

Abstract
When activated by proinflammatory stimuli, microglia release substantial levels of glutamate, and mounting evidence suggests this contributes to neuronal damage during neuroinflammation. Prior studies indicated a role for the Xc exchange system, an amino acid transporter that antiports glutamate for cystine. Because cystine is used for synthesis of glutathione (GSH), we hypothesized that glutamate release is an indirect consequence of GSH depletion by the respiratory burst, which produces superoxide from NADPH oxidase. Microglial glutamate release triggered by lipopolysaccharide was blocked by diphenylene iodonium chloride and apocynin, inhibitors of NADPH oxidase. This glutamate release was also blocked by vitamin E and elicited by lipid peroxidation products 4-hydroxynonenal and acrolein, suggesting that lipid peroxidation makes crucial demands on GSH. Although NADPH oxidase inhibitors also suppressed nitrite accumulation, vitamin E did not; moreover, glutamate release was largely unaffected by NO donors, inhibitors of NO synthase, or changes in gene expression. These findings indicate that a considerable degree of the neurodegenerative consequences of neuroinflammation may result from conversion of oxidative stress to excitotoxic stress. This phenomenon entails a biochemical chain of events initiated by a programmed oxidative stress and resultant mass-action amino acid transport. Indeed, some of the neuroprotective effects of antioxidants may be due to interference with these events rather than direct protection against neuronal oxidation.
Keywords: excitatory amino acid, inflammation, lipopolysaccharide, NADPH oxidase, nitric oxide, respiratory burst, Xc exchange
INTRODUCTION
Neuroinflammation exerts a degree of neurotoxicity that appears to contribute to clinical symptoms in a wide array of conditions, including multiple sclerosis, Alzheimer’s disease, brain abscess, ischemia, traumatic brain injury, and Parkinson’s disease. Much of the neuronal damage occurring in such conditions appears to arise from microglia. When stimulated by proinflammatory signals, microglia may undergo a reaction that includes a morphological transformation from ramified to amoeboid shape; production of prostanoids, cytokines, and chemokines; induction of surface markers, including members of the major histocompatibility complex family; elevated production of nitric oxide through expression of inducible NO synthase (iNOS); and initiation of an oxidative burst that produces superoxide from an orchestrated mechanism involving NADPH oxidase (Block and Hong 2005; Decoursey and Ligeti 2005). Many of these responses are likely irrelevant to neurotoxicity, and it is clear that states of activation occur in microglia that are actually neurotrophic or otherwise beneficial. A key determinant of microglial neurotoxicity is the release of excitotoxins, including glutamate (Piani et al. 1992; Barger and Basile 2001), quinolinate (Heyes et al. 1996), and D-serine (Wu et al. 2004).
The release of excitatory amino acids (EAA) from microglia is consistent with a growing body of evidence tying neuroinflammation to excitotoxic damage of neurons. Primarily mediated by the NMDA-class of glutamate receptors, excitotoxicity can be characterized as an ionic disturbance that results from excessive influx of cations—primarily sodium and calcium—through ionotropic glutamate receptors. Large sodium influxes cause rapid necrotic death, largely due to osmotic stress; prolonged excesses of intracellular calcium initiate signal transduction cascades and mitochondrial stress that often result in a delayed, caspase-independent form of programmed cell death (Choi 1988; Cheung et al. 2005). Lower levels of excitotoxicity can be restricted to synapses and dendrites (Hutchins and Barger 1998), resulting in pruning of these structures through a mechanism that may involve caspases (Mattson and Duan 1999). Considerable evidence indicates that these events participate in neurodegeneration in experimental models of neuroinflammation (Lipton 1996; Espey et al. 1998; Mascarucci et al. 1998; Willard et al. 2000; Groom et al. 2003; Bossuet et al. 2004; Takeuchi et al. 2005; Rosi et al. 2006). Furthermore, emerging evidence evokes these mechanisms in human disease (Lipton 1996; Reisberg et al. 2003; Feller et al. 2005; Bolton and Paul 2006).
The release of glutamate from malactivated microglia is chiefly conveyed by the Xc (a.k.a., SLC7A11 or CCBR1) exchange system. This antiporter exchanges glutamate for cystine, largely following the relative concentration gradients of each of these amino acids; Xc transport is virtually the sole means of delivering cystine—and thus, cysteine—into most cell types. Interference with the Xc exchanger by α-amino adipate or removal of extracellular cystine blocks evoked glutamate release from microglia (Piani and Fontana 1994; Barger and Basile 2001). In addition, Xc exchange was recently implicated in the microglial neurotoxicity elicited by Alzheimer amyloid β-peptide (Qin et al. 2006).
We have proposed that activation of the Xc exhanger in microglia is a function of mass-action demand for cysteine to replenish the reduced glutathione (GSH) that has been depleted in cellular self-defense against the oxidative burst effected by NADPH oxidase (Barger 2004). GSH is the primary antioxidant in the cytosol and participates in the reduction of hydrogen peroxide, itself generated by superoxide dismutase activity. Glutathione reductases can recycle oxidized glutathione, but GSH is consumed irreversibly in a covalent detoxification of lipid-peroxidation products such as 4-hydroxynonenal (4-HNE) and acrolein, a reaction catalyzed by specific isoforms of glutathione S-transferase (GST) (Awasthi et al. 2004). Thus, lipid peroxidation may make a larger contribution—vis-à-vis soluble oxidants such as hydrogen peroxide itself—to an oxidative increase in the demand for GSH and, thus, cysteine. We have tested this model through a series of experiments utilizing lipopolysaccharide (LPS) as a microglial stimulant and glutamate release as an endpoint. For comparison to more generalized aspects of activation, we also explored connections to NO/iNOS, a system that now appears to be independent from glutamate release.
