

Abstract
We describe an efficient method for generating highly functional membrane proteins with variant amino acids at defined positions that couples a modified site-saturation strategy with functional genetic selection. We applied this method to the production of a cysteine-less variant of the Crithidia fasciculata inosine-guanosine permease CfNT2, in order to facilitate biochemical studies using thiol-specific modifying reagents. Of ten endogenous cysteine residues in CfNT2, two cannot be replaced with serine or alanine without loss of function. High-quality single- and double-mutant libraries were produced by combining a previously reported site-saturation mutagenesis scheme based on the Quikchange method with a novel gel purification step that effectively eliminated template DNA from the products. Following selection for functional complementation in S. cerevisiae cells auxotrophic for purines, several highly functional non-cysteine substitutions were efficiently identified at each desired position, allowing the construction of cysteine-less variants of CfNT2 that retained wild-type affinity for inosine. This combination of an improved site-saturation mutagenesis technique and positive genetic selection provides a simple and efficient means to identify functional and perhaps unexpected amino acid variants at a desired position.
Keywords: cysteine-less, substituted cysteine accessibility method, membrane protein, site-saturation mutagenesis, genetic selection, Quikchange mutagenesis, equilibrative nucleoside transporter
A detailed understanding of how a protein functions must include structural information at some level of resolution. While x-ray crystal structures of thousands of soluble proteins have been determined to date, crystals of membrane proteins that diffract to high resolution are notoriously difficult to obtain [1]. In the absence of any high-resolution structural information for many membrane protein families, biochemical methods provide a valuable means to intuit structure. Several of these methods make use of strategically placed cysteine residues because of their unique chemistry among the natural amino acids. For example, cysteines engineered into two transmembrane helices that can be cross-linked with bifunctional thiol-reactive reagents of varying lengths provide information about the distance between the two helices in the folded structure of the protein [2]. Also, the steric and chemical environment of a cysteine residue engineered at a position of interest can be probed by the substituted cysteine accessibility method (SCAM), fluorescence resonance energy transfer (FRET), or site-directed spin labeling using a battery of thiol-reactive reagents of diverse sizes, chemical characteristics, and functionalities [3–6]. Such indirect measures of structure can provide important constraints in the computational modeling of membrane proteins in the absence of a crystal structure [1, 7] and provide information on dynamic movements in the presence of ligand or agonist that cannot be visualized by crystallography (e.g., [8]).
The construction of a functional variant of the membrane protein of interest that presents no reactive thiols is a necessary first step in any technique that probes the environment of genetically engineered cysteine residues. This generally requires that most or all naturally occurring cysteine residues in the protein be replaced with other amino acids without altering the structure of the protein, as assessed by comparing function to its wild-type counterpart. Mutation of cysteine residues to alanine or serine in many cases results in a cysteine-less protein retaining wild-type affinity for ligand (see [9–11]); however, in other proteins such substitutions result in a non-functional protein [12–14]. Identification of substitutions that are functional may not be straightforward in such cases, requiring a significant number of molecular biological and biochemical manipulations to construct and evaluate mutants that one thinks might be functional. Here we describe an unbiased and rapid approach that utilizes site-saturation mutagenesis based on the modified Stratagene Quikchange method of Zheng et al. [15] to create libraries of mutants of the Crithidia fasciculata inosine/guanosine transporter gene, CfNT2, lacking one or more cysteine codons, and the subsequent genetic selection of highly functional transporter mutants in a heterologous expression system. The identification of functional mutants is greatly facilitated by the addition of a gel-purification step to the Quikchange protocol, which effectively eliminates background of wild-type clones in the selection. This combination of cysteine-excluding site-saturation mutagenesis and positive genetic selection/screening provides an effective means to construct functional cysteine-less membrane protein mutants with a minimal investment of time and resources, provided that a method of screening or selecting for the function of the protein is available.
Materials and methods
Cell culture and reagents
E. coli were DH5α and TOP10 strains (Invitrogen Corp., Carlsbad, CA), and standard methods for recombinant DNA work were employed [16]. The purine auxotrophic S. cerevisiae strain YPH499 (MATa ura3-52 lys2-801 ade2-101 trp1-Δ63 leu2Δ1) was constructed by Sikorski and Hieter [17]. Synthetic complete (SC) media plates were prepared according to standard methods. Microbiological media were obtained from Fisher Scientific and MP Biomedicals (Irvine, CA). Yeast were transformed by either the rapid or the high-efficiency lithium acetate method [18]. Leishmania donovani cells in which the LdNT1 and LdNT2 genes (Δldnt1/Δldnt2 L. donovani) were deleted by targeted gene replacement and which are therefore null for purine nucleoside transport [19] were transfected with plasmid DNA according to the method of Robinson & Beverley [20]. The Δldnt1/Δldnt2 parasites were maintained in medium [21] supplemented with 5% fetal bovine serum (Invitrogen), 50 μg/ml hygromycin (Roche, Indianapolis, IN), 50 μg/ml phleomycin (RPI Research, Mt. Prospect, IL) and 25 μg/ml G418 (BioWhittaker, Walkersville, MD). Virtually all other chemicals were purchased from Sigma-Aldrich and were of the highest grade available. Oligonucleotides were obtained from Invitrogen.
Generation of cfnt2-Δ7Cys and cfnt2-Δ10Cys yeast expression plasmids
All expression of CfNT2 derivatives in yeast was driven from the high-copy pRS426-Cu yeast-E. coli shuttle vector [22], which allows copper inducible expression of the inserted gene and contains a URA3 selectable marker. The CfNT2 gene from the protozoan parasite C. fasciculata encodes a 502 amino acid inosine/guanosine equilibrative nucleoside transporter [23]. Seven of ten cysteine residues within the CfNT2 open reading frame were mutated to other amino acids by the Quikchange method (Stratagene, La Jolla, CA). The resulting plasmid encoding cfnt2-C149L, C217A, C354S, C428S, C455S, C458A, C489S was designated pRS426-Cu-cfnt2-Δ7Cys. Cysteine residues C158, C161 and C461 remained in this construct.
