

Abstract
The A and B type lamins are nuclear intermediate filament proteins that comprise the bulk of the nuclear lamina, a thin proteinaceous structure underlying the inner nuclear membrane. The A-type lamins are encoded by the lamin A gene (LMNA). Mutations in this gene have been linked to at least nine diseases, including the progeroid diseases Hutchinson-Gilford progeria and atypical Werner’s syndromes, striated muscle diseases including muscular dystrophies and dilated cardiomyopathies, lipodystrophies affecting adipose tissue deposition, diseases affecting skeletal development, and a peripheral neuropathy. To understand how different diseases arise from different mutations in the same gene, mouse lines carrying some of the same mutations found in the human diseases have been established. We, and others have generated mice with different mutations that result in progeria, muscular dystrophy, and dilated cardiomyopathy. To further our understanding of the functions of the lamins, we also created mice lacking lamin B1, as well as mice expressing only one of the A-type lamins. These mouse lines are providing insights into the functions of the lamina and how changes to the lamina affect the mechanical integrity of the nucleus as well as signaling pathways that, when disrupted, may contribute to the disease.
Keywords: Lamins, Nucleus, Laminopathies, Progeria
Introduction
In metazoans, the nuclear lamina—a thin proteinaceous meshwork—underlies the nuclear face of the inner nuclear membrane (INM). Because of its role in maintaining nuclear envelope (NE) integrity and providing anchoring sites for chromatin domains, the nuclear lamina is a significant determinant of interphase nuclear architecture and function. The principal components of the lamina are intermediate filament proteins—the nuclear lamins. Most adult mammalian somatic cells contain four major lamins, A, B1, B2 and C, as well as several minor lamins (AΔ10, C2 and B3). These various lamins are grouped into two classes, A-type (A, AΔ10, C and C2) and B-type (B1 and B2). Separate genes encode lamins B1 and B2, whereas a single gene, LMNA, encodes the A-type lamins, which arise through alternative splicing of a common pre-mRNA [1].
The nuclear lamina has important roles in regulating DNA synthesis, RNA transcription, and in the organization of chromatin [2]. Transcriptional cofactors also associate with the lamins, suggesting that the lamina and NE are important in transcriptional regulation [3]. The lamins are also developmentally regulated, with all cells expressing one or the other lamin B, whereas A-type lamins are absent in early embryonic development and in certain stem cell populations in adults [4–6].
The discovery that at least nine inherited diseases, ranging from muscular dystrophies to premature aging-like syndromes, are caused by LMNA mutations or by defective posttranslational processing of prelamin A has resulted in a reassessment of the function of the lamina and NE. The diseases associated with defects in the A-type lamins can be classified into the primary and secondary laminopathies. The primary laminopathies are caused by mutations in the LMNA gene. The secondary laminopathies are caused by mutations in the gene encoding the endoprotease ZMPSTE24, an enzyme essential for the conversion of farnesyl-prelamin A to mature lamin A.
The primary laminopathies are broadly classified into three groups: the first and largest group consists of diseases affecting striated muscles and includes the autosomal dominant form of Emery-Dreifuss muscular dystrophy (AD-EDMD), dilated cardiomyopathy (DCM) and limb-girdle muscular dystrophy 1B (LMG1B). We can also include in this group a peripheral neuropathy, Charcot-Marie-Tooth neuropathy type 2B (CMT2B), which results in demyelination of the motor nerves, and is inherited as a rare recessive mutation in LMNA. The second group of diseases has minimal if any effects on muscle but affect adipose tissue distribution. These two diseases are Dunnigan-type familial partial lipodystrophy (FPLD) and mandibuloacral dysplasia (MAD). MAD also results in craniofacial abnormalities and osteolysis of the digits. The third group of diseases is the progeroid syndromes, Hutchinson Gilford progeria syndrome (HGPS) and some cases of atypical Werner’s syndrome. The majority of these diseases are caused by amino acid substitutions in the A-type lamins and are inherited in an autosomal dominant manner. The exception is the most common mutation causing HGPS, is a C→T transition that leads to a splicing defect in exon 11 of LMNA [7, 8]. To date, some 200 mutations have been identified in LMNA; a database on the “nuclear envelopathies” can be found at http://www.umd.be.
Our approach to understanding how mutations in LMNA result in the different disorders has been to create mouse models of the different diseases, by introducing some of the same mutations that have been found in humans with laminopathies. Having a mouse model for each disease provides a valuable resource for understanding the physiological consequences of the different mutations and identifying the molecular basis of each disease. This article summarizes the insights that mouse mutants have provided, including potential therapies for treating some of the diseases.
The A-type Laminopathies
Laminopathies affecting striated muscle
The first mutations linked to the LMNA gene were those causing an autosomal dominant form of Emery-Dreifuss muscular dystrophy (AD-EDMD) [9]. As with the X-linked form, which is caused by mutations in the NE associated protein emerin (EMD), AD-EDMD results in dystrophy specific for certain muscle groups, as well as cardiac conduction defects [10]. LMNA mutations resulting in AD-EDMD display wide variability in the severity of the phenotype, as well as penetrance, with the conduction defects leading to death being more severe than observed in the X-linked form of the disease [11]. Shortly thereafter, mutations in LMNA were also associated with dilated cardiomyopathy with conduction system disease (DCM-CD1) with no apparent skeletal muscle dystrophy [12]. The conduction defects observed in DCM-CD1 are similar to those in patients with EDMD, suggesting the cardiomyopathy patients represent an extreme in a phenotypic spectrum in which skeletal muscle involvement is not observed. Dilation of heart chambers, hypertrophy, arrhythmic conduction defects and cardiac arrest are all found with LMNA-associated cardiomyopathy. At the other end of the spectrum, limb-girdle muscular dystrophy 1B (LGMD1B) is also caused by mutations in LMNA, and is associated with fewer cardiac complications and tendon contractures [13]. Muscle wasting in the proximal limbs is the main clinical feature of LGMD1B. The heterogeneity in disease phenotypes, even among members of a single family carrying the same LMNA mutation, suggests that these conditions represent a spectrum of diseases that may have a common underlying basis, the outcome of which is influenced by genetic or environmental modifiers [14–16]. To date, diseases affecting striated muscle comprise about 60% of the laminopathies. Of note, only ~50% of patients diagnosed with AD-EDMD or EDMD have EMD or LMNA mutations, indicating that mutations in other genes, perhaps those encoding proteins that interact with the lamins (e.g., LAP2), may account for the remaining 50% of patients [17, 18].
Four mouse lines have been established that to varying extent model the striated muscle laminopathies. The first is a mouse line that does not express lamin A or C proteins (Lmna−/−). These mice die at 6–7 weeks of age from muscular dystrophy and cardiomyopathy [19]. However, there is no report of a human patient completely lacking lamins A/C, apart from one patient that may have been haploinsufficient for LMNA, due to the presence of a nonsense mutation at codon 6 [9] and a fetus that died late in gestation that was homozygous for a premature stop codon in LMNA [20]. Interestingly, one-year-old mice that are heterozygous for the Lmna knockout develop atrioventricular (AV) conduction defects with atrial and ventricular arrhythmias analogous to those in humans with LMNA mutations. Lmna+/− myocytes have impaired cell and sarcomere contractility, and the AV node cells have abnormal nuclei, undergo apoptosis, which are replaced by fibroblasts (J. Seidman, personal communication).
The molecular basis as to how these mice become dystrophic is still unclear. The in vivo regeneration of cardiotoxin treated Lmna−/− muscles appears to proceed normally and primary Lmna−/− myoblast cultures do not show any impairment in their differentiation to myotubes [21]. However immortalized Lmna−/− myoblasts are impaired in their differentiation to myotubes, [22], suggesting that the process of immortalization affects the myoblasts dependency on having a functional lamina for differentiation to proceed.
Loss of the A-type lamins results in the redistribution of emerin away from the nucleus to the endoplasmic reticulum, suggesting that reduced levels of emerin at the nuclear envelope contribute to the etiology of AD- and X-linked EDMD [19]. Array analysis of regenerating Emd null muscle revealed abnormalities in cell-cycle parameters and delayed myogenic differentiation, which were associated with perturbations to transcriptional pathways regulated by the retinoblastoma (Rb1) and MyoD genes. Temporal activation of MyoD transcriptional targets was significantly delayed, whereas targets of the Rb1/E2F transcriptional repressor complex remained inappropriately active resulting in a delay in myoblast/myotube transition during regeneration. Cultured primary myoblasts from Emd null mice also showed a delay in fusing to form myotubes, which correlated with prolonged Rb1 hyperphosphorylation [21]. Despite these anomalies, mice (at least 1 year old) lacking emerin do not develop muscular dystrophy or cardiac conduction defects, suggesting that, in mice, loss of emerin may have few pathological consequences [21, 23].
A second line of mice was established that carries a missense mutation (histidine-to-proline substitution at amino acid 222 (H222P)], a mutation that was originally identified in a family with AD-EDMD. Adult male mice homozygous for this mutation exhibit reduced locomotion with an abnormal stiff walking posture. They develop cardiac fibrosis, chamber dilation, and hypokinesia with conduction defects, and die by 9 months of age. Female homozygotes also exhibit these pathologies but at a later stage and survive for longer. The LmnaH222P/H222P mice may represent a good model for studying laminopathies affecting striated muscles as they develop a dystrophic condition in both skeletal and cardiac musculature that is similar to the human disease [24].