MATERIALS AND METHODS
Materials
LPS was E. coli 026:B6 from Sigma-Aldrich (St. Louis MO). N-propyl-arginine, SP600125, U0126, caffeic acid phenethyl ester, and Bay 11-7082 were from Calbiochem/EMD Biosciences (San Diego CA); 1400W was from Alexis Biochemicals (San Diego CA). All other reagents were from Sigma-Aldrich (St. Louis MO).
Primary microglial cultures
Neonatal (P0-P1) Sprague-Dawley rats were utilized for the generation of mixed glia cultures as described previously (Wu et al. 2004). After 7–10 days in culture, microglia were removed from the astrocyte monolayer by vigorous lavage, collected by centrifugation, and replated in 24- or 96-well multiwell plates at 4 × 105 or 7 × 104 cells/well, respectively; for RNA extraction and qRT-PCR experiments, cultures were plated in 35-mm plates at 6 × 105 cells/plate. The plates were washed in growth medium (minimal essential medium, Earle’s salts [MEM]; 10% fetal bovine serum) 30 min after plating to reduce contamination by astrocytes and other slowly adhering cell types. The following day, cultures were washed to serum-free MEM and various pharmacological agents were applied at least 30 min prior to LPS stimulation.
Glutamate assay
Release of glutamate into the culture medium was determined by an enzymatic reaction of small aliquots with glutamate dehydrogenase. These determinations utilized an assay kit from r-biopharm, via Roche (Mannheim, Germany). Assays were conducted according to the manufacturer’s instructions except that volumes were reduced 25-fold from those recommended; endpoints of the reaction were taken at 15, 17, and 19 min to ensure linearity. Experimental values were interpolated within a standard curve of 0, 5, 50, and 500 μM glutamate.
Nitrate assays
The production of NO was monitored by determinations of its more stable, oxidized form: NO2−. Conventional Griess reactions were performed on aliquots of medium, and values were interpolated within a standard curve of 0, 5, 10, 15, 20, and 25 μM sodium nitrite, as described previously (Barger et al. 2000).
MTT assays
The potential effects of treatments on the viability or growth of the microglia were determined by assaying their ability to reduce 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to formazen. After removal of culture medium for the above assays, MTT was added to all wells at 125 μg/ml in growth medium and incubated at 37°C for 30 min. Then all medium was removed from each well, the formazen crystals were dissolved in 100 μL DMSO, and the plate was read at 540 nm in a spectrophotometer.
Quantitative RT-PCR
Two-step, real-time RT-PCR was performed on the unique subunit (xT) of the Xc transporter. RNA was prepared from microglial cultures with the RNAqueous kit (Ambion, Austin TX) according to the manufacturer’s instructions; residual DNA was removed by treatment with RNase-free DNase I (Roche, Mannheim Germany); RNA quality was assessed on the Agilent Bioanalyzer (Agilent, Palo Alto CA). The reverse-transcription (RT) reaction utilized 500 ng RNA and TaqMan Reverse Transcription Reagents (including random hexamers for priming). PCR was performed with the Power SYBR-Green PCR Master Mix (Applied Biosystems) in an ABI 7900HT Fast Real-time PCR System (Applied Biosystems, Foster City CA). The xCT signal was quantified relative to that for 18S rRNA. Equal amounts of RT-PCR from each sample were pooled to use for standard curve reactions with each primer set. A melting curve was generated for both xCT and 18S rRNA to ensure that a single peak of the predicted Tm was produced and no primer-dimer complexes were present. The relative standard curve method was used to calculate the amplification difference between the samples for each primer set. This is performed by correcting for signal concentration with the concentration of 18S signal for each sample (signal conc./18S conc.). The xCT primers used were as follows; forward: 5′-CCC AGA TAT GCA TCG TCC TT; reverse: 5′-ACA ACC ATG AAG AGG CAG GT. Primer sequences for 18S were forward: 5′-TTC GAA CGT CTG CCC TAT CAA-3′; reverse: 5′-ATG GTA GGC ACG GCG ACT A-3′.
Statistics
Dose-response curves were analyzed by ANOVA and Scheffe post-hoc test. Pairwise comparisons were made by Student’s t-test. Values of p less than or equal to 0.05 were taken to be significant.
RESULTS
Activation of primary microglia by LPS (10–100 ng/ml) induced a release of glutamate that reached concentrations on the order of 10−4 M within 16–24 h. Glutamate release was also triggered after application of LPS to the BV2 microglial cell line, though these cells exhibited a higher basal accumulation of glutamate and thus a smaller dynamic range of response (not shown). We tested the role of oxidation in LPS-evoked glutamate release by pretreating primary microglia with the lipid-soluble antioxidant vitamin E (α-tocopherol) or the water-soluble antioxidant N-acetylcysteine (NAC) (both of which are cell-permeant). At 500 μM NAC was ineffective (Fig. 1A). However, vitamin E completely blocked glutamate release with an EC50 of ~7 μM (Fig. 1B). The effectiveness of a lipid-soluble antioxidant suggested that lipid peroxidation contributes to glutamate release. This idea was supported by elevated glutamate release after direct application of the lipid peroxidation products 4-HNE and acrolein (Fig. 2).