Following genetic selection for plasmids mutant at C158 and C161 and for plasmids mutant at C461 (see text), fully cysteine-less constructs were engineered by replacing the 3′ end of the cfnt2 gene in a C158/161 mutant plasmid with that from a C461 mutant plasmid by subcloning. The resulting plasmids were designated pRS426-Cu-cfnt2-Δ10Cys, with the mutant residues encoded at positions 158, 161 and 461 indicated in parentheses; e.g. pRS426-Cu-cfnt2-Δ10Cys (VTS) is equivalent to pRS426-Cu-cfnt2-Δ7Cys, C158V, C161T, C461S.
Construction of site-saturation libraries at C158/C161 and C461
Randomization of cysteine codon 461 was achieved using an oligonucleotide pair that introduced “NHN” at this position (H = A, T, C; D = T, A, G): forward primer 5′-CGCGATCCACNHNCCACGCACGACGACGCTGCGC-3′, reverse primer 5′-CGTGCGTGGNDNGTGGATCGCGGCCATGGAGC-3′. To produce a library of plasmids mutant at codons 158 and 161, an oligonucleotide pair was designed for Quikchange mutagenesis according to the method of Zheng et al. [15] that introduced “NNG” at these two positions: forward primer 5′-CAAGGCGCTGNNGGACTCCNNGACGAACGCGCTGGTTGGC-3′, reverse primer 5′-GCGTTCGTCNNGGAGTCCNNCAGCGCCTTGGAGAAGCCAC-3′. Each primer pair was employed in a Quikchange PCR reaction (Stratagene) using pRS426-Cu-cfnt2-Δ7Cys as template: 50 ng template DNA was combined with 125 ng of each primer, 0.2 mM dNTPs, and 2.5 units Pfu Turbo (Stratagene) in Pfu Turbo buffer. The mixture was subjected to 17 cycles of 30 sec at 95°C, 1 min at 55°C, 17 min at 68°C. Reaction products were treated with 20 U DpnI (New England Biolabs, Ipswich, MA) for 1.5 hr. Following digestion, either (a) high molecular weight DNA in the reaction was purified from other components using the QIAprep PCR purification kit (Qiagen, Valencia, CA) or (b) the mixture was separated on a 0.8% agarose gel using TAE buffer supplemented with 1 mM guanosine to retard UV-induced DNA damage and maximize bacterial transformation efficiency [24], and the PCR-produced band was gel purified away from any remaining supercoiled template DNA using the Qiaex II kit (Qiagen). Resultant plasmid DNA was transformed into 50 μl chemically competent One Shot TOP10 cells (~1 ×109 cfu/μg DNA, Invitrogen). Colonies were pooled and library DNA prepared.
Genetic selection for functional cysteine-less mutants from DNA libraries
YPH499 yeast cells were transformed with library DNA by the high efficiency lithium acetate method [18] and 1–2×105 colony-forming units were immediately plated on minimal media that selected for both the presence of the plasmid and inosine transport function (SC ura– ade– + 100 μM inosine + 100 μM CuSO4). After six days of incubation at 30°C, a subset of large colonies were retested for robust growth on inosine by streaking on SC ura– ade– + 100 μM inosine + 100 μM CuSO4 plates versus yeast expressing cfnt2-Δ7Cys. Mutant plasmids were rescued from yeast cells using the QIAprep Spin Miniprep Kit (Qiagen) according to the suggested protocol for yeast (http://www1.qiagen.com/literature/protocols/pdf/PR04.pdf). Prepared DNA was transformed into chemically competent DH5α cells (~106 cfu/μg DNA, Invitrogen), and recovered plasmid DNA was sequenced over the mutated region of the cfnt2 gene.
Inosine uptake assays on live yeast cells
Yeast cells were grown to log phase in SC ura–, induced with 100 μM CuSO4 for 1 hr, and harvested at an optical density (OD600) of ~ 1.0. Cells were washed twice in assay buffer (30 mM MES, pH 5.6, 2% glucose), and resuspended at 20 OD600/ml in the same buffer. Uptake of [2,8-3H]-inosine (American Radiolabeled Chemicals, Inc., St. Louis, MO) was measured by the oil-stop method [25] using a 22:3 mixture of 550 oil (Dow Corning) to light paraffin oil (Fisher Scientific, cat no. O-119) as a cushion. Uptake by yeast cells expressing cysteine-less mutants was assessed over 30 or 60 sec (6 time points) in duplicate. Rate determinations were calculated by linear regression in Prism 4.0 (Graphpad Software, Inc.). Inosine uptake kinetics for CfNT2 and cfnt2-Δ10Cys (VTS) proteins were performed by measuring uptake at 5 sec in triplicate at various concentrations of inosine, and apparent Km values were calculated by fitting the data to the Michaelis-Menten equation in Prism 4.0.
Construction and characterization of L. donovani expressing cfnt2-Δ10Cys (VTS)
The L. donovani expression plasmid pALTneo-HA-cfnt2-Δ10Cys (VTS) was constructed as follows. The cfnt2 open reading frame from pRS426-Cu-cfnt2-Δ10Cys (VTS) was amplified by PCR using primers that added an in-frame NdeI site at the start codon and a BamHI site to the 3′ end, following the stop codon. Following cloning of this product into pCR-2.1-TOPO (Invitrogen) and sequence analysis of the entire open reading frame, the NdeI-BamHI fragment was ligated into the NdeI-BamHI site of pXG-HA-neo (Yates and Ullman, unpublished). The resulting plasmid produces a cfnt2 protein with an N-terminal HA tag. The HA-tagged open reading frame was removed by BglII/BamHI digestion and subcloned into the BamHI site of pALTneo [26]. pALTneo-HA-cfnt2-Δ10Cys (VTS) was transfected into Δldnt1/Δldnt2 L. donovani according to established protocols [21].
