

Abstract
The microsomal enzyme acyl-CoA:cholesterol acyltransferase (ACAT; EC 2.3.1.26) catalyzes the esterification of cellular cholesterol with fatty acids to form cholesterol esters. ACAT activity is found in many tissues, including macrophages, the adrenal glands, and the liver. In macrophages, ACAT is thought to participate in foam cell formation and thereby to contribute to atherosclerotic lesion development. Disruption of the gene for ACAT (Acact) in mice resulted in decreased cholesterol esterification in ACAT-deficient fibroblasts and adrenal membranes, and markedly reduced cholesterol ester levels in adrenal glands and peritoneal macrophages; the latter finding will be useful in testing the role of ACAT and macrophage foam cell formation in atherosclerosis. In contrast, the livers of ACAT-deficient mice contained substantial amounts of cholesterol esters and exhibited no reduction in cholesterol esterification activity. These tissue-specific reductions in cholesterol esterification provide evidence that in mammals this process involves more than one form of esterification enzyme.
Keywords: gene targeting, adrenal gland, macrophage, cholesterol ester
Cells of multicellular organisms are protected from excess free cholesterol toxicity in part by acyl-CoA:cholesterol acyltransferase (ACAT; EC 2.3.1.26), a microsomal enzyme that esterifies cholesterol with fatty acids to form cholesterol esters, which can then be stored in cytosolic droplets (1, 2, 3, 4). This process occurs in many tissues but is prominent in macrophages of atherosclerotic lesions, where large amounts of cholesterol from lipoproteins are taken up by receptor-mediated endocytosis (5), and in adrenocortical cells, where cholesterol to be used as substrate for steroidogenesis is stored as cytosolic cholesterol ester droplets. In macrophages, the esterification of cholesterol by ACAT gives rise to macrophage foam cells, a prominent feature of atherosclerotic lesions. Because of ACAT’s role in foam cell formation and its potential roles in intestinal cholesterol absorption (6) and hepatic lipoprotein synthesis (7), pharmaceutical inhibitors of ACAT have received considerable interest as possible therapies for atherosclerosis; although these agents have shown promise (8), their use in animals has been associated with adrenal gland toxicity (9, 10).
The recent cloning of a cDNA for human ACAT by Chang et al. (11) has facilitated a molecular genetic approach to determining ACAT’s in vivo functions. In this study, we used gene targeting in mouse embryonic stem (ES) cells to generate mice deficient in ACAT. The ACAT-deficient mice had tissue-specific reductions in cholesterol esterification, implying the existence either of multiple ACAT isoforms or of another not-yet-identified cholesterol esterification gene(s).
MATERIALS AND METHODS
Gene Targeting.
Degenerate primers derived from the human ACAT cDNA sequence (11) were used to amplify a 310-bp mouse cDNA fragment. Using this fragment as a probe, a 14-kb 129/Sv genomic λ clone was isolated, and sequencing of this clone identified a 5′ exon [amino acids 50–100 (12)]. To construct a targeting vector, a 700-bp fragment that terminated at amino acid 63 was amplified and cloned into the XhoI site, and a 9-kb downstream BamHI genomic fragment was ligated into the BamHI site of pPol2short-neobpA-HSVTK (13) (provided by S. Ishibashi, University of Texas Southwestern Medical School).
ES cells (RF8) were derived from 129/TerSv mice on feeder layers of SNL76/7 fibroblasts (provided by A. Bradley, Baylor College of Medicine, Houston) as described (14). Culture and electroporation of ES cells were as described (14, 15). For Southern blot analysis, ES cell genomic DNA was digested with EcoRI, and a ≈1-kb HindIII–SacI restriction fragment located upstream of the vector sequences was used as a probe. For PCR genotyping, genomic DNA was amplified with primer A (5′-CAGCTATGTACACACATATATTCATG-3′), which is located upstream of the targeting vector, primer B (5′-TTGGGAAGACAATAGCAGGCATGC-3′), which is located in the polyadenylylation signal of the neo gene (833-bp product, targeted allele), and primer C (5′-GGACTTTTCAATGAGGTTGGTCAC-3′), which is unique to the deleted region of the wild-type allele (790-bp fragment, wild-type allele). PCR conditions were 35 cycles, each of 30 sec at 96°C, 1 min at 50°C, and 2 min at 72°C. Microinjection of targeted clones and chimera generation were as described (15). The mice described herein were of a mixed C57BL/6 and 129/Sv genetic background. Mice were housed in a pathogen-free barrier facility operating on a 12-h light/12-h dark cycle and were fed either rodent chow (Ralston Purina) or a synthetic diet (ICN) containing 15% fat and 1.25% cholesterol (16).