Figure 1. Antioxidant effects on microglial glutamate release.

Primary microglia were treated with vitamin E or NAC for 30 min prior to application of LPS (100 ng/ml). After 20 h, medium was harvested for assay of nitrite (A only) and glutamate. In A, vitamin E was 100 μM and NAC was 500 μM. Values represent the means ± SEM of quadruplicate (A) or triplicate (B) cultures. Effects of vitamin E on glutamate were significant by ANOVA and Scheffe post hoc test (*p<0.01 vs. LPS alone).
Figure 2. Lipid peroxidation products evoke glutamate release.

Primary microglia were treated with acrolein or 4-HNE alone at the indicated concentrations; after 20 h glutamate levels were measured. Values represent mean ± SEM of quadruplicate cultures. All responses above 3 μM were significant (*p<0.0001, #p<0.002 vs. control) by ANOVA and Scheffe post hoc test.
Much of the oxidation resulting from inflammatory activation of microglia is generated by a programmed activation of NADPH oxidase. We tested the role of this system in the release of glutamate with inhibitors diphenylene iodonium chloride (DPIC) and apocynin (acetovanillone). Glutamate release triggered by LPS was inhibited by DPIC and apocynin in a dose-dependent manner (Fig. 3). In contrast to DPIC, the maximal efficacy of apocynin was incomplete. As with all our assays of glutamate release, we also examined viability of the cultures using MTT reduction. Neither DPIC nor apocynin produced a decrease in this index of viability.
Figure 3. Effect of NADPH oxidase inhibition in microglial glutamate release.
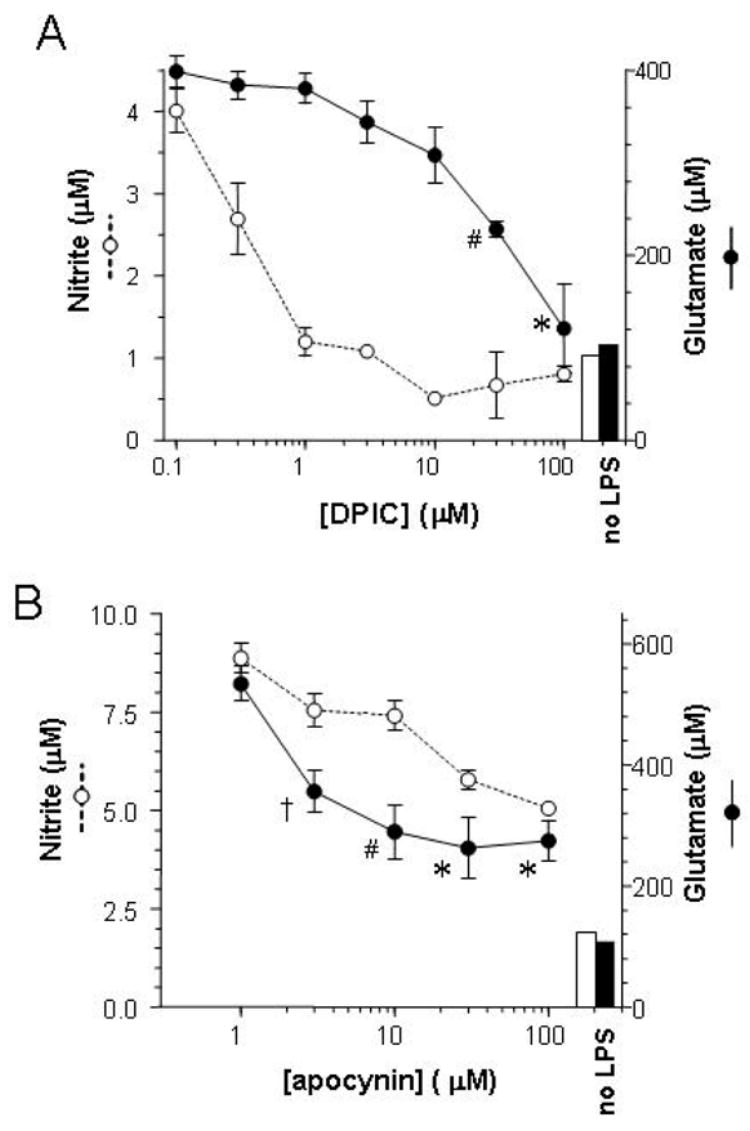
Primary microglia were treated with DPIC (A) or apocynin (B) for 30 min prior to application of LPS (100 ng/ml). After 20 h, medium was harvested for assay of nitrite and glutamate. Values represent the means ± SEM of quadruplicate (A) or triplicate (B) cultures. Bars represent levels of nitrite (open) and glutamate (filled) in untreated cultures. Effects of DPIC and apocynin on glutamate were significant by ANOVA and Scheffe post hoc test (*p<0.0002, #p<0.02, †p<0.05 vs. LPS alone).