Parasites expressing cfnt2-Δ10Cys (VTS) were analyzed for the ability to transport [3H]-inosine by the oil-stop method in PBS + 10 mM glucose using a 33:7 mixture of 550 oil to light paraffin oil as a cushion. Linear rates of uptake were determined over a 30 sec time course at various concentrations of [3H]-inosine, and the data were fit to the Michaelis-Menten equation in Prism 4.0.
Results
Heterologous expression of CfNT2 in yeast
CfNT2 is a Crithidia fasciculata inosine/guanosine-specific purine nucleoside permease of the equilibrative nucleoside transporter (ENT) family and contains ten cysteine residues in its primary sequence [23]. In order to study the structure of this protein using thiol-reactive reagents, such as those employed in SCAM or FRET, it is necessary to construct a functional, and therefore presumably properly folded, variant lacking all cysteines. Saccharomyces cerevisiae (yeast) was chosen as a heterologous expression system for the functional evaluation and genetic selection of cysteine-substituted variants of CfNT2 for several reasons. First, yeast have been successfully exploited as a heterologous expression system for genetic and biochemical characterization of a variety of human and protozoan equilibrative nucleoside transporters [23, 27–29]. Second, they have no endogenous purine nucleoside transport activity and when mutant for ade2 are also purine auxotrophs, enabling functional complementation with a purine nucleoside transporter to be evaluated merely by manipulating the purine content of the media. Finally, yeast have a short doubling time and are easily manipulated genetically and biochemically. Yeast auxotrophic for purines and expressing CfNT2 grew rapidly on media containing inosine as the sole purine source, while vector-transformed cells did not grow on inosine (Fig. 1A).
Figure 1.
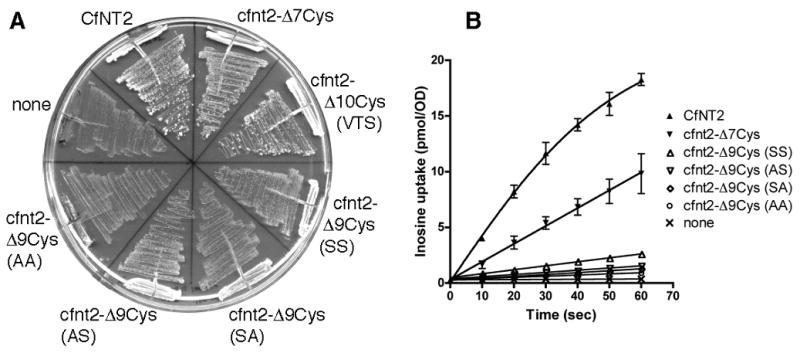
Characterization of serine and alanine substitutions for Cys158 and Cys161 of cfnt2-Δ7Cys in S. cerevisiae. (A) Purine auxotrophic yeast were transformed with plasmids carrying genes encoding the indicated cfnt2 mutant proteins. Cells were grown on SC ura– ade– media containing 100 μM inosine as the sole purine source at 30°C for 3 days. AA = C158A, C161A; AS = C158A, C161S; SA = C158S, C161A; SS = C158S, C161S; VTS: C158V, C161T, C461S. (B) Yeast strains were analyzed for the ability to take up 5 μM [3H]-inosine by the oil-stop method using duplicate time points. Data shown are representative of two independent experiments.
The Leishmania donovani inosine/guanosine transporter LdNT2 is 73% identical to CfNT2 [23], and shares seven cysteines with CfNT2. Four of these seven cysteines could be replaced with serine or alanine in LdNT2 without affecting transporter function (Arendt & Ullman, unpublished). The orthologous cysteines in CfNT2 tolerated identical changes (C354S, C428S, C458A, C489S) and a further three cysteines were changed to residues similar to those at corresponding positions in LdNT2 (C149L, C217A, C455S). The resulting cfnt2 mutant in which seven cysteine residues were replaced with alanine, serine and leucine (cfnt2-Δ7Cys) retained significant inosine transport function (Fig. 1).
Mutation of any of the remaining three cysteines in LdNT2, which are conserved in CfNT2, to serine, alanine or threonine residues resulted in loss of transporter function (Arendt & Ullman, unpublished). To quickly complete the design of a cfnt2 variant lacking all cysteines and retaining high levels of inosine transport function, we chose to use site-saturation mutagenesis at the positions of these three cysteine residues (Cys158, 161 and 461) combined with rapid functional selection in the ade2 yeast expression system (Fig. 2).
Figure 2.
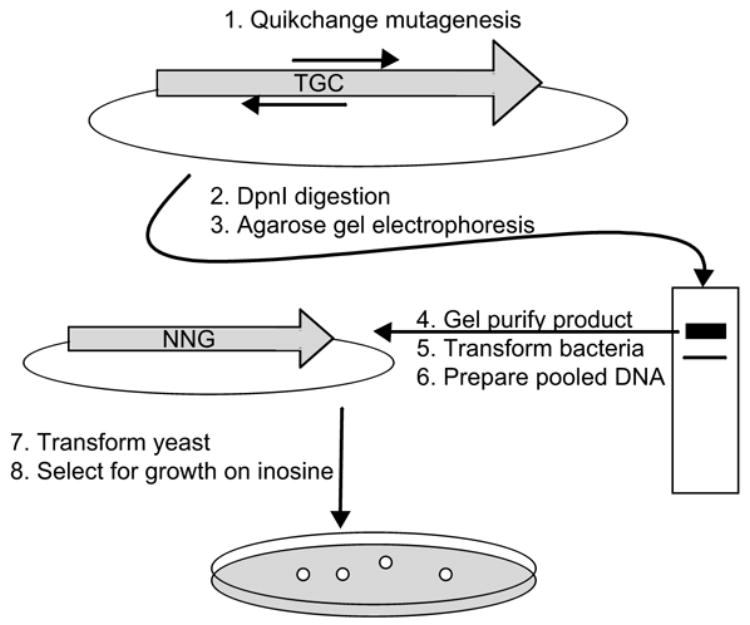
Diagram of steps in the production of a mutant DNA library replacing a given cysteine codon (TGC) with non-cysteine codons (NNG), followed by selection in purine auxotrophic yeast for ability to take up and grow on inosine as a purine source, which requires cfnt2 transport activity.