For Northern blotting, RNA was prepared from mouse tissues (17) and analyzed by standard techniques. Blots were probed with a 32P-labeled 501-bp ACAT cDNA fragment [nucleotides 804–1304 (12)] or an end-labeled 28S primer (18) and were exposed to x-ray film overnight. For immunoblotting, tissue homogenates were prepared as described (19), and proteins were separated by SDS/PAGE on 4–20% gradient gels; immunoblots were probed with polyclonal antibody DM10 (19) (generously provided by T. Y. Chang, Dartmouth University, Hanover, NH) and detected with an enhanced chemiluminescence system (Amersham).
Cholesterol Esterification Assays.
Murine embryonic fibroblast lines were generated (20), and fibroblasts (passages 3–5) were pulse-assayed for cholesterol esterification as described (21). For microsomal membrane assays, tissues were homogenized in buffer A (50 mM Tris·HCl, pH 7.8/1 mM EDTA/1 mM phenylmethylsulfonyl fluoride) with a Polytron homogenizer (Brinkmann, Westbury, NY); the microsomal membrane fractions (100,000 × g pellet) were isolated by sequential centrifugation. For the adrenal gland assays, total membrane homogenates (excluding nuclei) were used. The rate of incorporation of [14C]oleoyl-CoA (Amersham) into cholesterol esters was assayed essentially as described by Erickson et al. (22). Reactions were performed at 37°C for 5 min with 100–150 μg of protein homogenate and 5 nM oleoyl CoA (specific activity, ≈18 μCi/μmol; 1 Ci = 37 GBq). In some experiments, exogenous cholesterol (20 nmol) was added to the reaction by preincubating membranes for 30 min at 37°C with phosphatidylcholine/cholesterol (4:1 molar ratio) liposomes (23). CI-976 (a gift from R. Newton, Parke-Davis) was dissolved in dimethyl sulfoxide; equivalent amounts of dimethyl sulfoxide were added to control samples.
Plasma and Tissue Lipid Analysis.
Total cholesterol, high density lipoprotein (HDL) cholesterol, and triglycerides were determined by colorimetric assays (24). Peritoneal macrophages were isolated as described (25). Macrophages were cultured in serum-free Dulbecco’s minimal essential medium containing 50 μg/ml acetylated low density lipoproteins (LDL) (26). For tissue cholesterol and cholesterol ester determinations, lipids were extracted from tissue homogenates by the Bligh–Dyer method (27) and separated by thin-layer chromatography; extracted lipids were dissolved in isopropanol and aliquots were measured for cholesterol content using a colorimetric assay (Boehringer Mannheim). High-performance thin-layer chromatography (HPTLC) was as described (28).
Histologic Analysis.
Perfusion-fixed (3% paraformaldehyde in Tyrode’s solution) adrenal glands were frozen, sectioned with a vibratome, and stained with Oil red O (29).
Adrenocorticotropic Hormone (ACTH) Stimulation Tests.
ACTH stimulation tests were performed on 8- to 12-week-old male mice beginning at 9:00 a.m. (2 h into the light cycle). Mice were anesthetized with tribromoethyl alcohol, a baseline plasma sample was obtained, and mice were injected intraperitoneally with 1 unit of β1-24 ACTH (Cortrosyn, Organon). After 1 h, the anesthetized mice were killed, and blood was collected. Corticosterone was measured by a competitive protein-binding assay (30) after HPTLC (31).
Cholesterol Absorption.
Cholesterol absorption was measured with a fecal isotope ratio method that uses [14C]cholesterol and [3H]sitostanol (32).
Statistics.
Data are given as the mean ± SD. The t test, analysis of variance, and the Student Newman–Keuls test were used to compare means between groups.
RESULTS
Acact Gene Disruption.
A sequence-replacement vector using a positive-negative selection strategy (33) was used to disrupt the gene for ACAT (Acact) (Fig. 1A) in mouse ES cells and subsequently generate mice homozygous for the gene disruption (Acact−/−) (Fig. 1B). The Acact−/− mice were healthy, fertile, and grew normally. Northern blot analysis of RNA from preputial glands, which express high levels of ACAT (12), revealed that Acact−/− mice had low levels of an ACAT mRNA that was slightly smaller than the wild-type mRNA (Fig. 1C). Sequencing of reverse transcription–PCR products from Acact−/− mRNA revealed that the truncated mRNA arises from alternative splicing around the inserted neo gene, resulting in a frameshift and a premature stop codon (see legend to Fig. 1). Immunoblotting with an antibody (19) that detects the amino-terminal region of ACAT demonstrated no detectable ACAT protein in homogenates of preputial glands of Acact−/− mice (Fig. 1D) or in homogenates from ovaries and adrenals (not shown), which also express high levels of ACAT. No distinct band of the appropriate size for ACAT was found in liver homogenates of either wild-type or Acact−/− mice. The effect of the ACAT gene disruption on ACAT activity was initially assessed by measuring cholesterol esterification in embryonic fibroblasts (Fig. 1E). In contrast to that in wild-type fibroblasts, esterification activity in Acact−/− fibroblasts was not stimulated in response to cholesterol provided by human LDL; esterification activity was intermediate in heterozygous fibroblasts. In addition, esterification activity in Acact−/− fibroblasts was not stimulated by 25-hydroxycholesterol (not shown). In the Acact−/− fibroblasts, residual esterification activity was found, which decreased upon the addition of LDL to low but detectable levels (<10% versus wild-type) in most experiments.