In assays of glutamate release, we also measured nitrite accumulation as an index of general activation. Although vitamin E inhibited glutamate release, it had no effect on nitrite production (Fig. 1). DPIC and apocynin did inhibit nitrite accumulation, though with different concentration dependencies than those for glutamate release (Fig. 3). This led to consideration of a potential role for NO in glutamate release. We tested this possibility through both the application of NO donors (sufficiency) and treatment with LPS in the presence of NOS inhibitors (necessity). Among three NO donors tested, only sodium nitroprusside (NaNP) generated a significant elevation of extracellular glutamate (Fig. 4A). This appears to be an artifact of the oxidative stress generated by the ferricyanide anion that results after liberation of NO from NaNP, as potassium ferricyanide had a similar effect. To test whether NO was a necessary but insufficient component of the glutamate release, NOS inhibitors nitro-L-arginine, 1400W, and N-propyl-arginine were applied to microglia before exposure to LPS. None of these agents significantly inhibited glutamate release (Fig. 4B). Nitro-L-arginine and 1400W both provided effective inhibition of nitrite accumulation (not shown), further uncoupling this mechanism from glutamate release.
Figure 4. Tests of a role for nitric oxide in microglial glutamate release.

Primary microglia were treated with NO donors (A) or LPS (30 ng/ml) with and without inhibitors of NO synthase (B). After 20 h, medium was harvested for assay of glutamate. Values represent the means ± SEM of quadruplicate cultures. The glutamate levels in untreated cultures (open bar, B) or in cultures treated with LPS alone (black bars) are also indicated. Abbr.: NaNP, sodium nitroprusside; SNAP, S-Nitroso-N-acetylpenicillamine; SIN-1, 3-morpholinosydnonimine; nitro-Arg, Nω-nitro-L-arginine; N-propyl-Arg, N-propyl-L-arginine. No significant effects of the NO donors or NOS inhibitors was found by ANOVA and Scheffe post hoc test.
The data above are consistent with mechanism whereby glutamate release is driven by mass-action biochemical events creating a gradient of cystine across the cell membrane. However, the unique subunit of the Xc transporter, xCT, can undergo elevations of expression in response to proinflammatory stimulation (Sato et al. 2001). To determine if glutamate release from activated microglia required an increase in xCT expression, we treated cultures with α-amanitin and cycloheximide, inhibitors of transcription and translation, respectively. Neither compound blocked the ability of LPS to evoke glutamate release (Fig. 5A). In separate experiments, 30 μg/mL CHX was determined to block >95% of the 35S-methionine incorporation into TCA-precipitable protein (not shown). The relative depression of glutamate by α-amanitin was similar to its relative reduction of viability, as assessed by MTT (not shown). The remaining glutamate release was deduced to have come from live cells; while α-amanitin reduced viability by a similar degree in the absence of LPS, it did not elevate glutamate levels in this case. Cycloheximide did not influence viability over the time-frame of these experiments.
Figure 5. Glutamate release is independent of macromolecular synthesis.

A: Cycloheximide or α-amanitin was applied to primary microglia for 1 h prior to the application of LPS (30 ng/ml). After 16 h, medium was collected for assay of glutamate. The glutamate levels in untreated cultures (white bar) or in cultures treated with LPS alone (black bar) are also indicated. Values represent mean ± SEM of quadruplicate cultures. The effect of α-amanitin was significant by ANOVA and Scheffe post hoc (p<0.02). B: The indicated inhibitors were applied to primary microglia in triplicate for 1 h prior to the application of 30 ng/ml LPS; “JNK inh” = SP600125 (30 μM), “MEK inh” = U0126 (10 μM), α-AA = α-amino adipate (2.5 mM). After 20 h, medium was collected for assay of glutamate, and the cells were harvested for preparation of RNA. Solid bars represent glutamate values as mean ± SEM (# p<0.05 vs. LPS alone). Stipled bars indicate the level of xCT mRNA relative to 18S rRNA in each sample; values represent mean ± SEM (* p<0.02 vs. LPS alone).
To further test the potential contribution of xCT induction, we sought to correlate glutamate release with the relative levels of xCT mRNA in microglia under various treatment conditions. Jun N-terminal kinase (JNK) and extracellular signal-regulated kinases (ERK) mediate some aspects of LPS signal transduction, notably induction of iNOS and other events involving gene transcription. We tested an inhibitor of JNK (SP600125) and an inhibitor of the ERK pathway (U0126) alongside α-amino adipic acid, which blocks the Xc transporter. Microglia were pretreated with these compounds and then challenged with LPS. Culture medium was assayed for glutamate, and the cells were harvested for RNA preparation; xCT mRNA levels were compared by qRT-PCR. As shown in Figure 5B, effects of these agents on glutamate release were not directly correlated to their effects on xCT mRNA levels. Although LPS promoted the expected elevation of xCT, its levels were essentially unaffected by α-amino adipic acid. More importantly, a substantial glutamate release occurred in the presence of JNK and MEK inhibitors even though these agents dramatically attenuated the induction of xCT. We also tested caffeic acid phenethyl ester (CAPE) and Bay 11-7082 (inhibitors of NFκB activation) for their effects on glutamate release with negative results (not shown).