Construction of site-saturation library lacking Cys461 and selection for functional mutants
Using the pRS426-Cu-cfnt2-Δ7Cys plasmid as a starting point (see Materials and methods), a plasmid library in which the codon for Cys461 was randomly mutagenized was constructed by the Stratagene Quikchange method. Exclusion of template DNA from the library was desired as any yeast transformed with “wild-type” DNA would grow under the selection conditions, making the desired mutants more difficult to identify among the positive clones. Primers were designed that specified N-A/C/T-N at codon 461, which disallowed the wild-type Cys codon (TGC, TGT) at this position, but allowed 48 other codons encoding 16 amino acids (all Trp, Arg and Gly codons were also eliminated by this strategy). The introduction of stop codons was not specifically avoided, as any clones containing them would not be selected in the positive selection scheme. Attempts to construct plasmid libraries with mutagenic primers that were exact reverse complements except at the randomized positions, according to the standard Stratagene Quikchange protocol, resulted in an unacceptably high percentage of unproductive plasmids: template plasmids encoding cysteine, as well as insertion and frame shift mutants incorporating two copies of the primer sequence at the mutation site. Such aberrant clones are generally rare in Quikchange reactions not employing randomized codons and perhaps became abundant due to the necessity for the recipient bacteria to resolve mismatches between strands at the randomized positions.
To produce plasmid libraries that predominantly contained the desired site-saturation mutants, a minimum of aberrant plasmids, and as little template DNA as possible, the mutagenic primer pairs were redesigned according to the method of Zheng et al. [15]. Briefly, the 5′-end of each primer was shortened and 3′-end lengthened relative to the mutation site, such that the forward and reverse primers were not perfectly overlapping and would bind preferentially to the template rather than to each other during annealing. This modification significantly reduced the number of aberrant plasmids in the library.
To strongly bias against the survival of template (wild-type) plasmid following DpnI digestion, the amplification product was separated from traces of uncut supercoiled template by agarose gel electrophoresis and gel purified. Any circular template DNA would be expected to transform bacteria much more efficiently than the doubly-nicked amplification products, leading to a disproportionate representation of template plasmid in the resulting library. The ability of the gel-purification step to prevent the incorporation of wild-type plasmid into the site-saturation library was evaluated by comparing libraries constructed following bacterial transformation of DpnI-treated Quikchange reactions subjected to either (a) gel or (b) spin-column purification. While sequencing of ten bacterial clones from each of the two libraries revealed no cfnt2 genes encoding the wild-type cysteine at codon 461, sequencing of pooled library DNA showed that the proportion of the wild-type G nucleotide at position 2 of codon 461 present in the gel-purified library was closer to background levels than in the spin-column treated library (Fig. 3). The presence of low levels of unmutated template DNA in either of the libraries was expected to give rise to unwanted “positive” (wild-type) clones in the genetic selection for retention of CfNT2 function. DNA libraries derived from the spin-column purified and gel-purified reaction products were used to transform yeast auxotrophic for purines, and clones containing cfnt2 variants that were competent for inosine transport were selected by plating on media containing inosine as the sole purine source (Fig. 2). Use of the gel-purified library resulted in a much lower background of selected cfnt2 clones encoding the wild-type cysteine (0 of 10 positive clones sequenced) than did use of the spin-column treated library (4 of 10 clones). Inclusion of a gel-purification step following Quikchange mutagenesis therefore led to more efficient positive selection of mutant clones of interest.
Figure 3.
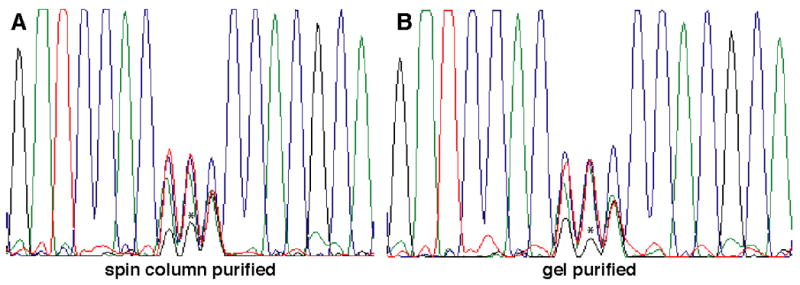
Sequencing of DNA pooled from transformation of bacteria with (A) spin-column purified or (B) agarose gel purified Quikchange mutagenesis products. Guanosine nucleotide at the second position, presumably derived from contaminating template plasmid encoding cysteine (TGC), is indicated by an asterisk. G: black, A: green, T: red, C: blue.
Of the sixteen mutant plasmids sequenced from the selections above, eight encoded serine at position 461, and all four possible serine codons were represented indicating a reasonable complexity of the mutant library. In addition, five plasmids encoded proline, two clones encoded phenylalanine, and one tyrosine, indicating that these three hydrophobic amino acids can functionally substitute for Cys461. The rate of uptake of [3H]-inosine over a 25 sec time course by yeast cells expressing each of the four selected cfnt2-Δ8Cys mutants was identical to that of cfnt2-Δ7Cys (data not shown).