Figure 1.
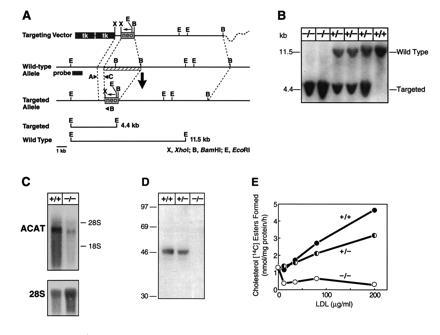
Generation of Acact−/− mice. (A) Targeting vector strategy. Homologous recombination of the vector results in the interruption of a 5′ early exon (amino acids 50–100) of ACAT by the neo gene (at amino acid 63 of 540) and causes a ≈2-kb deletion of genomic sequences (striped bar). Targeted clones were identified by a unique 4.4-kb EcoRI restriction fragment as detected by a ≈1-kb HindIII–SacI probe located upstream of the vector or by PCR analysis. Targeting frequency was 1 in 268. (B) Southern blot demonstrating Acact−/− mice. Of 226 offspring from heterozygous matings, 66 were wild-type mice, 104 were heterozygotes, and 56 were homozygotes (P = 0.573 by χ2 analysis versus the expected 1:2:1 Mendelian distribution). (C) Northern blot of preputial gland RNA in Acact−/− mice. Blots were probed with a mouse ACAT cDNA fragment and reprobed with a primer specific for 28S RNA to control for RNA sample loading. Reverse transcription–PCR analysis (not shown) revealed that the shorter ACAT mRNA in Acact−/− mice was caused by exon skipping around the inserted exon containing the neo mutation and the following exon, which was deleted in the targeting vector. The skipping of these two exons generates a 211-bp deletion and a frameshift resulting in a premature stop codon. (D) Immunoblot demonstrating lack of ACAT protein in preputial gland homogenates from Acact−/− mice. Each lane contains 100 μg of protein; membranes were incubated with DM10 (19). As a control, the same homogenates were tested for the LDL receptor–related protein, which was detectable at approximately equivalent amounts in each sample (not shown). (E) Cholesterol esterification activity in embryonic fibroblasts in response to LDL. Pulse assays were performed as described (21) on fibroblasts derived from Acact+/+, Acact+/−, or Acact−/− embryos. The experiment was repeated three times; data from a representative experiment are shown.
Cholesterol Ester Depletion in the Adrenal Glands and Macrophages.
In contrast to the opaque, whitish-yellow appearance of adrenals from wild-type mice (Fig. 2A), the adrenal glands of Acact−/− mice were translucent and brownish, suggesting the depletion of cholesterol esters. Staining with Oil red O demonstrated abundant lipid staining of the adrenal cortex in sections from wild-type mice, no lipid staining in Acact−/− mice, and intermediate lipid staining in heterozygous mice (Fig. 2B). Tissue lipid analysis revealed that the cholesterol ester content of adrenal glands was markedly reduced in the Acact−/− mice (Table 1). Additionally, adrenal cholesterol esterification activity was markedly lower in Acact−/− than in wild-type mice (40 ± 16 versus 296 ± 42; P < 0.01) (see Fig. 4A).
Figure 2.
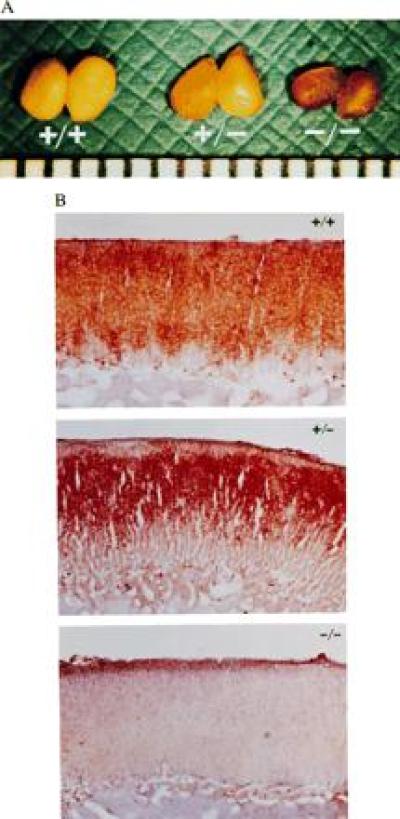
Adrenal lipid depletion in Acact−/− mice. (A) Gross appearance of adrenal glands from male Acact+/+, Acact+/−, and Acact−/− mice. The scale at the bottom of the photograph is in millimeters. (B) Oil red O-stained sections showing absence of neutral lipids in adrenal cortex of male Acact−/− mice. Identical findings were observed in female mice. Staining in the heterozygous mice was consistently confined to a smaller area of the zona fasciculata.