DISCUSSION
The release of glutamate from malactivated microglia has been documented by our studies and others to require the Xc exchange of cystine. Because of the role of cystine in antioxidant defenses, we proposed that this mechanism is directly dependent upon the respiratory burst instigated by NADPH oxidase. We have tested that hypothesis here by examining the sufficiency of oxidative stress and the necessity of NADPH oxidase and other mechanisms, namely nitric oxide and gene expression. The LPS-induced release of glutamate was mimicked qualitatively by oxidative stress, particularly products of lipid peroxidation, and was blocked by a lipophilic antioxidant and inhibitors of NADPH oxidase. Inhibitors of nitric oxide synthase or macromolecular synthesis did not substantially influence LPS-evoked glutamate release. Together, these data suggest a biochemical chain reaction whereby the activation of an oxidative burst, particularly lipid peroxidation, creates a need for additional GSH, the synthesis of which is dependent upon cystine import at the expense of glutamate antiport (Figure 6). In addition to lending support to the excitotoxic hypothesis of neuroinflammation, these findings imply that one benefit of antioxidants is the attenuation of glutamate release from microglia. These findings further indicate that it may be possible to ameliorate microglial neurotoxicity through strategies that do not rely upon manipulation of gene expression.
Figure 6. Hypothetical chain of events connecting NADPH oxidase to glutamate release.

Evidence from other studies indicates that PKC activation and phosphorylation of p47phox are important events in the activation of NADPH oxidase by proinflammatory stimuli. Once assembled at the membrane, p47phox and the other components of NADPH oxidase produce superoxide. This is dismutated by SOD to hydrogen peroxide. Some of the peroxide is reduced by glutathione peroxidase, consuming GSH. Some peroxide putatively initiates lipid oxidation, which must be halted by covalent conjugation of GSH to the lipid by glutathione S-transferase, particularly GSTA4-4. The depletion of GSH resulting from these events is replaced by de novo synthesis, requiring cysteine, imported into the cell as cystine via the Xc exchange mechanism. Though not required for the initiation of this mechanism, events mediated by JNK and ERK can also elevate expression of the antiporter’s xCT subunit. Pharmacological agents shown in the present work to block these events are enclosed in red boxes; entry into the chain of events at the point of lipid peroxidation is highlighted by the green box.
When exposed to proinflammatory stimuli, microglia initiate several responses that elevate the general stresses related to motility, energy utilization, and macromolecular synthesis. In addition to simple metabolic demands, activated microglia can produce cytotoxins that are responsible for collateral or “bystander” damage, and it is only reasonable that this could include autonomous damage. Indeed, activation of microglia has been reported to compromise their viability (Liu et al. 2001), and some of this vulnerability likely comes from the oxidative stress of the respiratory burst. Though much of the superoxide generated by NADPH oxidase is released to the cell’s exterior, intracellular production also occurs (Kobayashi et al. 2001). The superoxide and resulting hydrogen peroxide, per se, may not be particularly stressful to microglia’s autonomous antioxidant defenses. Catalase can help to reduce peroxide levels, and the oxidation of GSH by glutathione peroxidase can be reversed by glutathione reductase. However, the peroxide that slips these controls and oxidizes lipids creates products that can be exquisitely toxic (Keller and Mattson 1998). The primary cellular defense against lipid peroxidation products is covalent attachment of GSH by glutathione S-transferases (GSTs) such as GSTA4-4 and GST5.8 (Awasthi et al. 2004). Because this utilization of GSH is irreversible, it stands to make a larger impact on the steady-state levels of GSH. This hypothesis is consistent with two of our observations: the inhibition of glutamate release by vitamin E (a lipophilic antioxidant) and the elevation of glutamate release by 4-HNE and acrolein.
The actions of NADPH oxidase inhibitors indicate that the oxidative stress responsible for microglial glutamate release is generated by the respiratory burst. Apocynin (acetovanillone) was less efficacious than DPIC at maximal doses, but apocynin has been reported to stimulate γ-glutamylcysteine synthetase activity (Lapperre et al. 1999) which could create a cystine deficit on its own. The hypothesized involvement of NADPH oxidase in glutamate release is consistent with the findings of Nakamura et al. (Nakamura et al. 2003), who showed a requirement for protein kinase C (PKC) in LPS-evoked microglial glutamate release. A key rate-limiting component of NADPH oxidase, p47phox, has several PKC consensus phosphorylation sites, and PKC has been linked to the stimulation of NADPH oxidase (Nauseef et al. 1991; Korchak et al. 1998; Li et al. 1999). Nakamura et al. also tested for relationships between nitric oxide production and glutamate release. Whereas they did report inhibition by N-Arg, they also found that NO was unnecessary for stimulation of glutamate release by the PKC agonist phorbol myristolyl acetate; they showed that a NO donor was insufficient to elicit glutamate release, as well. The latter was confirmed by our tests of three different NO donors. Some studies have actually reported inhibition of the oxidative burst in macrophages by NO donors (Lee et al. 2002; Von Knethen and Brune 2002), even in vivo (Stehr et al. 2004). Indeed, evidence suggests that iNOS induction is secondary to the respiratory burst (Fries et al. 2003; Alblas et al. 2005). Interestingly, NO can trigger the release of vesicular glutamate from astrocytes via a transporter-independent mechanism (Bal-Price et al. 2002).
The poor correlation between microglial glutamate release and NO production may be extended to the difference between these phenomena with regard to their requirements for gene expression. Most of the NO produced upon activation of monocytic phagocytes results from the increased transcription of iNOS, resulting primarily from activation of NFκB. We previously showed that JNK was required for maximal induction of iNOS during microglial activation (Bodles and Barger 2005). If the “cystine deficit” model (described above and illustrated in Figure 6) is viable, there should be little requirement for gene induction in the release of glutamate. This is consistent with our demonstration that inhibitors of protein (cycloheximide) and RNA (α-amanitin) synthesis were largely ineffective in assays of LPS-stimulated glutamate release. Moreover, we found that glutamate release was poorly correlated with the effects of various pharmacological agents on xCT expression. LPS did indeed elevate xCT mRNA levels, consistent with prior reports (Sato et al. 2001). Therefore, a quantitative augmentation of glutamate release may result from the elevation of expression of the xCT subunit of the Xc antiporter. However, this induction does not appear to be qualitatively required for triggering glutamate release.