Simultaneous site-saturation and selection at two adjacent positions
Like Cys461 in CfNT2, most cysteine residues in membrane proteins can be replaced with serine or alanine without a loss of function. To evaluate the effect of such replacements of Cys158 and 161 of CfNT2, these two residues were mutated to any combination of Ala or Ser within the context of cfnt2-Δ7Cys, and function of the resulting proteins was evaluated in ade2 yeast. Serine substitutions at both C158 and C161 resulted in a slow growth phenotype and poor inosine uptake relative to CfNT2 and cfnt2-Δ7Cys expressing cells, while alanine mutations at one or both positions performed even less well (Fig. 1). These results suggest that serine and alanine substitutions at these two positions led to poor expression and/or poor function of the resulting cfnt2-Δ9Cys proteins.
In order to identify highly functional cfnt2-Δ9Cys mutants, the same site-saturation mutagenesis and genetic selection approach used above was adopted, this time eliminating two cysteine codons with a single set of mutagenic primers. Partially complementary primers were designed for Quikchange mutagenesis as before, specifying codons for 158 and 161 as “NNG” because cysteine is encoded only by TGC and TGT. While this approach also eliminated certain amino acids, e.g., Phe, Tyr, His, Ile, Asn and Asp, as possible substitutions at the mutagenized positions, it generated a library of 256 combinations of codons encoding 169 combinations of amino acids at the two positions. Following amplification, DNA was digested with DpnI for 1.5 hr, subjected to gel purification as described above, and transformed into E. coli. Approximately one thousand colonies were pooled, and sequencing of the resulting cfnt2-Δ9Cys mutant DNA library showed only a trace of the wild-type “C” at the third position of both randomized codons (data not shown), similar to the result obtained above. The cfnt2-Δ9Cys library was therefore effectively free of plasmids encoding cysteine at the two positions of interest and provided moderate coverage of all possible combinations of other codons at these positions.
To select for mutant cfnt2 genes that lacked cysteines at positions 158 and 161 but retained inosine transport function, the cfnt2-Δ9Cys library was transformed into ade2 yeast. Approximately 100,000 colony-forming units were plated onto media containing 100 μM inosine as the sole purine source, providing excellent coverage of the bacterial library. Ninety-six fast-growing colonies were picked and retested versus controls by streaking on 100 μM inosine media, and ultimately the 27 fastest-growing positive clones were chosen for plasmid sequencing. As shown in Table 1, none of these 27 cfnt2 plasmids retained the wild-type Cys, indicating that template DNA was present in only a very low percentage of the positive clones obtained. No positive clones contained Ala and few contained Ser, as expected (Fig. 1A). The sequenced mutations were instead almost entirely dominated by Val, Thr, and Met substitutions.
Table 1.
Sequences of functional cfnt2-Δ9Cys clones genetically selected for changes at amino acids 158 and 161.
| Cys158 | Cys161 | Number of clones |
|---|---|---|
| Val | Thr | 12 |
| Val | Ser | 4 |
| Thr | Met | 4 |
| Thr | Thr | 2 |
| Met | Met | 2 |
| Val | Met | 1 |
| Met | Leu | 1 |
| Thr | Val | 1 |
Serial mutagenesis of multiple codons is efficient only when gel purification is used
Because the introduction of the gel-purification step into the Quikchange procedure greatly decreased the amount of template plasmid contaminating the yeast clones, we tested whether four site-saturation mutations could be introduced serially prior to selection with a low background of unmutated clones, allowing functional selection on multiple positions in one step. Two libraries were generated: for the first, product DNA was purified on a spin-column after each Quikchange reaction, and for the second, the product DNA at each step was subjected to gel electrophoresis in the presence of 1 mM guanosine [24] followed by gel purification of the amplified band as described above.
Quikchange mutagenesis at positions 158 and 161, and the products were subjected to gel or spin column purification. Bacterial transformants from each library were pooled, and plasmid DNA collected. Each of the resulting DNA libraries was used as template in a second Quikchange reaction in which the codon for Cys428 was mutagenized to “NNG”. Finally, position 461 was mutagenized as described earlier. Pooled DNA was used to transform ade2 S. cerevisiae, and plasmid DNA was collected from clones that grew on inosine as the purine source.
Of the 18 clones sequenced from the spin-column purification library, only one had mutations at all three of the desired codons (Table 2), and several with wild-type codons at two or all positions were recovered. In contrast, eight of eighteen clones derived from the gel-purification library were mutant at all three positions, and nearly all others were wild-type at only one codon. The gel-purified library provided many more informative clones for the 158/161 two-codon mutant and at codon 461. Genetic selection for mutations at each of these two positions showed that only a limited number of non-cysteine codons can encode a functional transporter, and therefore the library that contained less contaminating DNA would be less likely to give rise to false positive clones expressing highly functional cysteine-containing variants. The data demonstrate that the inclusion of a gel-purification step in the Quikchange reaction after DpnI digestion greatly increased the efficiency of recovering these difficult to obtain mutagenized clones (Table 2). Both libraries performed equally well at producing non-cysteine variants of residue 428, a case in which many different amino acids could evidently give rise to a functional transporter (Table 2, 13 of 18 clones sequenced).
Table 2.