Table 1.
Cholesterol ester and free cholesterol content of tissues
| Tissue | Acact genotype | n | Lipid content, μg/mg
protein
|
|
|---|---|---|---|---|
| CE | FC | |||
| Adrenal | +/+ | 3 | 105.4 ± 9.7 | 11.5 ± 2.7 |
| +/− | 3 | 53.9 ± 5.1† | 10.3 ± 2.3 | |
| −/− | 3 | 3.3 ± 0.6† | 7.1 ± 0.6 | |
| Liver | +/+ | 5 | 2.7 ± 0.1 | 9.5 ± 1.7 |
| Chow diet | −/− | 5 | 1.9 ± 0.5† | 8.7 ± 2.5 |
| Liver | +/+ | 3 | 346.0 ± 119 | 30.5 ± 8.3 |
| High fat diet | −/− | 3 | 350.0 ± 42 | 31.0 ± 4.3 |
| Peritoneal | +/+ | 2* | 130.0 ± 33 | 36.1 ± 1.2 |
| macrophages | −/− | 3* | 7.9 ± 4.8† | 72.3 ± 23.8‡ |
Data are x̄ ± SD. Data are from female mice except for peritoneal macrophages, which were obtained from male mice. CE, cholesterol ester; FC, free cholesterol.
Each sample represents pooled lipid extracts of macrophages from four mice.
P < 0.01 vs. +/+.
P = 0.13 vs. +/+.
Figure 4.
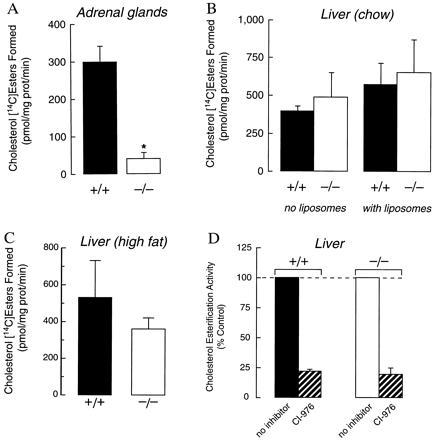
Cholesterol esterification activity in adrenal membranes and hepatic microsomes of wild-type and Acact−/− mice. (A) Adrenal membrane activity in female mice. Assays were performed with 100 μg of protein as described in Materials and Methods. Results are shown for three samples for each genotype; each sample contained pooled adrenals from two mice. Exogenous cholesterol (20 nmol) in the form of liposomes was added during the preincubation. ∗, P < 0.01 versus Acact+/+. (B) Hepatic membrane activity of female mice fed a chow diet. Assays were performed with 150 μg of protein in the presence or absence of 20 nmol of exogenous cholesterol provided as liposomes during preincubation (Acact+/+, n = 3; Acact−/−, n = 3). (C) Hepatic membrane activity of mice fed a high-fat, high-cholesterol diet. Assays were performed with 150 μg of protein without added cholesterol (Acact+/+, n = 3; Acact−/−, n = 3). P = 0.20 for the difference between means. (D) Effects of the ACAT inhibitor CI-976 (10 μg/ml) on cholesterol esterification activity of hepatic microsomes from chow-fed mice. Assays were performed after preincubation with exogenous cholesterol.
To determine if the adrenal cholesterol ester depletion in Acact−/− mice resulted in diminished glucocorticoid production in response to an acute stimulus, we measured serum corticosterone levels before and 1 h after ACTH stimulation in male wild-type and Acact−/− mice. We observed no significant differences in either baseline (Acact+/+, 16.9 ± 3.1 μg/dl, n = 6; Acact−/−, 17.7 ± 13.1 μg/dl, n = 5) or poststimulation (Acact+/+, 33.8 ± 4.0 μg/dl, n = 6; Acact−/−, 33.8 ± 8.1 μg/dl, n = 8) levels between the groups.
The effects of ACAT deficiency on macrophage foam cell formation were assessed by culturing peritoneal macrophages with acetylated LDL. HPTLC of lipid extracts demonstrated markedly lower cholesterol ester content in Acact−/− macrophages than in wild-type cells, although trace amounts were detectable (Fig. 3). Lipid quantitation confirmed the near absence of cholesterol esters in Acact−/− macrophages and demonstrated that the free cholesterol content tended to be higher in these cells (Table 1). Despite the marked reduction in cholesterol esters, neutral lipid droplets, presumably containing triglycerides, were observed in many of the Acact−/− macrophages as assessed by Nile red fluorescence (not shown).