A third line of mice was created to study dilated cardiomyopathy with conduction system disease (DCM-CD1). A missense mutation in LMNA (asparagine-to-lysine substitution at amino acid 195—N195K) acts in an autosomal-dominant manner and causes DCM in humans. A mouse line (LmnaN195K/N195K) homozygous for the same mutation shows characteristics consistent with DCM-CD1, with the mice dying at 3 months due to arrhythmia. The mice showed minimal or no indication of muscular dystrophy. The transcription factor Hf1b/Sp4 and the gap junction proteins connexin 40 and connexin 43 were misexpressed and/or mislocalized in the mutant hearts. Desmin staining revealed a loss of organization at sarcomeres and intercalated disks. These observations suggest that LMNA mutations may cause cardiomyopathy by disrupting the internal organization of the cardiomyocyte and/or altering the expression of transcription factors essential to normal cardiac development, aging and function [25].
In an alternative approach, transgenic mice were produced in which a mutant form lamin A, M371K (which causes EDMD), was specifically expressed in the heart with a heart-specific α-myosin heavy chain promoter. Mice expressing the mutant M371K lamin A were born at lower numbers than that expected, and those that were born died by 2–7 weeks of age. Histological analysis of the hearts revealed extensive pathology with disruption of the cardiomyocytes and abnormal nuclei. The early death made it difficult to establish a transgenic line of mice from which offspring carrying the transgene could be routinely derived. However, these results demonstrated that expression of a Lmna mutant that induces alterations in nuclear morphology can cause tissue and organ damage in mice that express the normal complement of endogenous lamins [26].
Laminopathies affecting axonal myelination
A single autosomal recessive change in the rod domain of A-type lamins results in the peripheral neuropathy Charcot-Marie-Tooth syndrome Type 2b (CMT2B1) [27, 28]. Families homozygous for the R298C lamin variant manifest absent deep-tendon reflexes, distal amyotrophy, motor deficits, and loss of large myelinated nerve fibers [27, 28]. The muscular weakening associated with this disease is probably a secondary result of loss of enervation and subsequent muscle atrophy. Lamin-associated CMT2B illustrates that nuclear lamina defects can utilize more than one mechanism to cause musculoskeletal defects. Intriguingly, neurons in the sciatic nerve of the Lmna null mice showed extensive demyelination [27]. A mouse line homozygous for the R298C mutation has also been derived, although to date we have not observed overt effect on myelination or locomotor activity (S. Kozlov and C.L. Stewart, unpublished observations)
Laminopathies affecting adipose and skeletal tissues
The second group of diseases, Dunnigan-type familial partial lipodystrophy (FPLD) and mandibuloacral dysplasia (MAD) do not affect muscle tissue [29–32]. FPLD is inherited as an autosomal dominant trait, with about 85% of the cases (from 217 affected individuals) being associated with a missense mutation at Arg482. FPLD is characterized by the loss of subcutaneous white adipose tissue from the limbs, gluteal region, and areas of the trunk, with a concomitant accumulation of white adipose tissue in the neck, face, and abdominal regions. These changes begin at puberty. Prior to adolescence, children are overtly normal, suggesting a possible hormonal influence on the initiation of disease phenotypes [33]. Progressive insulin resistance accompanies the redistribution or remodeling of adipose tissue, often resulting in frank type II diabetes mellitus. These patients frequently have hyperlipidemia and manifest an increased susceptibility to atherosclerotic coronary heart disease.
The aberrant adipose tissue redistribution in lipodystrophy may be a result of an autonomous defect in specific subsets of mesenchymal or adipocyte precursors. The body may attempt to compensate for this loss of fat in some areas by an accumulation of fat in others. Neither the levels of lamin A and C expression nor the ratio of these two A-type lamins to each other vary significantly in the subcutaneous, omental, and neck fat depots of normal individuals, suggesting that an intrinsic fat depot-specific pattern of A-type lamin expression does not underlie the fat depot abnormalities associated with FPLD [34]. Although Lmna−/− mice exhibit reduced stores of white fat, they were cachexic and did not exhibit the hallmark insulin and plasma lipid alterations found in humans with FPLD [35].
A rare autosomal recessive mutation in the carboxyl-terminal domain of A-type lamins is responsible for mandibuloacral dysplasia (MAD) [31, 32], with 94% cases having a missense mutation at residue 527 (R527H). MAD is a disease with many of the metabolic and fat depot redistribution phenotypes of lipodystrophy, but with an expanded set of skeletal abnormalities, including osteolytic lesions in the bones. The primary sites of skeletal malformations in MAD are the craniofacial region, terminal digits, and clavicles.
With these seemingly diverse diseases having tissue specific defects, one of the principal questions is how do so many different diseases arise from mutations in the same protein that is expressed in the majority of adult cell types? Structural studies on the lamin A protein have provided some clues as to how different diseases may arise. The mutations causing the muscular dystrophies and DCM are distributed throughout the Atype lamins. Many of these mutations disrupt assembly of the lamins into the lamina. These mutations also affect nuclear morphology and the localization of other proteins in the nuclear envelope, such as emerin or some of the nesprins—large proteins that connect the nuclear lamina to the cellular cytoskeleton via the LINC complex [36].
Comparative studies on cells derived from the lamin A–deficient mice and lamin B1 gene trap mice showed that the A-type lamins are the principal contributors to the biophysical properties of the lamina, providing mechanical strength and stiffness to the nuclei [37]. These observations would be consistent with the notion that mutations in LMNA weaken the nucleus, making it more prone to damage arising from physical stress, such as would occur in the contracting heart or skeletal musculature. However, Emd-deficient nuclei (at least in mice) do not exhibit such a physical weakening. Furthermore, LmnaN195K/N195K mice have either no or minimal dystrophy in their muscles. Yet LmnaN195K/N195K nuclei have severe morphological abnormalities and are just as weak as the Lmna−/− nuclei (J. Lammerding and C. L. Stewart, unpublished observations). Together, these findings suggest that it might be too simplistic to view physical weakening of the nuclei as the prime cause of striated muscle defects.
An alternative explanation may lie in alterations in the induction of specific signaling pathways, such as the stress induced NF-κB pathway. The NF-κB (nuclear factor-κB) pathway is activated in normal fibroblasts in response to mechanical-induced strain. Activation of the pathway results in the increased expression of some specific genes, such as iex-1 (immediate early response gene-1), which encodes a nuclear factor with an anti-apoptotic role in cardiomyocytes. In Lmna−/− fibroblasts, activation of the NF-κB was reduced, resulting in lower levels of iex-1, which correlated with higher levels of apoptosis and necrosis in Lmna−/− fibroblasts following mechanical strain [38]. Similarly, Emd-null fibroblasts, which have physically normal nuclei, show a reduced levels of iex-1 compared to normal cells and are more prone to apoptosis following mechanically induced strain [39].
Mutations resulting in FPLD and MAD are clustered in the carboxyl-terminal globular domain (at amino acids R482 and R527, respectively). They are predicted to have minimal effects on lamin structure and do not appear to disrupt lamin assembly or emerin localization [40, 41]. The molecular basis as to how these specific mutations result in these diseases remains obscure. It has been proposed that since they are both located on the outside of the carboxyl-terminal globular domain, they might perturb interactions between the A-type lamins and other nuclear proteins [1], such as the adipogenic factor SREBP1, rather than disrupting the structure and/or assembly of the lamins, as has been suggested for the striated muscle laminopathies [42]. Intriguingly, human cells carrying either an FPLD, MAD, or one of the mutations responsible for atypical Werner’s syndrome have been reported to have increased levels of prelamin A [43].
Progeroid Syndromes
Hutchinson-Gilford progeria syndrome (HGPS) is a rare dominantly inherited disease in which patients show symptoms reminiscent of premature aging, including severe growth retardation, loss of subcutaneous fat, alopecia, a reduction in bone density, and poor muscle development [44]. The average age of death in HGPS is 12 to 15 years, usually due to artherosclerosis resulting in myocardial infarction or stroke [44]. Artherosclerosis in the HGPS patients does not appear to be linked to abnormal systemic lipid levels [45], but might be linked to smooth muscle depletion in atherosclerotic aortas [46]. Also, HGPS individuals do not show any increase in tumor susceptibility, cataract formation, or cognitive degeneration, features often associated with normal aging. HGPS is therefore often classified as a segmental progeroid syndrome, as it incompletely reproduces the normal aging processes [47].
The majority of HGPS cases are associated with a splicing defect in exon 11 of the LMNA gene. This arises due to a de novo single-base substitution; a C-to-T transition at nucleotide 1824 of the coding sequence that results in a polymorphism at codon 608 within exon 11 (G608G). The G608G mutation introduces a cryptic donor splice site that results in a 150-bp deletion in the mRNA and a 50-amino acid internal deletion in prelamin A, with lamin C remaining unaffected [7, 8]. The mutant prelamin A in HGPS is often called “progerin.”
A second premature aging syndrome, Werner’s syndrome, is inherited as an autosomal recessive trait due to mutations in a 3′–5′ RecQ DNA helicase-exonuclease, which unwinds DNA and cleaves nucleotides from DNA termini. The disease exhibits a high incidence of cancers, early-onset cataracts, arthrosclerosis, diabetes, premature graying of hair, and early death, usually in the late 40’s, from myocardial infarction [48–50]. The majority (83%) of Werner’s patients have defects in the WRN locus. A few do not carry mutations in WRN. These are known as atypical cases, and mutations in LMNA are found in 15% of these cases [51]. These atypical Werner’s patients do not die in their teens, but have short stature, alopecia, osteoporosis, lipodystrophy, diabetes, and muscle atrophy [51]. Additional recessive missense mutations have also been identified in LMNA that result in syndromes resembling both progeria and MAD [52, 53].
The first gene-targeted Lmna mutant mouse line with progeria was created by the fortuitous introduction of a splicing defect, resulting in an in-frame deletion of exon 9 of Lmna, resulting in the removal of amino acids from the carboxyl-terminal globular domain. These mice share multiple phenotypes with children with HGPS [54]. Loss of subcutaneous fat, decreased bone density, osteoporosis, thin hyperkeratotic skin, growth retardation and death by 4 weeks of age were some of the most striking phenotypes in the mice.