Sequencing results of serial site-saturation mutagenesis followed by selection for functional complementation in yeast.
| Spin-column purification library | Gel-purification library | ||||
|---|---|---|---|---|---|
| 158/161 | 428 | 461 | 158/161 | 428 | 461 |
| V/V | S | S | V/T | I | S |
|
|
|||||
| C/C | Y | S | T/V | V | P |
| C/C | V | S | V/G | T | S |
| C/C | S | P | V/V | P | P |
| C/C | H | S | V/M | A | P |
| C/C | Y | S | L/T | S | S |
| C/C | V | S | M/S | L | P |
| C/C | L | S | M/G | H | S |
|
|
|||||
| V/S | C | S | T/C | A | S |
| V/S | A | C | T/C | L | P |
| A/L | Y | C | C/C | V | A |
| T/G | M | C | C/C | S | P |
|
|
|||||
| C/C | C | P | L/G | C | P |
| C/C | P | C | A/T | C | S |
| C/C | P | C | T/S | C | P |
| C/C | A | C | V/M | C | N |
| C/C | Y | C | V/M | T | C |
|
| |||||
| C/C | C | C | C/C | C | P |
Cysteine-less cfnt2 mutant proteins retain high affinity for inosine in both yeast and protozoan expression systems
Three completely cysteine-less cfnt2 mutants were constructed based on the results of the screening – cfnt2-Δ10Cys-C158V,C161T,C461S (VTS), cfnt2-Δ10Cys-C158V,C161T,C461P (VTP), and cfnt2-Δ10Cys-C158T,C161T,C461P (TTP). Each was expressed in ade2 yeast and characterized to determine if transporter function was retained. Initial rates of inosine uptake by the cysteine-less mutants were similar to those of CfNT2 and cfnt2-Δ7Cys (Fig. 4A). The cfnt2-Δ10Cys (VTS) mutant had consistently higher rates of uptake than cfnt2-Δ7Cys in these experiments and exhibited superior growth on inosine as well (Fig. 1A). This result may be due to increased stability of the cfnt2-Δ10Cys (VTS) mutant protein within yeast, resulting in higher amounts of transporter at the plasma membrane. The affinity of the cfnt2-Δ10Cys (VTS) mutant for inosine was determined by Michaelis-Menten analysis in yeast and compared favorably with wild-type CfNT2 (Fig. 4B & C). The apparent Km for cfnt2-Δ10Cys (VTS) was 1.66 ± 0.23 μM (n = 4), while that of CfNT2 was 1.16 ± 0.08 μM (n = 2). The cfnt2-Δ10Cys (VTP) and (TTP) mutant proteins exhibited similarly high affinity for inosine in yeast with apparent Km values of approximately 2 μM (data not shown).
Figure 4.
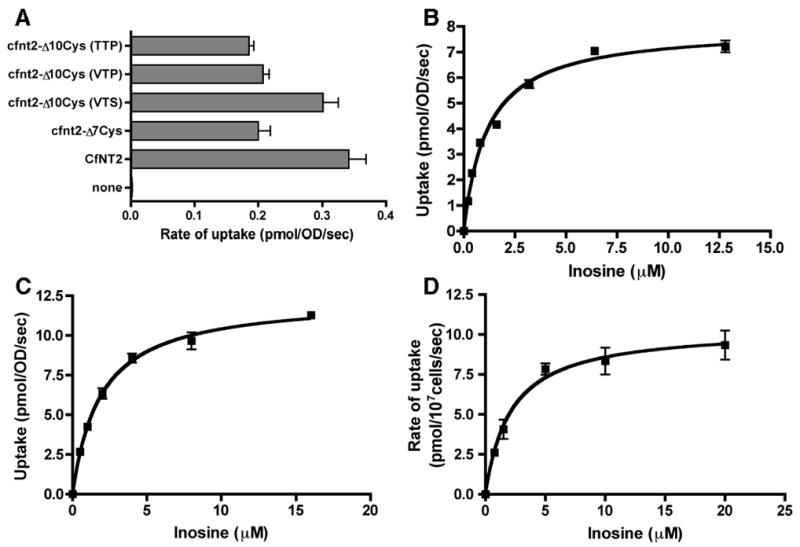
Biochemical characterization of functional cysteine-less cfnt2 variants. (A) Uptake of 5μM [3H]-inosine by yeast cells containing the indicated proteins was assayed in duplicate over a 30 sec time course, and linear regression was used to determine the initial rate. VTS: C158V, C161T, C461S; VTP: C158V, C161T, C461P; TTP: C158T, C161T, C461P. (B) Uptake of [3H]-inosine at various concentrations by yeast expressing CfNT2 was determined for triplicate 5 sec time points. (C) Uptake of [3H]-inosine at various concentrations by yeast expressing cfnt2- Δ10Cys (VTS) was determined for triplicate 5 sec time points. (D) Linear initial uptake rates at varying concentrations of [3H]-inosine were determined for Δldnt1/Δldnt2 L. donovani cells expressing cfnt2-Δ10Cys (VTS).
The cysteine-less cfnt2-Δ10Cys (VTS) derivative also displayed high affinity for inosine in L. donovani, a protozoan parasite phylogenetically similar to C. fasciculata. When expressed in Δldnt1/Δldnt2 L. donovani cells null for inosine transport activity[19], the cfnt2-Δ10Cys (VTS) protein demonstrated affinity for inosine (Km = 2.2 ± 0.18 μM, n = 2; Fig. 4D) similar to that reported for CfNT2 in L. donovani (~ 1 μM, [23]).
Discussion
A plethora of powerful biochemical techniques exist for measuring the structure and function of membrane proteins, and many of these require the site-specific derivatization of the protein, often via a thiol linkage to a unique cysteine residue. The value of a cysteine-less variant of such a protein, therefore, cannot be understated. However, a functional cysteine-less mutant retaining wild-type affinity and activity may not be produced using alanine, serine or other substitutions suggested by sequence alignment with related proteins. In such cases, genetic selection using a site-saturation library that excludes the wild-type codon can be an efficient way to identify substitutions that allow retention of wild-type function. Application of this approach to the CfNT2 purine nucleoside transporter resulted in the creation of cysteine-less cfnt2 variants that retained wild-type levels of inosine affinity in both yeast and L. donovani, validating the approach.