Figure 3.
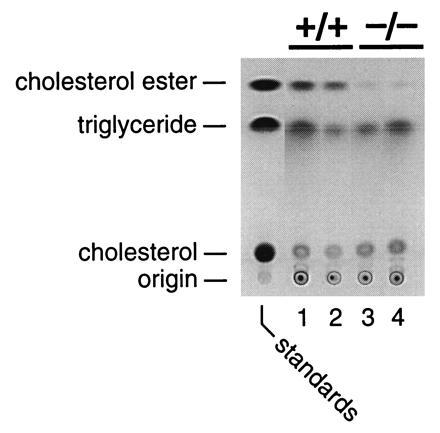
HPTLC of lipid extracts from wild-type or Acact−/− peritoneal macrophages demonstrating reduced macrophage cholesterol ester accumulation in Acact−/− mice. Macrophages were isolated from male mice and incubated with acetylated LDL (50 μg/ml) for 36 h. Lipid extracts were pooled and analyzed by HPTLC (28). Equivalent amounts of lipid extract (from ≈3 μg of protein homogenate) were loaded in each lane; each lane represents lipids from four mice.
Hepatic Cholesterol Ester Metabolism.
In contrast to the adrenal glands, the livers of Acact−/− mice contained substantial amounts of cholesterol esters (Table 1). Hepatic cholesterol ester levels were 30% lower in Acact−/− mice than in wild-type mice that were fed a chow diet but were similar in wild-type and Acact−/− mice fed a high-fat diet (Table 1). Also, cholesterol esterification in hepatic Acact−/− microsomes was not reduced compared with wild-type microsomes isolated from mice fed either chow (Fig. 4B) or a high-fat diet (Fig. 4C). The residual cholesterol esterification activity responded identically in Acact−/− and wild-type hepatic microsomes incubated with the ACAT inhibitor CI-976 (Fig. 4D).
Cholesterol Absorption and Plasma Lipid Levels.
Cholesterol absorption was not significantly different in Acact−/− (n = 9) and wild-type mice (n = 9) that consumed a chow diet (74 ± 15% versus 82 ± 9%, respectively; P = 0.20) or in wild-type (n = 2) and Acact−/− mice (n = 3) that consumed a high-fat diet (37% ± 0.7 and 41% ± 5, respectively). When fed a chow or high-fat diet, Acact−/− mice had higher plasma levels of total and HDL cholesterol than wild-type mice (Table 2). The increase in plasma HDL cholesterol in chow-fed mice was confirmed by measuring the cholesterol concentration of lipoproteins separated by gel filtration chromatography (not shown). Plasma triglyceride levels were not different in Acact−/− and wild-type mice.
Table 2.
Plasma lipids of wild-type and ACAT-deficient mice
| Diet | Acact genotype | n | Plasma lipid concentration,
mg/dl
|
||
|---|---|---|---|---|---|
| TC | HDL-C | TG | |||
| Chow* | +/+ | 8 | 57 ± 19 | 48 ± 12 | 60 ± 20 |
| +/− | 5 | 70 ± 19 | 52 ± 13 | 57 ± 7 | |
| −/− | 7 | 123 ± 29‡§ | 83 ± 19‡§ | 63 ± 22 | |
| High-fat† | +/+ | 8 | 170 ± 61 | 54 ± 18 | 35 ± 17 |
| +/− | 10 | 207 ± 56 | 66 ± 21 | 35 ± 12 | |
| −/− | 10 | 240 ± 48‡ | 89 ± 21‡§ | 29 ± 9 | |
Data are x̄ ± SD. TC, total cholesterol; HDL-C, HDL cholesterol; TG, triglycerides.
Female mice fed standard chow for 3–5 months.
Male and female mice fed a high-fat, high-cholesterol diet for 6–10 weeks.
P < 0.05 vs. +/+.
P < 0.05 vs. +/−.
DISCUSSION
In this study, the disruption of the 5′ region of Acact yielded mice with tissue-specific reductions in cholesterol esters and cholesterol esterification. In Acact−/− mice, cholesterol esters were markedly diminished in the adrenocortical cells and cultured peritoneal macrophages, and cholesterol esterification was markedly reduced in embryonic fibroblasts and adrenal membranes; however, Acact−/− livers contained substantial amounts of cholesterol esters, and hepatic cholesterol esterification was not diminished. In addition, the gene disruption had no detectable effect on intestinal cholesterol absorption, a process thought to involve a cholesterol esterification enzyme (6).