Mice expressing progerin have also been established. In these mice, the last 150 nucleotides of exon 11 plus all of introns 10 and 11 were deleted. Eliminating intron 10 meant that no lamin C could be produced. The sole product of this allele (LmnaHG) was progerin [55]. Fibroblasts heterozygous for the HG allele (LmnaHG/+) expressed large amounts of progerin and had an increased frequency of misshapen nuclei. LmnaHG/+ mice appeared normal at birth and weighed almost as much as wild-type littermates at weaning. However, both male and females gained weight slowly after weaning. The LmnaHG/+mice and humans with HGPS have many phenotypes in common, including retarded growth, osteoporosis, alopecia, micrognathia, reduced subcutaneous fat, and osteolysis of the clavicle. These phenotypes were progressive, and the mice died by 6–7 months of age. Interestingly, the LmnaHG/+ mice did not have significant muscle weakness. Homozygous mice (LmnaHG/HG) had severe osteoporosis and spontaneous bone fractures and died before weaning. No arterial lesions were found in the aortas of the LmnaHG/+or LmnaHG/HGmice [56].
In addition to the two knock-in lines of mice, a transgenic mouse line was created from a 164-kb human bacterial artificial chromosome spanning four genes (RAB25, UBQLN4, MAPBPIP, and LMNA)—with LMNA engineered to contain the exon 11 point mutation causing HGPS [57]. The amount of progerin expressed in the tissues of the BAC transgenic mice, relative to human lamin A and lamin C and mouse lamin A and lamin C, was not established. The transgenic mice did not manifest any of the early hallmarks of progeria, such as retarded growth or bone disease, nor did they exhibit atherosclerotic lesions in the intima layer of large vessels. However, they did show a loss of smooth muscle cells in the media of the aorta, a feature noted in the autopsies of some progeric patients [46].
Mice with Different Lamina Compositions
Lamin C–Only Mice
Lamin C–only mice (LmnaLCO)—mice that synthesize exclusively lamin C but no prelamin A or lamin A—have also been established [58]. Normally, lamin A and lamin C transcripts are present in similar amounts in wild-type fibroblasts, as judged by northern blot analysis. In LmnaLCO/LCO cells, all of the transcription of Lmna is diverted to the production of lamin C. Western blots of LmnaLCO/LCO fibroblasts with a lamin A/C antibody revealed lamin C and absolutely no lamin A, prelamin A, or progerin. The amount of lamin C protein in LmnaLCO/LCO and LmnaLCO/+ fibroblasts was higher than in wild-type cells [58].
Whereas Lmna−/− mice exhibit growth retardation and muscle weakness and die by 4–7 weeks of age [19], LmnaLCO/LCO showed none these phenotypes. The LmnaLCO/LCO mice appeared normal for a full two years of observation, and necropsy and histological studies did not uncover any histological abnormalities.
All of the Lmna gene transcription in LmnaLCO/LCO mice is channeled into the production of lamin C; hence, LmnaLCO/LCO mice produce roughly twice the normal amount of lamin C. Fong et al. considered the possibility that the increased lamin C synthesis in LmnaLCO/LCO mice somehow compensated for the loss of lamin A, thereby preventing the emergence of obvious disease phenotypes. However, LmnaLCO/− mice appeared entirely normal, and their growth rate, grip strength, and bones were indistinguishable from wild-type and LmnaLCO/LCO mice. Nuclei from LmnaLCO/LCO cells showed only a slight increase in abnormal morphology, compared to normal nuclei [58]. Also emerin targeting to the nuclear envelope was entirely normal in LmnaLCO/LCO cells, despite earlier reports that had suggested that lamin A may play a key role in directing emerin to the nuclear envelope [41, 59]. Clearly, there is much to learn about the unique and overlapping roles of lamin C and lamin A in the nuclear lamina.
The mechanical properties of the LmnaLCO/LCO nuclei were also analyzed by measuring the extent of nuclear deformation in response to biaxial strain. Measurements of nuclear strain overlapped substantially in LmnaLCO/LCO and wild-type cells; however, a small increase in normalized nuclear strain was noted in LmnaLCO/LCO but much less than the strain increase found in the Lmna−/− nuclei [58].
The absence of discernible disease phenotypes in LmnaLCO/LCO mice and any overt abnormality in the LmnaLCO/LCO cells in culture suggest that prelamin A and lamin A are dispensable. However, the authors emphasized that they were not suggesting that prelamin A and lamin A have no purpose or function. The fact that the two lamin isoforms have been highly conserved through evolution strongly favors the view that the two proteins have unique (and as-yet-unidentified) roles [58].
Lamin B1–deficient Mice
Inhibition of lamin B1 expression in cultured HeLa cells results in growth cessation and their eventual apoptosis [60]. Likewise, expression of lamin B1 with a deletion in the rod domain results in a patchwork accumulation of the lamins, with the nuclei becoming lobulated, together with the clustering of nuclear pore complexes and other NE proteins [61].
The in vivo function of lamin B1 has been investigated in mice carrying an insertional mutation in Lmnb1. The gene-trap insertion, located in intron 5 of Lmnb1, resulted in the production of an mRNA fusion transcript containing the first five Lmnb1 exons fused in-frame to a βgeo reporter gene. The mutant lamin B1 protein contains the amino-terminal head domain and a truncated α-helical central rod domain fused to βgeo, but lacked the carboxyl-terminal 273 amino acids. The 273-amino acid deletion eliminates one of two known phosphorylation sites, the nuclear localization signal, and the carboxyl-terminal CaaX motif required for protein farnesylation [62].
Heterozygous Lmnb1 gene-trap mice are viable but the homozygotes died a few minutes after birth. The Lmnb1−/− mice were smaller than wild-type mice, exhibited craniofacial dysmorphology, and a variety of skeletal abnormalities. The perinatal mortality in Lmnb1−/− mice may have been caused by respiratory failure, as many of the Lmnb1−/− mice embryos had abnormal lung histology with fewer inflated alveoli than in wild-type mice.
Fibroblasts from Lmnb1−/− embryos had grossly misshapen nuclei with frequent blebs, reduced replication rates, impaired differentiation, increased polyploidy, and premature senescence [62]. However, the mechanical properties of the nuclei of Lmnb1−/− mouse embryonic fibroblasts (MEFs) in response to defined nuclear strain were normal [37]. These experiments revealed that loss of lamin B1 may cause local disturbances in NE structure without causing generalized defects in nuclear organization, stiffness, and shape stability. It is possible, of course, that the B-type lamins play an important role in regulating nuclear stiffness, but that lamin B1 and lamin B2 have largely redundant roles and the impact of lamin B1 deficiency is masked by the expression of lamin B2. In the future, this issue needs to be addressed by creating and analyzing lamin B1/lamin B2 double knockout cells.
The fact that the Lmnb1−/− mice survived until birth and the fact that it was possible to culture and immortalize Lmnb1−/− fibroblasts was surprising, as knockdown experiments with siRNAs [60] had suggested that LMNB1 was an essential gene, at least in human cells. It is conceivable that the truncated lamin B1–βgeo fusion produced in the mouse Lmnb1−/− cells retains some biological activity, perhaps preventing an even more severe phenotype, or that lamin B2 compensated for the loss of lamin B1. Determining the precise phenotype of a lamin B1 knockout mouse with a true null mutation (no protein product at all) will be of interest.
Laminopathies Associated with Mutations in the B-type Lamins
To date, no missense mutations in lamin B1 have been linked to any disease. However, autosomal-dominant leukodystrophy, a neurodegenerative disease caused by myelin loss in the central nervous system, was recently found to be associated with a duplication of the lamin B1 gene. It is not clear how duplication of how the lamin B1 gene results in such a tissue-specific disease, although it was suggested that such an alteration may lead to the generation of autoantibodies resulting in demyelination [63].
With lamin B2, 3 rare mutations (including 2 resulting in amino acid substitutions) have been described in the LMNB2 gene of four patients with Acquired Partial Lipodystrophy (APL). As other patients with APL has no detectable mutations in the LMNB2 gene the relationship of the mutations in the LMNB2 gene to the etiology of the disease has not been fully established [64].
The Secondary Laminopathies
Following their synthesis, lamins A, B1 and B2 undergo a series of sequential posttranslational modifications that are of significance in relation to understanding the involvement of the lamins in some diseases. Each of these proteins contains a carboxyl-terminal CaaX motif, (“C” is cysteine, “a” is commonly an aliphatic amino acid, and “X” can be many different amino acids). The CaaX motif sequence is CSIM for prelamin A and CAIM for lamin B1; lamin C does not have a CaaX motif. Posttranslational modification of the lamins starts with the addition of a farnesyl lipid to the cysteine, a reaction carried out by the cytosolic enzyme protein farnesyltransferase [65, 66]. Subsequently, the –aaX amino acids are removed in the endoplasmic reticulum (ER) by specific endoproteases, with RCE1 cleaving lamin B1. The cleavage of the –aaX from prelamin A cleavage is likely a redundant function of ZMPSTE24 and RCE1. The isoprenylated cysteine is then methylated in the ER by a membrane methyltransferase, ICMT. These three modifications render the carboxyl-terminal domains of the lamins more hydrophobic, and that this enhanced hydrophobicity could facilitate their subsequent association with the INM, as assembly of lamins A, B1 and 2 into the lamina is compromised in the absence of a functional CaaX motif. In contrast to the B-type lamins, prelamin A undergoes a second endoproteolytic cleavage, by ZMPSTE24, that removes an additional 15 amino acids, including the farnesylated and methylated cysteine, to produce the mature lamin A. This cleavage event occurs 30–60 minutes post-synthesis—after assembly into the nuclear lamina [67–69].
