin-1: Possible role in SCA1 pathogenesis. Neuroscience Letters. 2006;409:5–9. doi: 10.1016/j.neulet.2006.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vig PJS, Subramony SH, Burright EN, Fratkin JD, McDaniel DO, Desaiah D, Qin Z. Reduced immunoreactivity to calcium-binding proteins in Purkinje cells precedes onset of ataxia in spinocerebellar ataxia-1. Neurology. 1998;50:106–113. doi: 10.1212/wnl.50.1.106. [DOI] [PubMed] [Google Scholar]
- 15.Vig PJS, Subramony SH, Qin Z, McDaniel DO, Fratkin J. Relationship between ataxin-1 nuclear inclusions and Purkinje cell specific proteins in SCA-1 transgenic mice. J Neurol Sci. 2000;174:100–110. doi: 10.1016/s0022-510x(00)00262-8. [DOI] [PubMed] [Google Scholar]
- 16.Xu H, Somers ZB, Robinson ML, 2nd, Hebert MD. Tim50a, a nuclear isoform of the mitochondrial Tim50, interacts with proteins involved in snRNP biogenesis. BMC Cell Biol. 2005;6:29. doi: 10.1186/1471-2121-6-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vig PJS, Desaiah D, Subramony SH, Fratkin JD. Developmental changes in cerebellar endothelin-1 receptor binding in the neurologic mouse lurcher mutant. Res Comm Mol Pathol & Pharmacol. 1995;89:307–316. [PubMed] [Google Scholar]
- 18.Nagao Y, Ishiguro H, Nukina N. DMSO and glycerol reduce bacterial death induced by expression of truncated N-terminal huntingtin with expanded polyglutamine tracts. Biochim Biophys Acta. 2000;1502:246–256. doi: 10.1016/s0925-4439(00)00047-8. [DOI] [PubMed] [Google Scholar]
- 19.Orr HT. Qs in the nucleus. Neuron. 2001;31:875–876. doi: 10.1016/s0896-6273(01)00435-4. [DOI] [PubMed] [Google Scholar]
- 20.Takeuchi Y, Ohashi H, Birckbichler PJ, Ikejima T. Nuclear translocation of tissue type transglutaminase during sphingosine-induced cell death: A novel aspect of the enzyme with DNA hydrolytic activity. Z Naturforsch [C] 1998;53:352–358. doi: 10.1515/znc-1998-5-609. [DOI] [PubMed] [Google Scholar]
- 21.Lesort M, Attanavanich K, Zhang J, Johnson GV. Distinct nuclear localization and activity of tissue transglutaminase. J Biol Chem. 1998;273:11991–11994. doi: 10.1074/jbc.273.20.11991. [DOI] [PubMed] [Google Scholar]
- 22.Howell JL, Truant R. Live-cell nucleocytoplasmic protein shuttle assay utilizing laser confocal microscopy and FRAP. Biotechniques. 2002;32:80–87. doi: 10.2144/02321st04. [DOI] [PubMed] [Google Scholar]
- 23.Irwin S, Vandelft M, Pinchev D, Howell JL, Graczyk J, Orr HT, Truant R. RNA association and nucleocytoplasmic shuttling by ataxin-1. J Cell Sci. 2005;118:233–242. doi: 10.1242/jcs.01611. [DOI] [PubMed] [Google Scholar]
- 24.German DC, Ng MC, Liang CL, McMahon A, Iacopino AM. Calbindin-D28k in nerve cell nuclei. Neuroscience. 1997;81:735–743. doi: 10.1016/s0306-4522(97)00206-6. [DOI] [PubMed] [Google Scholar]
- 25.Sayer R, Turnbull C, Hubbard M. Calbindin 28kDa is specifically associated with extranuclear constituents of the dense particulate fraction. Cell Tissue Res. 2000;302:171–180. doi: 10.1007/s004410000285. [DOI] [PubMed] [Google Scholar]
- 26.Brederman PJ, Wasserman RH. Chemical composition affinity for calcium, and some related properties of the vitamin D dependent calcium-binding protein. Biochemistry. 1974;13:1687–1694. doi: 10.1021/bi00705a021. [DOI] [PubMed] [Google Scholar]
- 27.Baudier J, Mochly-Rosen D, Newton A, Lee SH, Koshland DE, Cole RD. Comparison of S100b protein with calmodulin: interactions with melittin and microtubule-associated tau proteins and inhibition of phosphorylation of tau proteins by protein kinase C. Biochemistry. 1987;26:2886–2893. doi: 10.1021/bi00384a033. [DOI] [PubMed] [Google Scholar]
- 28.Celio MR. Calbindin D-28k and parvalbumin in the rat nervous system. Neuroscience. 1990;35:375–475. doi: 10.1016/0306-4522(90)90091-h. [DOI] [PubMed] [Google Scholar]
- 29.Kojetin DJ, Venters RA, Kordys DR, Thompson RJ, Kumar R, Cavanagh J. Structure, binding interface and hydrophobic transitions of Ca2+-loaded calbindin-D(28K) Nat Struct Mol Biol. 2006;13:641–647. doi: 10.1038/nsmb1112. [DOI] [PubMed] [Google Scholar]
- 30.Berggard T, Szczepankiewicz O, Thulin E, Linse S. Myo-inositol monophosphatase is an activated target of calbindin D28k. J Biol Chem. 2002;277:41954–41959. doi: 10.1074/jbc.M203492200. [DOI] [PubMed] [Google Scholar]
- 31.Schmidt H, Schwaller B, Eilers J. Calbindin D28k targets myo-inositol monophosphatase in spines and dendrites of cerebellar Purkinje neurons. Proc Natl Acad Sci U S A. 2005;102:5850–5855. doi: 10.1073/pnas.0407855102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin X, Antalffy B, Kang D, Orr HT, Zoghbi HY. Polyglutamine expansion down-regulates specific neuronal genes before pathologic changes in SCA1. Nature Neurosci. 2000;3:157–163. doi: 10.1038/72101. [DOI] [PubMed] [Google Scholar]
- 33.Robinson NA, Eckert RL. Identification of transglutaminase-reactive residues in S100A11. J Biol Chem. 1998;273:2721–2728. doi: 10.1074/jbc.273.5.2721. [DOI] [PubMed] [Google Scholar]
- 34.Vanbelle C, Halgand F, Cedervall T, Thulin E, Akerfeldt KS, Laprevote O, Linse S. Deamidation and disulfide bridge formation in human calbindin D28k with effects on calcium binding. Protein Sci. 2005;14:968–979. doi: 10.1110/ps.041157705. [DOI] [PMC free article] [PubMed] [Google Scholar]