In summary, we have analyzed various aspects of the glutamate release triggered in malactivated microglia. The results are consistent with the hypothesis that the respiratory burst initiates a biochemical deficit for GSH and, by equilibrium-dependent transfer, cystine; the chain of chemical reactions and transport means that these events may occur independent of changes in gene expression. This is not to say that other aspects of microglial activation make no contribution to neurotoxicity. Indeed, neurological deficits may even arise independently of neurotoxicity via the physiological effects of cytokines (e.g., interleukin-1 and tumor necrosis factor) on functional parameters of synapses (Curran et al. 2003; Griffin et al. 2006). Furthermore, proinflammatory cytokines can interfere with astrocytic glutamate uptake (Wang et al. 2003; Korn et al. 2005), which would otherwise attenuate the effects of microglial glutamate release. Nevertheless, our hypothesis suggests that a significant portion of the oxidative stress arising during neuroinflammation may be most relevant in the cell of origin, microglia, rather than in the neurons that are eventually harmed. Piani et al. (Piani et al. 1992) found that microglial neurotoxicity could be ameliorated by glutamate-receptor antagonists but not by the extracellular application of antioxidant enzymes. Together, these findings reinforce that the supposition that the neurotoxicity exhibited by microglia is not mediated by ROS themselves; instead, the microglial oxidative stress seems to be converted to an excitotoxic stress, manifest through the liberation of a neurotransmitter that is stable and diffusible enough to impact neurons over intermediate distances.
Acknowledgments
This work was supported by funds from the NIH (R01AG17498 and P01AG12411).
References
- Alblas J, Honing H, de Lavalette CR, Brown MH, Dijkstra CD, van den Berg TK. Signal regulatory protein alpha ligation induces macrophage nitric oxide production through JAK/STAT- and phosphatidylinositol 3-kinase/Rac1/NAPDH oxidase/H2O2-dependent pathways. Mol Cell Biol. 2005;25:7181–7192. doi: 10.1128/MCB.25.16.7181-7192.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awasthi YC, Yang Y, Tiwari NK, Patrick B, Sharma A, Li J, Awasthi S. Regulation of 4-hydroxynonenal-mediated signaling by glutathione S-transferases. Free Radic Biol Med. 2004;37:607–619. doi: 10.1016/j.freeradbiomed.2004.05.033. [DOI] [PubMed] [Google Scholar]
- Bal-Price A, Moneer Z, Brown GC. Nitric oxide induces rapid, calcium-dependent release of vesicular glutamate and ATP from cultured rat astrocytes. Glia. 2002;40:312–323. doi: 10.1002/glia.10124. [DOI] [PubMed] [Google Scholar]
- Barger SW. An unconventional hypothesis of oxidation in Alzheimer’s disease: intersections with excitotoxicity. Front Biosci. 2004;9:3286–3295. doi: 10.2741/1481. [DOI] [PubMed] [Google Scholar]
- Barger SW, Basile AS. Activation of microglia by secreted amyloid precursor protein evokes release of glutamate by cystine exchange and attenuates synaptic function. J Neurochem. 2001;76:846–854. doi: 10.1046/j.1471-4159.2001.00075.x. [DOI] [PubMed] [Google Scholar]
- Barger SW, Chavis JA, Drew PD. Dehydroepiandrosterone inhibits microglial nitric oxide production in a stimulus-specific manner. J Neurosci Res. 2000;62:503–509. doi: 10.1002/1097-4547(20001115)62:4<503::AID-JNR4>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol. 2005;76:77–98. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Bodles AM, Barger SW. Secreted beta-amyloid precursor protein activates microglia via JNK and p38-MAPK. Neurobiol Aging. 2005;26:9–16. doi: 10.1016/j.neurobiolaging.2004.02.022. [DOI] [PubMed] [Google Scholar]
- Bolton C, Paul C. Glutamate receptors in neuroinflammatory demyelinating disease. Mediators Inflamm. 2006;2006:93684. doi: 10.1155/MI/2006/93684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossuet C, Vaufrey F, Conde F, Chretien F, Pichon J, Hantraye P, Le Grand R, Dormont D, Gras G. Up-regulation of glutamate concentration in the putamen and in the prefrontal cortex of asymptomatic SIVmac251-infected macaques without major brain involvement. J Neurochem. 2004;88:928–938. doi: 10.1046/j.1471-4159.2003.02237.x. [DOI] [PubMed] [Google Scholar]
- Cheung EC, Melanson-Drapeau L, Cregan SP, Vanderluit JL, Ferguson KL, McIntosh WC, Park DS, Bennett SA, Slack RS. Apoptosis-inducing factor is a key factor in neuronal cell death propagated by BAX-dependent and BAX-independent mechanisms. J Neurosci. 2005;25:1324–1334. doi: 10.1523/JNEUROSCI.4261-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW. Calcium-mediated neurotoxicity: relationship to specific channel types and role in ischemic damage. Trends Neurosci. 1988;11:465–469. doi: 10.1016/0166-2236(88)90200-7. [DOI] [PubMed] [Google Scholar]