Compound heterozygous and homozygous missense mutations in ZMPSTE24 have been reported to result in a case of mandibuloacral disease (MAD) and a progeroid-like phenotype in another individual [70, 71]. Recessive mutations resulting in complete absence of ZMPSTE24 cause restrictive dermopathy, which is characterized by intrauterine growth retardation, rigid or tight skin with prominent superficial vessels, defects in bone mineralization, dysplastic clavicles, and early postnatal death [72, 73]. Hypomorphic ZMPSTE24 alleles can lead to the accumulation of some unprocessed prelamin A in addition to mature lamin A, indicating residual activity of the mutated ZMPSTE24 protein. In ZMPSTE24-null cells, lamin C and prelamin A are present, but no mature lamin A is produced.
Zmpste24-deficient mice
Zmpste24 deficient mice (Zmpste24−/−) were created by two groups [68, 69, 74]. Intriguingly, mice with complete loss of Zmpste24 do not develop the severe disease in human patients with restrictive dermopathy that is associated with perinatal death [73]. A study of Zmpste24−/− mice revealed that ZMPSTE24 is absolutely required for the processing of farnesyl-prelamin A to mature lamin A [68, 69]. In Zmpste24−/− cells, no mature lamin A is produced, and farnesyl-prelamin A accumulates at the nuclear rim [68, 75]. The presence of farnesyl-prelamin A at the nuclear rim interferes with the integrity of the nuclear lamina, and Zmpste24−/− cells have an increased frequency of misshapen nuclei. The percentage of nuclear shape abnormalities in Zmpste24−/− MEFs was low in early-passage MEFs but substantially higher in later-passage cells [68, 75].
Zmpste24−/− mice are normal at birth and weigh almost as much as wild-type littermates at weaning. Thereafter, they grow slowly and develop incisor abnormalities, alopecia, kyphosis, and a slow arthritic gait. After 3–4 months of age, body weight begins to decline. One of the most striking bone abnormalities is osteolytic lesions in the ribs near the costovertebral junction, which ultimately lead to rib fractures. By ~20 weeks of age, nearly every rib in Zmpste24−/− mice is broken near the costovertebral joint. The fractures, which are surrounded by fracture callus, do not heal. All of the bone abnormalities in Zmpste24−/− mice are age-related; the bones are entirely normal at weaning (as judged by μCT scans) but bone phenotypes are prominent by 3–4 months of age [69].
Bone density is reduced in 3-month-old Zmpste24−/− mice, but there is no evidence for increased bone turnover [69]. Urinary levels of deoxypyridinoline, a bone collagen breakdown product [76], were similar in Zmpste24−/− and Zmpste24+/+ mice, and their bones had similar numbers of osteoclasts. The plasma levels of calcium and phosphate were normal, and plasma alkaline phosphatase was not elevated [69]. Examination of the thoracic vertebrae of 3-month-old Zmpste24−/− mice by electron microscopy revealed vacuolated osteoblasts lacking the typical stacks of rough endoplasmic reticulum, suggesting that defects in the osteoblast lineage maybe the cause of osteoporosis, osteolysis, and poor healing of fractures [77].
Zmpste24−/− mice also develop muscle weakness. At about 8 weeks of age, they began to hobble, dragging their hind limbs, and lacked the ability to hang upside down from a grid. Some dystrophic changes in the skeletal muscle of Zmpste24−/− mice were observed [68], but in another independently derived line, no histological abnormalities were observed [69]. It seems possible that the muscle weakness phenotype could be due, at least in part to a neuropathy, as the R298C LMNA missense mutation can cause neuropathy in humans, and neuropathy has also been documented in Lmna−/− mice [27]. It seems unlikely that the muscle weakness is simply due to bone disease because the muscle weakness occasionally precedes the onset of significant bone disease. Also, LmnaHG/+ mice develop bone disease that is very similar to that of Zmpste24−/− mice without developing significant muscle weakness [56].
One study reported that Zmpste24−/− cells showed enhanced senescence in culture, which appeared to be associated with an upregulation of genes controlled by the tumor suppressor p53 [68]. That study, as well as another study [74], suggested that the loss of ZMPSTE24 results in increased DNA damage and activation of a cell senescence program, and that this could be partly ameliorated by deleting p53 and by reducing synthesis of farnesyl-prelamin A.
Eliminating Disease Phenotypes in Zmpste24−/− Mice by Reducing Prelamin A Synthesis
The accumulation of farnesyl-prelamin A at the nuclear envelope and the increased number of abnormally shaped nuclei in Zmpste24−/− cells suggested that farnesyl-prelamin A is toxic, accounting for the disease phenotypes in Zmpste24−/− mice [75]. If the disease phenotypes were indeed caused by defective prelamin A processing and the toxic accumulation of farnesyl-prelamin A, then the disease phenotypes would be ameliorated by reducing the levels of prelamin A.
Zmpste24−/− mice were compared with Zmpste24−/− mice heterozygous for Lmna (Zmpste24−/−Lmna+/−) [75]. Western analysis revealed lower amounts of prelamin A in the Zmpste24−/−Lmna+/− cells. Interestingly, the Zmpste24−/−Lmna+/− mice appeared to be completely normal. Their growth rate and muscle strength were the same as wild-type mice, and they were free of bone disease. Also, Zmpste24−/−Lmna+/− MEFs exhibited significantly fewer misshapen nuclei [75]. In a related experiment using human cell lines, inhibition of ZMPSTE24 expression resulted in severe nuclear abnormalities, with blebbing and lamin accumulation in the nuclei. However, reducing lamin A expression prior to inhibiting ZMPSTE24 expression prevented the nuclei from developing severe morphological abnormalities [78].
Taken together, these results suggested that it is the levels of and persistence of farnesylated prelamin A that is toxic to cells, and a 50% reduction in lamin A levels (by removing one allele) results in a subthreshold level of toxicity.
In follow-up experiments, Fong et al. predicted that it also might be possible to fully rescue Zmpste24−/− mice, at both the cellular and whole-animal levels, by introducing a LmnaLCO allele, inasmuch as this intervention would reduce the accumulation of prelamin A in cells. Indeed, Zmpste24−/−LmnaLCO/+ and Zmpste24−/−LmnaLCO/LCO MEFs had far fewer misshapen nuclei than MEFs from littermate Zmpste24−/− embryos. At the “whole-animal” level, the impact of the LmnaLCO allele was dramatic. Zmpste24−/−LmnaLCO/+ and Zmpste24−/−LmnaLCO/LCO mice were entirely normal, with normal growth rates, muscle strength, bones, fertility, and lifespan [58].
The studies with Zmpste24−/−LmnaLCO/+ mice showed that adding a single LmnaLCO allele to Zmpste24−/− mice resulted in improved nuclear shape and disease phenotypes. Those data, along with the absence of discernible phenotypes in LmnaLCO/LCO mice, suggested the possibility at improving nuclear shape in Zmpste24−/− cells by blocking the synthesis of prelamin A. To test this idea, Fong et al. screened 78 mouse prelamin A-specific antisense oligonucleotides for their capacity to reduce prelamin A synthesis in mouse fibroblasts. Eleven of the oligonucleotides reduced prelamin A mRNA levels by more than 75%, and one (ISIS 359445) reduced prelamin A mRNA levels by more than 95%. Zmpste24−/− cells treated with ISIS 359445 had significantly fewer misshapen nuclei than untreated cells [79].
FTI Treatment Inhibits Lamin A Biogenesis in Cultured Cells
The genetic experiments suggested that it was the farnesylated form of prelamin A that caused the disease phenotypes associated with progeroid syndromes. Therefore, it was reasonable to postulate that the disease phenotypes might be improved by blocking prelamin A farnesylation with an inhibitor of protein farnesyltransferase (FTI).
FTIs were developed to inhibit the targeting of mutated and activated Ras proteins to the plasma membrane, thereby mislocalizing the Ras proteins away from their signaling partners [80]. In the case of progeria, the rationale for FTIs was similar—to eliminate the farnesyl lipid anchor and thereby mislocalize the disease-causing protein (progerin) away from the NE. The first attempts at determining the efficacy of FTIs were performed on cells from the HGPS knock-in mouse model [55]. FTI treatment of LmnaHG/HG cells relocated progerin away from the NE to the nucleoplasm, as well as improving nuclear shape abnormalities in LmnaHG/+ MEFs. In subsequent studies, Toth et al. tested the effect of an FTI on multiple lines of Zmpste24−/− mouse fibroblasts, ZMPSTE24-deficient human cells, and human HGPS fibroblasts [81]. In all of these cells, the FTI treatment significantly reduced nuclear shape abnormalities. Other laboratories [82–84] also reported that FTIs improved nuclear shape in human HGPS fibroblasts and/or cells that had been transfected with a progerin cDNA.
The block to protein farnesylation can be assessed by performing a western blot for HDJ-2, an unrelated farnesylated CaaX protein [55]. In the cell culture studies, virtually 100% of the HDJ-2 in FTI-treated cells was nonfarnesylated [55]. Also, the FTI dramatically blocked prelamin A processing, leading to an accumulation of the nonfarnesylated form of prelamin A. Interestingly, the FTI also significantly reduced the total amount of lamin A/prelamin A (the sum of the two proteins) in MEFs, presumably because nonfarnesylated prelamin A turns over more rapidly [55, 81]. A similar FTI-induced reduction in lamin A/prelamin A is not apparent in human fibroblasts [55].