The Quikchange method provides a simple way to produce site-saturation libraries using a double-stranded template and standard molecular biological reagents and methods. However, using primers that were exact reverse complements, as the standard protocol dictates, made it nearly impossible to create a library containing a majority of non-aberrant mutants. Using primers that were not completely complementary [15] provided good coverage of all possible variants at the desired position, and decreased the number of aberrant plasmids obtained, but did not completely remove wild-type template plasmids from the reaction products, thereby complicating the use of the libraries for the positive genetic selection of functional mutants. However, modification of the Quikchange protocol to include a gel-purification step prior to bacterial transformation allowed the production of high quality plasmid libraries essentially free of unmutated template DNA and provided good coverage of all possible variants at the desired position, two conditions required for the success of this approach. This modification significantly increased the efficiency of the positive selection method, as nearly all yeast clones selected contained mutant plasmids, and fewer clones had to be sequenced to obtain meaningful information.
The unbiased nature of the mutagenesis and selection method was evident in the results obtained in both selections performed. For instance, while serine substitutes well for cysteine in many cases due to the similarity in size and chemistry of the two amino acids, selection on the codon461 library also resulted in functional variants containing proline, phenylalanine and tyrosine residues, substitutions that would not otherwise have been intuited or attempted. Similarly, simultaneous selection for two mutations (at positions 158 and 161) was successful and yielded unexpected functional substitutions –Val, Thr and Met, among others (Table 1). This result may provide information about the environment of these two positions in the protein. All three amino acids are larger than Cys in van der Waal’s radius [30], and Val and Thr have a poorer helical propensity relative to Met and Cys [31]. In contrast, Ala and Ser are smaller and have a greater predilection to form helices than Cys. Cys158 and 161 are predicted to lie near or beyond the intracellular end of TM4 in CfNT2 [23], and the result that Val and Thr were more effective alterations than Ala and Ser at these positions may suggest that Cys158 and 161 are in fact not part of this helix.
When a systematic testing of all amino acid substitutions for a desired amino acid (Cys, Trp, etc.) is not necessary, and substitution with chemically similar amino acids fails to preserve function (e.g., Ala and Ser for Cys), genetic selection for functional mutants at the site of interest may save time and resources. This method can be especially advantageous when multiple neighboring residues are to be changed. In the second test case described above, the two cysteine residues to be substituted were located within one helical turn of one another. This proximity raised the possibility that mutation of one cysteine might influence the residues that could substitute at the other, and therefore a simultaneous search for functional variants at both positions was ideal. Using systematic site-directed mutagenesis would have required up to 192 primer pairs and associated molecular biological manipulations to construct and assay all possible combinations of non-cysteine amino acids at the two sites. Instead, our site-saturation mutagenesis and selection method allowed nearly all combinations of two amino acids to be sampled in a single, simple growth assay. Indeed the total time required from the beginning of the Quikchange reaction to the receipt of sequences from the selected yeast clones was as little as two weeks, with a minimum of hands-on time. Inclusion of a gel purification step greatly increased the efficiency of obtaining non-cysteine-containing variants, especially at positions with few functional amino acid possibilities, and even allowed the selection of mutants in which four mutations were introduced serially. The low cost and robustness of this method recommend its use in the generation of all manner of function- or structure-conserving amino acid variants of any protein for which an appropriate selection or screen can be devised.
Acknowledgments
We wish to thank Jonathan Galazka for help in the development of the mutagenesis protocol, and Svetlana Lutsenko and members of the Ullman laboratory for comments on the manuscript. This work was supported in part by grants RO1 AI23682, AI51507 and AI44138 from the National Institutes of Health (BU) and a postdoctoral grant (PF-02-097-01-CSM) from the American Cancer Society (CSA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fleishman SJ, Unger VM, Ben-Tal N. Transmembrane protein structures without X-rays. Trends Biochem Sci. 2006;31:106–13. doi: 10.1016/j.tibs.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 2.Frillingos S, Kaback HR. The role of helix VIII in the lactose permease of Escherichia coli: II. Site-directed sulfhydryl modification. Protein Sci. 1997;6:438–443. doi: 10.1002/pro.5560060221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karlin A, Akabas MH. Substituted-cysteine accessibility method. Methods Enzymol. 1998;293:123–145. doi: 10.1016/s0076-6879(98)93011-7. [DOI] [PubMed] [Google Scholar]
- 4.Bogdanov M, Zhang W, Xie J, Dowhan W. Transmembrane protein topology mapping by the substituted cysteine accessibility method (SCAM™): Application to lipid-specific membrane protein topogenesis. Methods. 2005;36:148–171. doi: 10.1016/j.ymeth.2004.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Columbus L, Hubbell WL. A new spin on protein dynamics. Trends Biochem Sci. 2002;27:288–95. doi: 10.1016/s0968-0004(02)02095-9. [DOI] [PubMed] [Google Scholar]