The adrenal lipid depletion phenotype in Acact−/− mice is similar to that in apolipoprotein (apo) AI knockout mice reported recently by Plump et al. (34). In apo-AI-deficient mice, the lack of apo-AI-containing HDL particles presumably prevents adrenocortical cell uptake of cholesterol esters via the “selective uptake” pathway (35), resulting in the depletion of the cytosolic cholesterol ester droplets. Our findings suggest that cholesterol molecules taken up by the selective uptake pathway as cholesterol esters are hydrolyzed and subsequently re-esterified by ACAT for storage as cytosolic cholesterol ester droplets. Interestingly, we (36) and others (12) recently mapped the mouse ACAT gene to a region of chromosome 1 that contains ald (37), a naturally occurring adrenal lipid depletion locus that occurs as an autosomal recessive trait in AKR inbred mice (38). Studies to determine if the ald phenotype is due to an ACAT mutation are in progress. To our knowledge, a similar adrenal lipid depletion disorder in humans has not been reported.
The dramatic cholesterol ester depletion in Acact−/− adrenocortical cells had no discernible effect on adrenal steroidogenesis in response to an acute ACTH stimulus in male Acact−/− mice. In apo-AI-deficient mice, plasma corticosterone responses to ACTH stimulation or swimming stress tests were blunted, especially in males (34). The different responses in the apo-AI-deficient mice and the Acact−/− mice may reflect different degrees of stress or stimulation; alternatively, the provision of cholesterol through the selective uptake pathway may be more critical for steroidogenesis than cytosolic cholesterol ester pools during acute stress. In both mouse models, however, adrenal glucocorticoid production responded remarkably well to acute stimulation despite the dramatic depletion of cholesterol esters in the cells.
Cholesterol esters were also decreased in cultured peritoneal macrophages from Acact−/− mice. When incubated with acetylated LDL, macrophages from the Acact−/− mice had a marked reduction in their ability to accumulate the cholesterol esters that are characteristic of macrophage foam cells in atherosclerotic lesions. It will be of interest to determine whether ACAT deficiency and reduced foam cell formation will protect against the development of atherosclerotic lesions. This could be assessed by breeding the Acact−/− mice with atherosclerosis-susceptible strains, such as apo-E-deficient (39, 40) or LDL receptor-deficient mice (41). With regard to atherosclerosis studies, it is noteworthy that the Acact−/− mice had increased plasma total and HDL cholesterol levels. The mechanism for this unexpected finding remains to be determined. The increased HDL cholesterol levels could be due to the effects of ACAT deficiency on cholesterol metabolism, but could also result from the effect of a strain 129-derived allele linked to the targeted locus [e.g. Ath-1 (42) affects plasma HDL levels and maps close to Acact].
In contrast to its effects on adrenal glands and macrophages, the gene disruption had little effect on hepatic cholesterol ester metabolism. Although the livers of Acact−/− mice fed a chow diet had a modest decrease in cholesterol ester content, this was not the case when the mice were fed a cholesterol-rich diet. Further, hepatic cholesterol esterification activity was not reduced in Acact−/− mice. These results are consistent with the low level of ACAT mRNA in hepatocytes (12, 43) and our inability to detect ACAT in immunoblots of mouse hepatic tissue. Taken together, these findings suggest that mouse liver contains a form of cholesterol esterification enzyme distinct from the enzyme we inactivated.
At least two possible explanations could account for an alternative cholesterol esterification enzyme. First, there may be another, not-yet-identified cholesterol esterification enzyme(s). Both biochemical (44) and ACAT inhibitor (45) data have suggested that there is more than one esterification enzyme in mammals, and Yang et al. (46) have recently identified two sterol esterification enzymes in yeast, both of which must be inactivated to abolish sterol esterification. Of note, both we and Yang et al. (46) have identified human cDNAs from expressed sequence tag data bases that are ACAT homologues and that share extensive regions of identity with ACAT in the carboxyl terminus. These cDNAs appear to be members of an ACAT gene family, but it remains to be determined if the proteins they encode can esterify cholesterol. Second, it is possible that a currently unrecognized alternative splicing of ACAT mRNA occurs in the liver, and that the gene disruption did not inactivate a liver-specific ACAT isoform. Although we and others (12) have no evidence supporting the encoding of different ACAT isoforms from the Acact locus, this remains a possibility.
In summary, our study provides evidence suggesting that more than one form of cholesterol esterification enzyme exists in mammals. Such a redundancy in the cellular cholesterol esterification pathway may have contributed to the healthy phenotype we observed in Acact−/− mice. Interestingly, the gene we disrupted was cloned from macrophages (11), and the two tissues in which we observed a phenotype—the adrenal gland and macrophages—both process large cholesterol loads and are characterized by large cytosolic cholesterol ester droplets. Insights into the molecular aspects of different cholesterol esterification enzymes could lead to the development of more selective cholesterol esterification enzyme inhibitors of specific processes, such as macrophage foam cell formation or hepatic lipoprotein production.