Improving Disease Phenotypes in Zmpste24−/− and LmnaHG/+ Mice by FTI treatment
Subsequent experiments tested the ability of a potent FTI, ABT-100 [85], to ameliorate the disease phenotypes in Zmpste24−/− mice. In these studies, the FTI inhibited protein farnesylation in vivo, as judged by western blots for HDJ-2, with 10–50% of the HDJ-2 exhibiting a retarded electrophoretic mobility (i.e., was nonfarnesylated). The FTI treatment also resulted in the appearance of prelamin A in the tail extracts from wild-type mice (with western blots employing prelamin A–specific antibodies), again indicating that the FTI was active in vivo. However, nonfarnesylated prelamin A was almost undetectable with a lamin A/C antibody, suggesting that the absolute level of inhibition in lamin A processing was small [79].
The most striking phenotype in humans with HGPS is retarded growth and reduced body weight [44]; retarded growth is also a prominent feature of Zmpste24−/− mice [75]. FTI treatment significantly improved the body weight curves of both male and female Zmpste24−/− mice [79]. In contrast, the FTI reduced the body weights of wild-type female mice and tended to reduce body weight in males. The reduced weight gain in the FTI-treated wild-type mice was presumably due to inhibition of protein farnesylation or to a toxic effect of the FTI. FTI treatment of Zmpste24−/− mice also significantly improved their muscle strength, improved the longevity of Zmpste24−/− mice, and significantly reduced the incidence of rib fractures that are a hallmark of Zmpste24−/− mice [79].
These studies strongly suggested that an FTI does more than simply improve the morphology of nuclei of progeria cells—FTI may actually improve disease phenotypes. However, FTI treatment was not successful in “curing” the Zmpste24−/− mice. Although they were clearly improved, the FTI-treated Zmpste24−/− mice still had an abnormal growth rate; grip abnormality, and some still developed a few rib fractures. Additional studies will be required to determine if further benefit could be obtained with a higher dose of an FTI (with more complete inhibition of protein farnesylation). With the water delivery method, peak drug levels fluctuate with activity levels. It would not be surprising if maximal efficacy of FTI therapy in progeria required the continuous administration and maintenance of a high drug concentration.
Similar experiments, using the same FTI (ABT-100), were performed on LmnaHG/+ mice. The FTI interfered with the biogenesis of mature lamin A from the wild-type (LmnaHG/+) allele, resulting in the appearance of nonfarnesylated prelamin A in the livers of treated mice, as judged by western blotting with a prelamin A–specific antibody. Once again, however, the absolute levels of nonfarnesylated prelamin A in cells were fairly small, as judged by lamin A/C western blots [56].
In Lmna+/+ female mice, FTI treatment tended to reduce body weight, compared with Lmna+/+ mice given the vehicle alone. In contrast, the FTI improved body-weight curves in both female and male LmnaHG/+ mice. FTI treatment also increased the weight of the major fat pads in LmnaHG/+mice and increased the amount of subcutaneous fat. The FTI treatment also significantly reduced bone pathology in the LmnaHG/+ mice. The degree of kyphosis was reduced as well as the number of rib fractures, and bone mineralization and cortical thickness were improved [56].
FTI therapy clearly reduced the pathologies and abnormalities found in the Zmpste24−/− and LmnaHG/+ mice. In the future, it will be interesting to determine if FTIs are useful for treating children with progeria. A trial of FTIs in human patients with HGPS should begin soon (http://www.progeriaresearch.org/). The mouse studies provide a reason for optimism, but the mouse studies simply showed that an FTI can prevent disease—not that an FTI can reverse disease [56]. Humans afflicted with HGPS will obviously be hoping that FTIs can reverse existing disease phenotypes.
Conclusion and Future Directions
Mutations within the LMNA gene are unique in that no other gene is known which when mutated results in so many different diseases. One of the fascinations in studying the laminopathies, as well as other diseases linked to mutations in proteins found within the nuclear envelope, are the insights they are providing to understanding the architecture of the nucleus in regulating nuclear functions, such as the organization of chromatin and the interaction between the nucleus and cytoskeleton. It has been somewhat surprising that the A-type lamins, as structural IF proteins, have so many roles. Much interest is now focusing on what function does physical linkage of the lamina with the cytoplasmic cytoskeleton have in cells? Does a breakdown in this link contribute to disease and can this link affect chromatin and gene expression?
We clearly need to understand more about the role of the lamina (and NE) in regulating signaling pathways. We also need to understand whether alterations in the signaling pathways are an important contributing factor to the various laminopathies, as these pathways maybe more amenable to drug treatment. In pursuing these objectives mouse models of the laminopathies will have a significant role.
Figure 1. Immunofluoresence images showing the distribution of progerin in untreated and FTI-treated LmnaHG/HG cells.
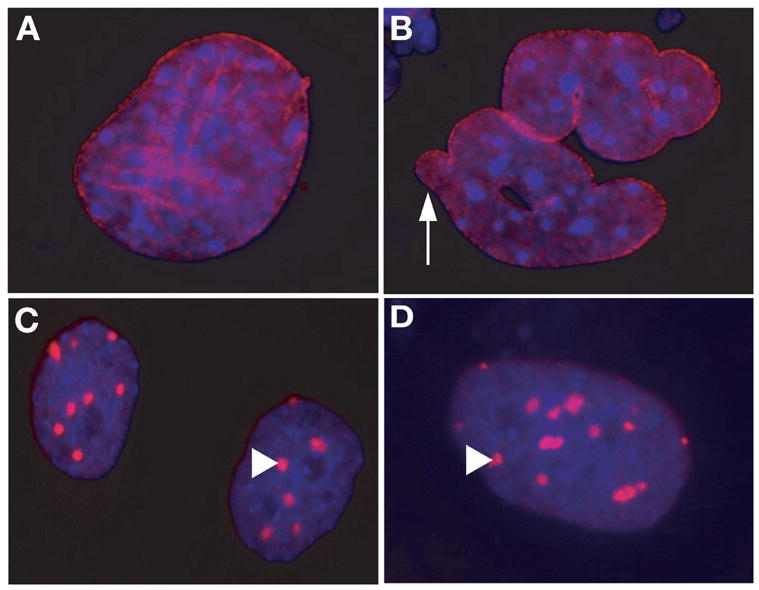
DNA was visualized with DAPI (blue), and progerin was visualized with an antibody against lamin A (red). A and B, Untreated LmnaHG/HG cells, showing progerin along the nuclear envelope. Misshapen nuclei were common (white arrow). C and D, FTI-treated LmnaHG/HG cells, revealing intensely staining progerin aggregates (white arrowheads) in the nucleoplasm. Reproduced, with permission, from The Proceedings of the National Academy of Science USA [55].
Figure 2. A positive effect of FTIs on disease phenotypes in LmnaHG/+ mice.
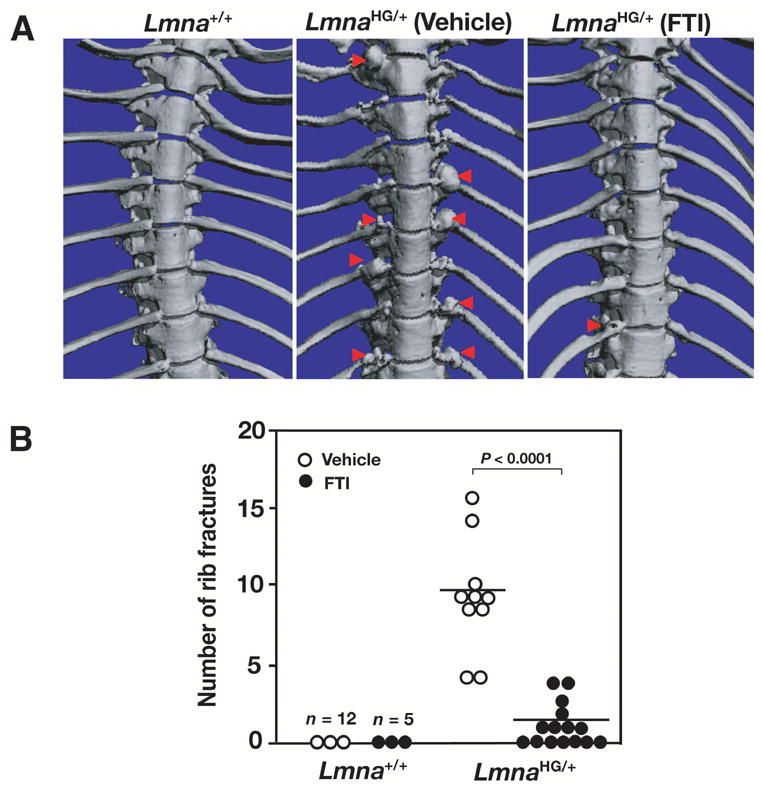
A, μCT scans illustrating reduced numbers of rib fractures in FTI-treated LmnaHG/+ mice. Red arrowheads indicate rib fractures and surrounding callus. In the FTI-treated mouse, there is thinning of one rib along with a small amount of callus. B, Reduced number of rib fractures in FTI-treated LmnaHG/+ mice. After 24 weeks, surviving mice were euthanized, and the number of rib fractures was counted. The number of rib fractures in the FTI-treated LmnaHG/+ mice was lower than in vehicle-treated LmnaHG/+ mice (P < 0.0001). Reproduced, with permission, from The Journal of Clinical Investigation [56].
Table 1.