- 6.Johnson AE. Fluorescence approaches for determining protein conformations, interactions and mechanisms at membranes. Traffic. 2005;6:1078–92. doi: 10.1111/j.1600-0854.2005.00340.x. [DOI] [PubMed] [Google Scholar]
- 7.Sorgen PL, Hu Y, Guan L, Kaback HR, Girvin ME. An approach to membrane protein structure without crystals. Proc Natl Acad Sci U S A. 2002;99:14037–40. doi: 10.1073/pnas.182552199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leighton BH, Seal RP, Watts SD, Skyba MO, Amara SG. Structural rearrangements at the translocation pore of the human glutamate transporter, EAAT1. J Biol Chem. 2006;281:29788–96. doi: 10.1074/jbc.M604991200. [DOI] [PubMed] [Google Scholar]
- 9.Loo TW, Clarke DM. Membrane topology of a cysteine-less mutant of human P-glycoprotein. J Biol Chem. 1995;270:843–8. doi: 10.1074/jbc.270.2.843. [DOI] [PubMed] [Google Scholar]
- 10.Valdes R, Vasudevan G, Conklin D, Landfear SM. Transmembrane domain 5 of the LdNT1.1 nucleoside transporter is an amphipathic helix that forms part of the nucleoside translocation pathway. Biochemistry. 2004;43:6793–802. doi: 10.1021/bi049873m. [DOI] [PubMed] [Google Scholar]
- 11.Zhang J, Tackaberry T, Ritzel MW, Raborn T, Barron G, Baldwin SA, Young JD, Cass CE. Cysteine-accessibility analysis of transmembrane domains 11–13 of human concentrative nucleoside transporter 3. Biochem J. 2006;394:389–98. doi: 10.1042/BJ20051476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaback HR. Molecular biology of active transport: from membrane to molecule to mechanism. Harvey Lect. 1987;83:77–105. [PubMed] [Google Scholar]
- 13.Menick DR, Sarkar HK, Poonian MS, Kaback HR. Cys154 is important for lac permease activity in Escherichia coli. Biochem Biophys Res Commun. 1985;132:162–70. doi: 10.1016/0006-291x(85)91002-2. [DOI] [PubMed] [Google Scholar]
- 14.Cui L, Aleksandrov L, Hou YX, Gentzsch M, Chen JH, Riordan JR, Aleksandrov AA. The role of cystic fibrosis transmembrane conductance regulator phenylalanine 508 side chain in ion channel gating. J Physiol. 2006;572:347–58. doi: 10.1113/jphysiol.2005.099457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng L, Baumann U, Reymond JL. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 2004;32:e115. doi: 10.1093/nar/gnh110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current Protocols in Molecular Biology. John Wiley & Sons, Inc; New York, NY: 1993. [Google Scholar]
- 17.Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gietz RD, Woods RA. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002;350:87–96. doi: 10.1016/s0076-6879(02)50957-5. [DOI] [PubMed] [Google Scholar]
- 19.Liu W, Boitz JM, Galazka J, Arendt CS, Carter NS, Ullman B. Functional characterization of nucleoside transporter gene replacements in Leishmania donovani. Mol Biochem Parasitol. 2006;150:300–7. doi: 10.1016/j.molbiopara.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robinson KA, Beverley SM. Improvements in transfection efficiency and tests of RNA interference (RNAi) approaches in the protozoan parasite Leishmania. Mol Biochem Parasitol. 2003;128:217–28. doi: 10.1016/s0166-6851(03)00079-3. [DOI] [PubMed] [Google Scholar]
- 21.Goyard S, Segawa H, Gordon J, Showalter M, Duncan R, Turco SJ, Beverley SM. An in vitro system for developmental and genetic studies of Leishmania donovani phosphoglycans. Mol Biochem Parasitol. 2003;130:31–42. doi: 10.1016/s0166-6851(03)00142-7. [DOI] [PubMed] [Google Scholar]
- 22.Labbe S, Thiele DJ. Copper ion inducible and repressible promoter systems in yeast. Methods Enzymol. 1999;306:145–153. doi: 10.1016/s0076-6879(99)06010-3. [DOI] [PubMed] [Google Scholar]
- 23.Liu W, Arendt CS, Gessford SK, Ntaba D, Carter NS, Ullman B. Identification and characterization of purine nucleoside transporters from Crithidia fasciculata. Mol Biochem Parasitol. 2005;140:1–12. doi: 10.1016/j.molbiopara.2004.11.018. [DOI] [PubMed] [Google Scholar]
- 24.Grundemann D, Schomig E. Protection of DNA during preparative agarose gel electrophoresis against damage induced by ultraviolet light. Biotechniques. 1996;21:898–903. doi: 10.2144/96215rr02. [DOI] [PubMed] [Google Scholar]
- 25.Aronow B, Kaur K, McCartan K, Ullman B. Two high affinity nucleoside transporters from Leishmania donovani. Mol Biochem Parasitol. 1987;22:29–37. doi: 10.1016/0166-6851(87)90066-1. [DOI] [PubMed] [Google Scholar]
- 26.Laban A, Tobin JF, Curotto de Lafaille MA, Wirth DF. Stable expression of the bacterial neor gene in Leishmania enriettii. Nature. 1990;343:572–4. doi: 10.1038/343572a0. [DOI] [PubMed] [Google Scholar]
- 27.Arastu-Kapur S, Arendt CS, Purnat T, Carter NS, Ullman B. Second-site suppression of a non-functional mutation within the Leishmania donovani inosine-guanosine transporter. J Biol Chem. 2005;280:2213–2219. doi: 10.1074/jbc.M408224200. [DOI] [PubMed] [Google Scholar]
- 28.Endres CJ, SenGupta DJ, Unadkat JD. Mutation of leucine-92 selectively reduces the apparent affinity of inosine, guanosine, NBMPR [S6-(4-nitrobenzyl)-mercaptopurine riboside] and dilazep for the human equilibrative nucleoside transporter, hENT1. Biochem J. 2004;380:131–138. doi: 10.1042/BJ20031880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maeser P, Suetterlin C, Kralli A, Kaminsky R. A nucleoside transporter from Trypanosoma brucei involved in drug resistance. Science. 1999;285:242–244. doi: 10.1126/science.285.5425.242. [DOI] [PubMed] [Google Scholar]
- 30.Richards FM. The interpretation of protein structures: total volume, group volume distributions and packing density. J Mol Biol. 1974;82:1–14. doi: 10.1016/0022-2836(74)90570-1. [DOI] [PubMed] [Google Scholar]
- 31.O’Neil KT, DeGrado WF. A thermodynamic scale for the helix-forming tendencies of the commonly occurring amino acids. Science. 1990;250:646–51. doi: 10.1126/science.2237415. [DOI] [PubMed] [Google Scholar]