Acknowledgments
We thank T. Y. Chang for advice and for antibody DM10; Sandra Ruland and Dale Newland for technical assistance; Sandra Erickson and David Sanan for advice and methods; Shun Ishibashi for the neo-TK plasmid; Roger Newton and Parke-Davis for providing CI-976; Angela Chen for manuscript preparation; Amy Corder, Brian Clark, and John Carroll for graphics; and Gary Howard and Stephen Ordway for editorial assistance. Corticosterone measurements were performed in the Core Laboratory of the General Clinical Research Center at San Francisco General Hospital (RR-83). This work was funded by the Gladstone Institutes and a Grant-in-Aid (95-239) from the American Heart Association, California Affiliate.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: ACAT, acyl-CoA:cholesterol acyltransferase; ACTH, adrenocorticotropic hormone; ES, embryonic stem; apo, apolipoprotein; HDL, high density lipoprotein; LDL, low density lipoprotein.
References
- 1.Goodman D S. Physiol Rev. 1965;45:747–839. doi: 10.1152/physrev.1965.45.4.747. [DOI] [PubMed] [Google Scholar]
- 2.Goldstein J L, Dana S E, Brown M S. Proc Natl Acad Sci USA. 1974;71:4288–4292. doi: 10.1073/pnas.71.11.4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang T-Y, Doolittle G M. In: The Enzymes. Boyer P D, editor. Vol. 16. New York: Academic; 1983. pp. 523–539. [Google Scholar]
- 4.Suckling K E, Stange E F. J Lipid Res. 1985;26:647–671. [PubMed] [Google Scholar]
- 5.Brown M S, Goldstein J L. Annu Rev Biochem. 1983;52:223–261. doi: 10.1146/annurev.bi.52.070183.001255. [DOI] [PubMed] [Google Scholar]
- 6.Wilson M D, Rudel L L. J Lipid Res. 1994;35:943–955. [PubMed] [Google Scholar]
- 7.Thompson G R, Naoumova R P, Watts G F. J Lipid Res. 1996;37:439–447. [PubMed] [Google Scholar]
- 8.Sliskovic D R, White A D. Trends Pharmacol Sci. 1991;12:194–199. doi: 10.1016/0165-6147(91)90546-5. [DOI] [PubMed] [Google Scholar]
- 9.Dominick M A, Bobrowski W A, MacDonald J R, Gough A W. Toxicol Pathol. 1993;21:54–62. doi: 10.1177/019262339302100107. [DOI] [PubMed] [Google Scholar]
- 10.Wolfgang G H I, MacDonald J R, Vernetti L A, Pegg D G, Robertson D G. Life Sci. 1995;56:1089–1093. doi: 10.1016/0024-3205(95)00045-8. [DOI] [PubMed] [Google Scholar]
- 11.Chang C C Y, Huh H Y, Cadigan K M, Chang T Y. J Biol Chem. 1993;268:20747–20755. [PubMed] [Google Scholar]
- 12.Uelmen P J, Oka K, Sullivan M, Chang C C Y, Chang T Y, Chan L. J Biol Chem. 1995;270:26192–26201. doi: 10.1074/jbc.270.44.26192. [DOI] [PubMed] [Google Scholar]
- 13.Ishibashi S, Brown M S, Goldstein J L, Gerard R D, Hammer R E, Herz J. J Clin Invest. 1993;92:883–893. doi: 10.1172/JCI116663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robertson E J. In: Teratocarcinomas and Embryonic Stem Cells: A Practical Approach. Robertson E J, editor. Oxford: IRL; 1987. pp. 71–112. [Google Scholar]
- 15.Ramírez-Solis R, Davis A C, Bradley A. Methods Enzymol. 1993;225:855–878. doi: 10.1016/0076-6879(93)25054-6. [DOI] [PubMed] [Google Scholar]
- 16.Purcell-Huynh D A, Farese R V, Jr, Johnson D F, Flynn L M, Pierotti V, Newland D L, Linton M F, Sanan D A, Young S G. J Clin Invest. 1995;95:2246–2257. doi: 10.1172/JCI117915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chirgwin J M, Przybyla A E, MacDonald R J, Rutter W J. Biochemistry. 1979;18:5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- 18.Barbu V, Dautry F. Nucleic Acids Res. 1989;17:7115. doi: 10.1093/nar/17.17.7115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang C C Y, Chen J, Thomas M A, Cheng D, Del Priore V A, Newton R S, Pape M E, Chang T-Y. J Biol Chem. 1995;270:29532–29540. doi: 10.1074/jbc.270.49.29532. [DOI] [PubMed] [Google Scholar]
- 20.Willnow T E, Herz J. J Cell Sci. 1994;107:719–726. [PubMed] [Google Scholar]