- Curran BP, Murray HJ, O’Connor JJ. A role for c-Jun N-terminal kinase in the inhibition of long-term potentiation by interleukin-1beta and long-term depression in the rat dentate gyrus in vitro. Neuroscience. 2003;118:347–357. doi: 10.1016/s0306-4522(02)00941-7. [DOI] [PubMed] [Google Scholar]
- Decoursey TE, Ligeti E. Regulation and termination of NADPH oxidase activity. Cell Mol Life Sci. 2005;62:2173–2193. doi: 10.1007/s00018-005-5177-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espey MG, Kustova Y, Sei Y, Basile AS. Extracellular glutamate levels are chronically elevated in the brains of LP-BM5-infected mice: a mechanism of retrovirus-induced encephalopathy. J Neurochem. 1998;71:2079–2087. doi: 10.1046/j.1471-4159.1998.71052079.x. [DOI] [PubMed] [Google Scholar]
- Feller L, Jadwat Y, Bouckaert M. Herpes zoster post-herpetic neuralgia. Sadj. 2005;60:432, 436–437. [PubMed] [Google Scholar]
- Fries DM, Paxinou E, Themistocleous M, Swanberg E, Griendling KK, Salvemini D, Slot JW, Heijnen HF, Hazen SL, Ischiropoulos H. Expression of inducible nitric-oxide synthase and intracellular protein tyrosine nitration in vascular smooth muscle cells: role of reactive oxygen species. J Biol Chem. 2003;278:22901–22907. doi: 10.1074/jbc.M210806200. [DOI] [PubMed] [Google Scholar]
- Griffin R, Nally R, Nolan Y, McCartney Y, Linden J, Lynch MA. The age-related attenuation in long-term potentiation is associated with microglial activation. J Neurochem. 2006;99:1263–1272. doi: 10.1111/j.1471-4159.2006.04165.x. [DOI] [PubMed] [Google Scholar]
- Groom AJ, Smith T, Turski L. Multiple sclerosis and glutamate. Ann N Y Acad Sci. 2003;993:229–275. doi: 10.1111/j.1749-6632.2003.tb07533.x. discussion 287–228. [DOI] [PubMed] [Google Scholar]
- Heyes MP, Achim CL, Wiley CA, Major EO, Saito K, Markey SP. Human microglia convert L-tryptophan into the neurotoxin quinolinic acid. Biochem J. 1996;320:595–597. doi: 10.1042/bj3200595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchins JB, Barger SW. Why neurons die: cell death in the nervous system. New Anatomist. 1998;253:79–90. doi: 10.1002/(SICI)1097-0185(199806)253:3<79::AID-AR4>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Keller JN, Mattson MP. Roles of lipid peroxidation in modulation of cellular signaling pathways, cell dysfunction, and death in the nervous system. Rev Neurosci. 1998;9:105–116. doi: 10.1515/revneuro.1998.9.2.105. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Tsunawaki S, Seguchi H. Evaluation of the process for superoxide production by NADPH oxidase in human neutrophils: evidence for cytoplasmic origin of superoxide. Redox Rep. 2001;6:27–36. doi: 10.1179/135100001101536003. [DOI] [PubMed] [Google Scholar]
- Korchak HM, Rossi MW, Kilpatrick LE. Selective role for beta-protein kinase C in signaling for O-2 generation but not degranulation or adherence in differentiated HL60 cells. J Biol Chem. 1998;273:27292–27299. doi: 10.1074/jbc.273.42.27292. [DOI] [PubMed] [Google Scholar]
- Korn T, Magnus T, Jung S. Autoantigen specific T cells inhibit glutamate uptake in astrocytes by decreasing expression of astrocytic glutamate transporter GLAST: a mechanism mediated by tumor necrosis factor-alpha. Faseb J. 2005;19:1878–1880. doi: 10.1096/fj.05-3748fje. [DOI] [PubMed] [Google Scholar]
- Lapperre TS, Jimenez LA, Antonicelli F, Drost EM, Hiemstra PS, Stolk J, MacNee W, Rahman I. Apocynin increases glutathione synthesis and activates AP-1 in alveolar epithelial cells. FEBS Lett. 1999;443:235–239. doi: 10.1016/s0014-5793(98)01723-2. [DOI] [PubMed] [Google Scholar]
- Lee MC, Shoji H, Komatsu T, Yoshino F, Ohmori Y, Zweier JL. Inhibition of superoxide generation from fMLP-stimulated leukocytes by high concentrations of nitric oxide or peroxynitrite: characterization by electron spin resonance spectroscopy. Redox Rep. 2002;7:271–275. doi: 10.1179/135100002125000776. [DOI] [PubMed] [Google Scholar]
- Li Q, Subbulakshmi V, Fields AP, Murray NR, Cathcart MK. Protein kinase calpha regulates human monocyte O-2 production and low density lipoprotein lipid oxidation. J Biol Chem. 1999;274:3764–3771. doi: 10.1074/jbc.274.6.3764. [DOI] [PubMed] [Google Scholar]
- Lipton SA. Similarity of neuronal cell injury and death in AIDS dementia and focal cerebral ischemia: potential treatment with NMDA open-channel blockers and nitric oxide-related species. Brain Pathol. 1996;6:507–517. doi: 10.1111/j.1750-3639.1996.tb00879.x. [DOI] [PubMed] [Google Scholar]