Summary of Mouse Models
| Mutations introduced into Mouse Lamin genes | |||
|---|---|---|---|
| Mutation | Description | Phenotype | Ref |
| Lmna−/− | Lamin A and C Null | Postnatal lethality associated with muscular dystrophy and cardiomyopathy | [19] |
| LmnaN195K/N195K | Mis-sense mutation | Postnatal death associated with cardiomyopathy | [25] |
| LmnaH222P/H222P | Mis-sense mutation | Postnatal death associated with muscular dystrophy and cardiomyopathy | [24] |
| LmnaΔ9/Δ9 | Splicing mutation and inframe deletion of exon 9 | Early postnatal lethality and a model for progeria | [54] |
| LmnaHG/HG | A-type lamins are replaced by Progerin | Heterozygotes die at 6 months with osteoporosis, alopecia. Homozygotes severely retarded postnatal growth, death at 3 weeks | [55] |
| LmnaLCO/LCO | Lamin C only mice with no Lamin A | Overtly normal | [58] |
| Lmnb1−/− | Gene trap insertion into Lmnb1 | Perinatal lethal possibly due to respiratory failure | [62] |
| Mutations in proteins associated with Lamin A | |||
|
| |||
| Emd−/− | Null | Mice overtly normal but with slightly retarded muscle regeneration | [21, 23] |
| Zmpste24−/− | Null | Mice retain farnesylated pre-lamin A. They die at 6–7 months and have rib fractures, osteoporosis, muscle weakness | [69, 74] |
| Transgenic Lines | |||
|
| |||
| Lmna M371K | cDNA with missense mutation | Expressed in heart resulting in cardiomyopathy and early postnatal lethality | [26] |
| Lmna BAC G608G | hBAC with G606g base change | Mice show progressive loss of smooth muscle cells in medial layer of large arteries | [57] |
Abbreviations
- IF
Intermediate filament
- INM
Inner Nuclear Membrane
- ONM
Outer Nuclear Membrane
- NE
Nuclear Envelope
- LMNA
Lamin A/C
- LMNB
Lamin B
- EDMD
Emery Dreifuss Muscular Dystrophy (X-linked)
- AD-EDMD
Autosomal Dominant EDMD
- LMG1B
Limb Girdle Muscular Dystrophy 1B
- DCM
Dilated Cardiomyopathy
- MAD
Mandibuloacral dysplasia
- FPLD
Familial Partial Lipodystrophy
- CMT2B1
Charcot-Marie-Tooth neuropathy
- HGPS
Hutchinson-Gilford Progeria
- ZMPSTE24
Zinc Metallopeptidase (STE24 homolog, S. cerevisiae)
- RCE1
Prenyl protein peptidase (homolog S. cerevisiae)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Burke B, Stewart CL. Life at the edge: the nuclear envelope and human disease. Nat Rev Mol Cell Biol. 2002;3:575–85. doi: 10.1038/nrm879. [DOI] [PubMed] [Google Scholar]
- 2.Goldman RD, Gruenbaum Y, Moir RD, Shumaker DK, Spann TP. Nuclear lamins: building blocks of nuclear architecture. Genes Dev. 2002;16:533–47. doi: 10.1101/gad.960502. [DOI] [PubMed] [Google Scholar]
- 3.Burke B, Stewart CL. The laminopathies: the functional architecture of the nucleus and its contribution to disease. Annu Rev Genomics Hum Genet. 2006;7:369–405. doi: 10.1146/annurev.genom.7.080505.115732. [DOI] [PubMed] [Google Scholar]
- 4.Stewart C, Burke B. Teratocarcinoma stem cells and early mouse embryos contain only a single major lamin polypeptide closely resembling lamin B. Cell. 1987;51:383–392. doi: 10.1016/0092-8674(87)90634-9. [DOI] [PubMed] [Google Scholar]
- 5.Rober RA, Weber K, Osborn M. Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: a developmental study. Development. 1989;105:365–78. doi: 10.1242/dev.105.2.365. [DOI] [PubMed] [Google Scholar]
- 6.Rober RA, Sauter H, Weber K, Osborn M. Cells of the cellular immune and hemopoietic system of the mouse lack lamins A/C: distinction versus other somatic cells. J Cell Sci. 1990;95 (Pt 4):587–98. doi: 10.1242/jcs.95.4.587. [DOI] [PubMed] [Google Scholar]
- 7.De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, Levy N. Lamin a truncation in Hutchinson-Gilford progeria. Science. 2003;300:2055. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- 8.Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–8. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonne G, Di Barletta MR, Varnous S, Becane HM, Hammouda EH, Merlini L, Muntoni F, Greenberg CR, Gary F, Urtizberea JA, Duboc D, Fardeau M, Toniolo D, Schwartz K. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet. 1999;21:285–8. doi: 10.1038/6799. [DOI] [PubMed] [Google Scholar]
- 10.Bione S, Maestrini E, Rivella S, Mancini M, Regis S, Romeo G, Toniolo D. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet. 1994;8:323–7. doi: 10.1038/ng1294-323. [DOI] [PubMed] [Google Scholar]
- 11.Morris GE. The role of the nuclear envelope in Emery-Dreifuss muscular dystrophy. Trends Mol Med. 2001;7:572–7. doi: 10.1016/s1471-4914(01)02128-1. [DOI] [PubMed] [Google Scholar]
- 12.Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ, Jr, Spudich S, De Girolami U, Seidman JG, Seidman C, Muntoni F, Muehle G, Johnson W, McDonough B. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341:1715–24. doi: 10.1056/NEJM199912023412302. [DOI] [PubMed] [Google Scholar]
- 13.Muchir A, Bonne G, van der Kooi AJ, van Meegen M, Baas F, Bolhuis PA, de Visser M, Schwartz K. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B) Hum Mol Genet. 2000;9:1453–9. doi: 10.1093/hmg/9.9.1453. [DOI] [PubMed] [Google Scholar]
- 14.Brodsky GL, Muntoni F, Miocic S, Sinagra G, Sewry C, Mestroni L. Lamin A/C gene mutation associated with dilated cardiomyopathy with variable skeletal muscle involvement. Circulation. 2000;101:473–6. doi: 10.1161/01.cir.101.5.473. [DOI] [PubMed] [Google Scholar]
- 15.Canki-Klain N, Recan D, Milicic D, Llense S, Leturcq F, Deburgrave N, Kaplan JC, Debevec M, Zurak N. Clinical variability and molecular diagnosis in a four-generation family with X-linked Emery-Dreifuss muscular dystrophy. Croat Med J. 2000;41:389–95. [PubMed] [Google Scholar]
- 16.Vytopil M, Ricci E, Dello Russo A, Hanisch F, Neudecker S, Zierz S, Ricotti R, Demay L, Richard P, Wehnert M, Bonne G, Merlini L, Toniolo D. Frequent low penetrance mutations in the Lamin A/C gene, causing Emery Dreifuss muscular dystrophy. Neuromuscul Disord. 2002;12:958–63. doi: 10.1016/s0960-8966(02)00178-5. [DOI] [PubMed] [Google Scholar]
- 17.Ben Yaou R, Muchir A, Arimura T, Massart C, Demay L, Richard P, Bonne G. Genetics of laminopathies. Novartis Found Symp. 2005;264:81–90. discussion 90–97, 227–30. [PubMed] [Google Scholar]
- 18.Taylor MR, Slavov D, Gajewski A, Vlcek S, Ku L, Fain PR, Carniel E, Di Lenarda A, Sinagra G, Boucek MM, Cavanaugh J, Graw SL, Ruegg P, Feiger J, Zhu X, Ferguson DA, Bristow MR, Gotzmann J, Foisner R, Mestroni L. Thymopoietin (lamina-associated polypeptide 2) gene mutation associated with dilated cardiomyopathy. Hum Mutat. 2005;26:566–74. doi: 10.1002/humu.20250. [DOI] [PubMed] [Google Scholar]
- 19.Sullivan T, Escalante-Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K, Stewart CL, Burke B. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol. 1999;147:913–20. doi: 10.1083/jcb.147.5.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Engelen BG, Muchir A, Hutchison CJ, van der Kooi AJ, Bonne G, Lammens M. The lethal phenotype of a homozygous nonsense mutation in the lamin A/C gene. Neurology. 2005;64:374–6. doi: 10.1212/01.WNL.0000149763.15180.00. [DOI] [PubMed] [Google Scholar]
- 21.Melcon G, Kozlov S, Cutler DA, Sullivan T, Hernandez L, Zhao P, Mitchell S, Nader G, Bakay M, Rottman JN, Hoffman EP, Stewart CL. Loss of emerin at the nuclear envelope disrupts the Rb1/E2F and MyoD pathways during muscle regeneration. Hum Mol Genet. 2006;15:637–51. doi: 10.1093/hmg/ddi479. [DOI] [PubMed] [Google Scholar]
- 22.Frock RL, Kudlow BA, Evans AM, Jameson SA, Hauschka SD, Kennedy BK. Lamin A/C and emerin are critical for skeletal muscle satellite cell differentiation. Genes Dev. 2006;20:486–500. doi: 10.1101/gad.1364906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ozawa R, Hayashi YK, Ogawa M, Kurokawa R, Matsumoto H, Noguchi S, Nonaka I, Nishino I. Emerin-lacking mice show minimal motor and cardiac dysfunctions with nuclear-associated vacuoles. Am J Pathol. 2006;168:907–17. doi: 10.2353/ajpath.2006.050564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arimura T, Helbling-Leclerc A, Massart C, Varnous S, Niel F, Lacene E, Fromes Y, Toussaint M, Mura AM, Keller DI, Amthor H, Isnard R, Malissen M, Schwartz K, Bonne G. Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum Mol Genet. 2005;14:155–69. doi: 10.1093/hmg/ddi017. [DOI] [PubMed] [Google Scholar]