- 21.Goldstein J L, Basu S K, Brown M S. Methods Enzymol. 1983;98:241–260. doi: 10.1016/0076-6879(83)98152-1. [DOI] [PubMed] [Google Scholar]
- 22.Erickson S K, Shrewsbury M A, Brooks C, Meyer D J. J Lipid Res. 1980;21:930–941. [PubMed] [Google Scholar]
- 23.Erickson S K, Fielding P E. J Lipid Res. 1986;27:875–883. [PubMed] [Google Scholar]
- 24.Farese R V, Jr, Ruland S L, Flynn L M, Stokowski R P, Young S G. Proc Natl Acad Sci USA. 1995;92:1774–1778. doi: 10.1073/pnas.92.5.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edelson P J, Cohn Z A. In: In Vitro Methods in Cell-Mediated and Tumor Immunity. Bloom B R, David J R, editors. New York: Academic; 1976. pp. 333–340. [Google Scholar]
- 26.Pitas R E, Innerarity T L, Mahley R W. Arteriosclerosis. 1983;3:2–12. doi: 10.1161/01.atv.3.1.2. [DOI] [PubMed] [Google Scholar]
- 27.Bligh E G, Dyer W J. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 28.Pitas R E, Mahley R W. In: Lipoprotein Analysis: A Practical Approach. Converse C A, Skinner E R, editors. Oxford: Oxford Univ. Press; 1992. pp. 215–242. [Google Scholar]
- 29.Coalson R E. In: Staining Procedures. Clark G, editor. Baltimore: Williams & Wilkins; 1981. pp. 228–229. [Google Scholar]
- 30.Murphy B E P. J Clin Endocrinol Metab. 1967;27:973–990. doi: 10.1210/jcem-27-7-973. [DOI] [PubMed] [Google Scholar]
- 31.Kater C E, Biglieri E G, Brust N, Chang B, Hirai J, Irony I. Endocr Rev. 1989;10:149–164. doi: 10.1210/edrv-10-2-149. [DOI] [PubMed] [Google Scholar]
- 32.Turley S D, Daggy B P, Dietschy J M. Gastroenterology. 1994;107:444–452. doi: 10.1016/0016-5085(94)90170-8. [DOI] [PubMed] [Google Scholar]
- 33.Mansour S L, Thomas K R, Capecchi M R. Nature (London) 1988;336:348–352. doi: 10.1038/336348a0. [DOI] [PubMed] [Google Scholar]
- 34.Plump A S, Erickson S K, Weng W, Partin J S, Breslow J L, Williams D L. J Clin Invest. 1996;97:2660–2671. doi: 10.1172/JCI118716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glass C, Pittman R C, Weinstein D B, Steinberg D. Proc Natl Acad Sci USA. 1983;80:5435–5439. doi: 10.1073/pnas.80.17.5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Welch C L, Xia Y-R, Shechter I, Farese R, Mehrabian M, Mehdizadeh S, Warden C H, Lusis A J. J Lipid Res. 1996;37:1406–1421. [PubMed] [Google Scholar]
- 37.Taylor B A, Meier H. Genet Res. 1975;26:307–312. doi: 10.1017/s0016672300016104. [DOI] [PubMed] [Google Scholar]
- 38.Arnesen K. Acta Pathol Microbiol Scand. 1963;58:212–218. [Google Scholar]
- 39.Plump A S, Smith J D, Hayek T, Aalto-Setälä K, Walsh A, Verstuyft J G, Rubin E M, Breslow J L. Cell. 1992;71:343–353. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- 40.Zhang S H, Reddick R L, Piedrahita J A, Maeda N. Science. 1992;258:468–471. doi: 10.1126/science.1411543. [DOI] [PubMed] [Google Scholar]
- 41.Ishibashi S, Goldstein J L, Brown M S, Herz J, Burns D K. J Clin Invest. 1994;93:1885–1893. doi: 10.1172/JCI117179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paigen B, Mitchell D, Reue K, Morrow A, Lusis A J, LeBoeuf R C. Proc Natl Acad Sci USA. 1987;84:3763–3767. doi: 10.1073/pnas.84.11.3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pape M E, Schultz P A, Rea T J, DeMattos R B, Kieft K, Bisgaier C L, Newton R S, Krause B R. J Lipid Res. 1995;36:823–838. [PubMed] [Google Scholar]
- 44.Kinnunen P M, DeMichele A, Lange L G. Biochemistry. 1988;27:7344–7350. doi: 10.1021/bi00419a025. [DOI] [PubMed] [Google Scholar]
- 45.Maduskuie T P, Jr, Wilde R G, Billheimer J T, Cromley D A, Germain S, Gillies P J, Higley C A, Johnson A L, Pennev P, Shimshick E J, Wexler R R. J Med Chem. 1995;38:1067–1083. doi: 10.1021/jm00007a004. [DOI] [PubMed] [Google Scholar]
- 46.Yang H, Bard M, Bruner D A, Gleeson A, Deckelbaum R J, Aljinovic G, Pohl T M, Rothstein R, Sturley S L. Science. 1996;272:1353–1356. doi: 10.1126/science.272.5266.1353. [DOI] [PubMed] [Google Scholar]