- Liu B, Wang K, Gao HM, Mandavilli B, Wang JY, Hong JS. Molecular consequences of activated microglia in the brain: overactivation induces apoptosis. J Neurochem. 2001;77:182–189. doi: 10.1046/j.1471-4159.2001.t01-1-00216.x. [DOI] [PubMed] [Google Scholar]
- Mascarucci P, Perego C, Terrazzino S, De Simoni MG. Glutamate release in the nucleus tractus solitarius induced by peripheral lipopolysaccharide and interleukin-1 beta. Neuroscience. 1998;86:1285–1290. doi: 10.1016/s0306-4522(98)00105-5. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Duan W. Apoptotic” biochemical cascades in synaptic compartments: roles in adaptive plasticity and neurodegenerative disorders. J Neurosci Res. 1999;58:152–166. [PubMed] [Google Scholar]
- Nakamura Y, Ohmaki M, Murakami K, Yoneda Y. Involvement of protein kinase C in glutamate release from cultured microglia. Brain Res. 2003;962:122–128. doi: 10.1016/s0006-8993(02)03979-3. [DOI] [PubMed] [Google Scholar]
- Nauseef WM, Volpp BD, McCormick S, Leidal KG, Clark RA. Assembly of the neutrophil respiratory burst oxidase. Protein kinase C promotes cytoskeletal and membrane association of cytosolic oxidase components. J Biol Chem. 1991;266:5911–5917. [PubMed] [Google Scholar]
- Piani D, Fontana A. Involvement of the cystine transport system xc- in the macrophage- induced glutamate-dependent cytotoxicity to neurons. J Immunol. 1994;152:3578–3585. [PubMed] [Google Scholar]
- Piani D, Spranger M, Frei K, Schaffner A, Fontana A. Macrophage-induced cytotoxicity of N-methyl-D-aspartate receptor positive neurons involves excitatory amino acids rather than reactive oxygen intermediates and cytokines. Eur J Immunol. 1992;22:2429–2436. doi: 10.1002/eji.1830220936. [DOI] [PubMed] [Google Scholar]
- Qin S, Colin C, Hinners I, Gervais A, Cheret C, Mallat M. System Xc- and apolipoprotein E expressed by microglia have opposite effects on the neurotoxicity of amyloid-beta peptide 1–40. J Neurosci. 2006;26:3345–3356. doi: 10.1523/JNEUROSCI.5186-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ, Group MS. Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med. 2003;348:1333–1341. doi: 10.1056/NEJMoa013128. [DOI] [PubMed] [Google Scholar]
- Rosi S, Vazdarjanova A, Ramirez-Amaya V, Worley PF, Barnes CA, Wenk GL. Memantine protects against LPS-induced neuroinflammation, restores behaviorally-induced gene expression and spatial learning in the rat. Neuroscience. 2006 doi: 10.1016/j.neuroscience.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Sato H, Kuriyama-Matsumura K, Hashimoto T, Sasaki H, Wang H, Ishii T, Mann GE, Bannai S. Effect of oxygen on induction of the cystine transporter by bacterial lipopolysaccharide in mouse peritoneal macrophages. J Biol Chem. 2001;276:10407–10412. doi: 10.1074/jbc.M007216200. [DOI] [PubMed] [Google Scholar]
- Stehr SN, Weber S, Heller SC, Weikel J, Hubler M, Koch T, Heller AR. N(omega)-nitro-L-arginine methyl ester effects on neutrophil function and bacterial clearance. Shock. 2004;22:180–185. doi: 10.1097/01.shk.0000132487.89800.15. [DOI] [PubMed] [Google Scholar]
- Takeuchi H, Mizuno T, Zhang G, Wang J, Kawanokuchi J, Kuno R, Suzumura A. Neuritic beading induced by activated microglia is an early feature of neuronal dysfunction toward neuronal death by inhibition of mitochondrial respiration and axonal transport. J Biol Chem. 2005;280:10444–10454. doi: 10.1074/jbc.M413863200. [DOI] [PubMed] [Google Scholar]
- Von Knethen A, Brune B. Activation of peroxisome proliferator-activated receptor gamma by nitric oxide in monocytes/macrophages down-regulates p47phox and attenuates the respiratory burst. J Immunol. 2002;169:2619–2626. doi: 10.4049/jimmunol.169.5.2619. [DOI] [PubMed] [Google Scholar]
- Wang Z, Pekarskaya O, Bencheikh M, Chao W, Gelbard HA, Ghorpade A, Rothstein JD, Volsky DJ. Reduced expression of glutamate transporter EAAT2 and impaired glutamate transport in human primary astrocytes exposed to HIV-1 or gp120. Virology. 2003;312:60–73. doi: 10.1016/s0042-6822(03)00181-8. [DOI] [PubMed] [Google Scholar]
- Willard LB, Hauss-Wegrzyniak B, Danysz W, Wenk GL. The cytotoxicity of chronic neuroinflammation upon basal forebrain cholinergic neurons of rats can be attenuated by glutamatergic antagonism or cyclooxygenase-2 inhibition. Exp Brain Res. 2000;134:58–65. doi: 10.1007/s002210000446. [DOI] [PubMed] [Google Scholar]
- Wu S-Z, Bodles AM, Porter MM, Griffin WST, Basile AS, Barger SW. Induction of serine racemase expression and D-serine release from microglia by amyloid β-peptide. J Neuroinflammation. 2004;1:2–12. doi: 10.1186/1742-2094-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]