- 25.Mounkes LC, Kozlov SV, Rottman JN, Stewart CL. Expression of an LMNA-N195K variant of A-type lamins results in cardiac conduction defects and death in mice. Hum Mol Genet. 2005;14:2167–80. doi: 10.1093/hmg/ddi221. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, Herron AJ, Worman HJ. Pathology and nuclear abnormalities in hearts of transgenic mice expressing M371K lamin A encoded by an LMNA mutation causing Emery-Dreifuss muscular dystrophy. Hum Mol Genet. 2006;15:2479–89. doi: 10.1093/hmg/ddl170. [DOI] [PubMed] [Google Scholar]
- 27.De Sandre-Giovannoli A, Chaouch M, Kozlov S, Vallat JM, Tazir M, Kassouri N, Szepetowski P, Hammadouche T, Vandenberghe A, Stewart CL, Grid D, Levy N. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot- Marie-Tooth disorder type 2) and mouse. Am J Hum Genet. 2002;70:726–36. doi: 10.1086/339274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chaouch M, Allal Y, De Sandre-Giovannoli A, Vallat JM, Amer-el-Khedoud A, Kassouri N, Chaouch A, Sindou P, Hammadouche T, Tazir M, Levy N, Grid D. The phenotypic manifestations of autosomal recessive axonal Charcot-Marie-Tooth due to a mutation in Lamin A/C gene. Neuromuscul Disord. 2003;13:60–7. doi: 10.1016/s0960-8966(02)00196-7. [DOI] [PubMed] [Google Scholar]
- 29.Cao H, Hegele RA. Nuclear lamin A/C R482Q mutation in canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum Mol Genet. 2000;9:109–12. doi: 10.1093/hmg/9.1.109. [DOI] [PubMed] [Google Scholar]
- 30.Shackleton S, Lloyd DJ, Jackson SN, Evans R, Niermeijer MF, Singh BM, Schmidt H, Brabant G, Kumar S, Durrington PN, Gregory S, O’Rahilly S, Trembath RC. LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat Genet. 2000;24:153–6. doi: 10.1038/72807. [DOI] [PubMed] [Google Scholar]
- 31.Novelli G, Muchir A, Sangiuolo F, Helbling-Leclerc A, D’Apice MR, Massart C, Capon F, Sbraccia P, Federici M, Lauro R, Tudisco C, Pallotta R, Scarano G, Dallapiccola B, Merlini L, Bonne G. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am J Hum Genet. 2002;71:426–31. doi: 10.1086/341908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simha V, Agarwal AK, Oral EA, Fryns JP, Garg A. Genetic and phenotypic heterogeneity in patients with mandibuloacral dysplasia-associated lipodystrophy. J Clin Endocrinol Metab. 2003;88:2821–4. doi: 10.1210/jc.2002-021575. [DOI] [PubMed] [Google Scholar]
- 33.Vigouroux C, Magre J, Vantyghem MC, Bourut C, Lascols O, Shackleton S, Lloyd DJ, Guerci B, Padova G, Valensi P, Grimaldi A, Piquemal R, Touraine P, Trembath RC, Capeau J. Lamin A/C gene: sex-determined expression of mutations in Dunnigan-type familial partial lipodystrophy and absence of coding mutations in congenital and acquired generalized lipoatrophy. Diabetes. 2000;49:1958–62. doi: 10.2337/diabetes.49.11.1958. [DOI] [PubMed] [Google Scholar]
- 34.Lelliott CJ, Logie L, Sewter CP, Berger D, Jani P, Blows F, O’Rahilly S, Vidal-Puig A. Lamin expression in human adipose cells in relation to anatomical site and differentiation state. J Clin Endocrinol Metab. 2002;87:728–34. doi: 10.1210/jcem.87.2.8256. [DOI] [PubMed] [Google Scholar]
- 35.Cutler DA, Sullivan T, Marcus-Samuels B, Stewart CL, Reitman ML. Characterization of adiposity and metabolism in Lmna-deficient mice. Biochem Biophys Res Commun. 2002;291:522–7. doi: 10.1006/bbrc.2002.6466. [DOI] [PubMed] [Google Scholar]
- 36.Crisp M, Liu Q, Roux K, Rattner JB, Shanahan C, Burke B, Stahl PD, Hodzic D. Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol. 2006;172:41–53. doi: 10.1083/jcb.200509124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lammerding J, Fong LG, Ji JY, Reue K, Stewart CL, Young SG, Lee RT. Lamins A and C but not lamin B1 regulate nuclear mechanics. J Biol Chem. 2006;281:25768–80. doi: 10.1074/jbc.M513511200. [DOI] [PubMed] [Google Scholar]
- 38.Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, Stewart CL, Lee RT. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest. 2004;113:370–8. doi: 10.1172/JCI19670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lammerding J, Hsiao J, Schulze PC, Kozlov S, Stewart CL, Lee RT. Abnormal nuclear shape and impaired mechanotransduction in emerin-deficient cells. J Cell Biol. 2005;170:781–91. doi: 10.1083/jcb.200502148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ostlund C, Bonne G, Schwartz K, Worman HJ. Properties of lamin A mutants found in Emery-Dreifuss muscular dystrophy, cardiomyopathy and Dunnigan-type partial lipodystrophy. J Cell Sci. 2001;114:4435–45. doi: 10.1242/jcs.114.24.4435. [DOI] [PubMed] [Google Scholar]
- 41.Raharjo WH, Enarson P, Sullivan T, Stewart CL, Burke B. Nuclear envelope defects associated with LMNA mutations cause dilated cardiomyopathy and Emery-Dreifuss muscular dystrophy. J Cell Sci. 2001;114:4447–57. doi: 10.1242/jcs.114.24.4447. [DOI] [PubMed] [Google Scholar]
- 42.Lloyd DJ, Trembath RC, Shackleton S. A novel interaction between lamin A and SREBP1: implications for partial lipodystrophy and other laminopathies. Hum Mol Genet. 2002;11:769–77. doi: 10.1093/hmg/11.7.769. [DOI] [PubMed] [Google Scholar]
- 43.Capanni C, Mattioli E, Columbaro M, Lucarelli E, Parnaik VK, Novelli G, Wehnert M, Cenni V, Maraldi NM, Squarzoni S, Lattanzi G. Altered pre-lamin A processing is a common mechanism leading to lipodystrophy. Hum Mol Genet. 2005;14:1489–502. doi: 10.1093/hmg/ddi158. [DOI] [PubMed] [Google Scholar]
- 44.Sarkar PK, Shinton RA. Hutchinson-Guilford progeria syndrome. Postgrad Med J. 2001;77:312–7. doi: 10.1136/pmj.77.907.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gordon LB, Harten IA, Patti ME, Lichtenstein AH. Reduced adiponectin and HDL cholesterol without elevated C-reactive protein: clues to the biology of premature atherosclerosis in Hutchinson-Gilford Progeria Syndrome. J Pediatr. 2005;146:336–41. doi: 10.1016/j.jpeds.2004.10.064. [DOI] [PubMed] [Google Scholar]
- 46.Stehbens WE, Delahunt B, Shozawa T, Gilbert-Barness E. Smooth muscle cell depletion and collagen types in progeric arteries. Cardiovasc Pathol. 2001;10:133–6. doi: 10.1016/s1054-8807(01)00069-2. [DOI] [PubMed] [Google Scholar]
- 47.Martin GM. Genetic modulation of the senescent phenotype in Homo sapiens. Genome. 1989;31:390–7. doi: 10.1139/g89-059. [DOI] [PubMed] [Google Scholar]
- 48.Oshima J. The Werner syndrome protein: an update. Bioessays. 2000;22:894–901. doi: 10.1002/1521-1878(200010)22:10<894::AID-BIES4>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 49.Fry M. The Werner syndrome helicase-nuclease--one protein, many mysteries. Sci Aging Knowledge Environ 2002. 2002:re2. doi: 10.1126/sageke.2002.13.re2. [DOI] [PubMed] [Google Scholar]
- 50.Hickson ID. RecQ helicases: caretakers of the genome. Nat Rev Cancer. 2003;3:169–78. doi: 10.1038/nrc1012. [DOI] [PubMed] [Google Scholar]
- 51.Chen L, Lee L, Kudlow BA, Dos Santos HG, Sletvold O, Shafeghati Y, Botha EG, Garg A, Hanson NB, Martin GM, Mian IS, Kennedy BK, Oshima J. LMNA mutations in atypical Werner’s syndrome. Lancet. 2003;362:440–5. doi: 10.1016/S0140-6736(03)14069-X. [DOI] [PubMed] [Google Scholar]
- 52.Csoka AB, Cao H, Sammak PJ, Constantinescu D, Schatten GP, Hegele RA. Novel lamin A/C gene (LMNA) mutations in atypical progeroid syndromes. J Med Genet. 2004;41:304–8. doi: 10.1136/jmg.2003.015651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Plasilova M, Chattopadhyay C, Pal P, Schaub NA, Buechner SA, Mueller H, Miny P, Ghosh A, Heinimann K. Homozygous missense mutation in the lamin A/C gene causes autosomal recessive Hutchinson-Gilford progeria syndrome. J Med Genet. 2004;41:609–14. doi: 10.1136/jmg.2004.019661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mounkes LC, Kozlov S, Hernandez L, Sullivan T, Stewart CL. A progeroid syndrome in mice is caused by defects in A-type lamins. Nature. 2003;423:298–301. doi: 10.1038/nature01631. [DOI] [PubMed] [Google Scholar]
- 55.Yang SH, Bergo MO, Toth JI, Qiao X, Hu Y, Sandoval S, Meta M, Bendale P, Gelb MH, Young SG, Fong LG. Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutation. Proc Natl Acad Sci U S A. 2005;102:10291–6. doi: 10.1073/pnas.0504641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang SH, Meta M, Qiao X, Frost D, Bauch J, Coffinier C, Majumdar S, Bergo MO, Young SG, Fong LG. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J Clin Invest. 2006;116:2115–21. doi: 10.1172/JCI28968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Varga R, Eriksson M, Erdos MR, Olive M, Harten I, Kolodgie F, Capell BC, Cheng J, Faddah D, Perkins S, Avallone H, San H, Qu X, Ganesh S, Gordon LB, Virmani R, Wight TN, Nabel EG, Collins FS. Progressive vascular smooth muscle cell defects in a mouse model of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2006;103:3250–5. doi: 10.1073/pnas.0600012103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fong LG, Ng JK, Lammerding J, Vickers TA, Meta M, Cote N, Gavino B, Qiao X, Chang SY, Young SR, Yang SH, Stewart CL, Lee RT, Bennett CF, Bergo MO, Young SG. Prelamin A and lamin A appear to be dispensable in the nuclear lamina. J Clin Invest. 2006;116:743–52. doi: 10.1172/JCI27125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vaughan A, Alvarez-Reyes M, Bridger JM, Broers JL, Ramaekers FC, Wehnert M, Morris GE, Whitfield WGF, Hutchison CJ. Both emerin and lamin C depend on lamin A for localization at the nuclear envelope. J Cell Sci. 2001;114:2577–90. doi: 10.1242/jcs.114.14.2577. [DOI] [PubMed] [Google Scholar]
- 60.Harborth J, Elbashir SM, Bechert K, Tuschl T, Weber K. Identification of essential genes in cultured mammalian cells using small interfering RNAs. J Cell Sci. 2001;114:4557–65. doi: 10.1242/jcs.114.24.4557. [DOI] [PubMed] [Google Scholar]
- 61.Schirmer EC, Guan T, Gerace L. Involvement of the lamin rod domain in heterotypic lamin interactions important for nuclear organization. J Cell Biol. 2001;153:479–89. doi: 10.1083/jcb.153.3.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vergnes L, Peterfy M, Bergo MO, Young SG, Reue K. Lamin B1 is required for mouse development and nuclear integrity. Proc Natl Acad Sci U S A. 2004;101:10428–33. doi: 10.1073/pnas.0401424101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Padiath QS, Saigoh K, Schiffmann R, Asahara H, Yamada T, Koeppen A, Hogan K, Ptacek LJ, Fu YH. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat Genet. 2006;38:1114–23. doi: 10.1038/ng1872. [DOI] [PubMed] [Google Scholar]
- 64.Hegele RA, Cao H, Liu DM, Costain GA, Charlton-Menys V, Rodger NW, Durrington PN. Sequencing of the reannotated LMNB2 gene reveals novel mutations in patients with acquired partial lipodystrophy. Am J Hum Genet. 2006;79:383–9. doi: 10.1086/505885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Holtz D, Tanaka RA, Hartwig J, McKeon F. The CaaX motif of lamin A functions in conjunction with the nuclear localization signal to target assembly to the nuclear envelope. Cell. 1989;59:969–77. doi: 10.1016/0092-8674(89)90753-8. [DOI] [PubMed] [Google Scholar]
- 66.Sinensky M, Fantle K, Trujillo M, McLain T, Kupfer A, Dalton M. The processing pathway of prelamin A. J Cell Sci. 1994;107 (Pt 1):61–7. doi: 10.1242/jcs.107.1.61. [DOI] [PubMed] [Google Scholar]
- 67.Gerace L, Comeau C, Benson M. Organization and modulation of nuclear lamina structure. J Cell Sci Suppl. 1984;1:137–60. doi: 10.1242/jcs.1984.supplement_1.10. [DOI] [PubMed] [Google Scholar]
- 68.Pendas AM, Zhou Z, Cadinanos J, Freije JM, Wang J, Hultenby K, Astudillo A, Wernerson A, Rodriguez F, Tryggvason K, Lopez-Otin C. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet. 2002;31:94–9. doi: 10.1038/ng871. [DOI] [PubMed] [Google Scholar]
- 69.Bergo MO, Gavino B, Ross J, Schmidt WK, Hong C, Kendall LV, Mohr A, Meta M, Genant H, Jiang Y, Wisner ER, Van Bruggen N, Carano RA, Michaelis S, Griffey SM, Young SG. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc Natl Acad Sci U S A. 2002;99:13049–54. doi: 10.1073/pnas.192460799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Agarwal AK, Fryns JP, Auchus RJ, Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet. 2003;12:1995–2001. doi: 10.1093/hmg/ddg213. [DOI] [PubMed] [Google Scholar]
- 71.Shackleton S, Smallwood DT, Clayton P, Wilson LC, Agarwal AK, Garg A, Trembath RC. Compound heterozygous ZMPSTE24 mutations reduce prelamin A processing and result in a severe progeroid phenotype. J Med Genet. 2005;42:e36. doi: 10.1136/jmg.2004.029751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moulson CL, Go G, Gardner JM, van der Wal AC, Smitt JH, van Hagen JM, Miner JH. Homozygous and Compound Heterozygous Mutations in ZMPSTE24 Cause the Laminopathy Restrictive Dermopathy. J Invest Dermatol. 2005;125:913–9. doi: 10.1111/j.0022-202X.2005.23846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Navarro CL, Cadinanos J, De Sandre-Giovannoli A, Bernard R, Courrier S, Boccaccio I, Boyer A, Kleijer WJ, Wagner A, Giuliano F, Beemer FA, Freije JM, Cau P, Hennekam RC, Lopez-Otin C, Badens C, Levy N. Loss of ZMPSTE24 (FACE-1) causes autosomal recessive restrictive dermopathy and accumulation of Lamin A precursors. Hum Mol Genet. 2005;14:1503–13. doi: 10.1093/hmg/ddi159. [DOI] [PubMed] [Google Scholar]
- 74.Liu B, Wang J, Chan KM, Tjia WM, Deng W, Guan X, Huang JD, Li KM, Chau PY, Chen DJ, Pei D, Pendas AM, Cadinanos J, Lopez-Otin C, Tse HF, Hutchison C, Chen J, Cao Y, Cheah KS, Tryggvason K, Zhou Z. Genomic instability in laminopathy-based premature aging. Nat Med. 2005;11:780–5. doi: 10.1038/nm1266. [DOI] [PubMed] [Google Scholar]
- 75.Fong LG, Ng JK, Meta M, Cote N, Yang SH, Stewart CL, Sullivan T, Burghardt A, Majumdar S, Reue K, Bergo MO, Young SG. Heterozygosity for Lmna deficiency eliminates the progeria-like phenotypes in Zmpste24-deficient mice. Proc Natl Acad Sci U S A. 2004;101:18111–6. doi: 10.1073/pnas.0408558102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cahill R, Smith R, Westall RG. A detailed study of the urinary peptides in a patient with osteomalacia and hyperparathyroidism. Clin Sci. 1970;38:519–32. doi: 10.1042/cs0380519. [DOI] [PubMed] [Google Scholar]
- 77.Young SG, Fong LG, Michaelis S. Prelamin A, Zmpste24, misshapen cell nuclei, and progeria--new evidence suggesting that protein farnesylation could be important for disease pathogenesis. J Lipid Res. 2005;46:2531–58. doi: 10.1194/jlr.R500011-JLR200. [DOI] [PubMed] [Google Scholar]
- 78.Gruber J, Lampe T, Osborn M, Weber K. RNAi of FACE1 protease results in growth inhibition of human cells expressing lamin A: implications for Hutchinson-Gilford progeria syndrome. J Cell Sci. 2005;118:689–96. doi: 10.1242/jcs.01652. [DOI] [PubMed] [Google Scholar]
- 79.Fong LG, Frost D, Meta M, Qiao X, Yang SH, Coffinier C, Young SG. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science. 2006;311:1621–3. doi: 10.1126/science.1124875. [DOI] [PubMed] [Google Scholar]
- 80.Sebti SM, Hamilton AD. Farnesyltransferase and geranylgeranyltransferase I inhibitors and cancer therapy: lessons from mechanism and bench-to-bedside translational studies. Oncogene. 2000;19:6584–93. doi: 10.1038/sj.onc.1204146. [DOI] [PubMed] [Google Scholar]
- 81.Toth JI, Yang SH, Qiao X, Beigneux AP, Gelb MH, Moulson CL, Miner JH, Young SG, Fong LG. Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc Natl Acad Sci U S A. 2005;102:12873–8. doi: 10.1073/pnas.0505767102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Capell BC, Erdos MR, Madigan JP, Fiordalisi JJ, Varga R, Conneely KN, Gordon LB, Der CJ, Cox AD, Collins FS. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2005;102:12879–84. doi: 10.1073/pnas.0506001102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Glynn MW, Glover TW. Incomplete processing of mutant lamin A in Hutchinson-Gilford progeria leads to nuclear abnormalities, which are reversed by farnesyltransferase inhibition. Hum Mol Genet. 2005;14:2959–69. doi: 10.1093/hmg/ddi326. [DOI] [PubMed] [Google Scholar]
- 84.Mallampalli MP, Huyer G, Bendale P, Gelb MH, Michaelis S. Inhibiting farnesylation reverses the nuclear morphology defect in a HeLa cell model for Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2005;102:14416–21. doi: 10.1073/pnas.0503712102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gu WZ, Joseph I, Wang YC, Frost D, Sullivan GM, Wang L, Lin NH, Cohen J, Stoll VS, Jakob CG, Muchmore SW, Harlan JE, Holzman T, Walten KA, Ladror US, Anderson MG, Kroeger P, Rodriguez LE, Jarvis KP, Ferguson D, Marsh K, Ng S, Rosenberg SH, Sham HL, Zhang H. A highly potent and selective farnesyltransferase inhibitor ABT-100 in preclinical studies. Anticancer Drugs. 2005;16:1059–69. doi: 10.1097/00001813-200511000-00004. [DOI] [PubMed] [Google Scholar]