

Abstract
Although pain is regarded traditionally as neuronally mediated, recent progress shows an important role of spinal glial cells in persistent pain sensitization. Mounting evidence has implicated spinal microglia in the development of chronic pain (e.g. neuropathic pain after peripheral nerve injury). Less is known about the role of astrocytes in pain regulation. However, astrocytes have very close contact with synapses and maintain homeostasis in the extracellular environment. In this review, we provide evidence to support a role of spinal astrocytes in maintaining chronic pain. In particular, c-Jun N-terminal kinase (JNK) is activated persistently in spinal astrocytes in a neuropathic pain condition produced by spinal nerve ligation. This activation is required for the maintenance of neuropathic pain because spinal infusion of JNK inhibitors can reverse mechanical allodynia, a major symptom of neuropathic pain. Further study reveals that JNK is activated strongly in astrocytes by basic fibroblast growth factor (bFGF), an astroglial activator. Intrathecal infusion of bFGF also produces persistent mechanical allodynia. After peripheral nerve injury, bFGF might be produced by primary sensory neurons and spinal astrocytes because nerve injury produces robust bFGF upregulation in both cell types. Therefore, the bFGF/JNK pathway is an important signalling pathway in spinal astrocytes for chronic pain sensitization. Investigation of signaling mechanisms in spinal astrocytes will identify new molecular targets for the management of chronic pain.
Keywords: neuropathic pain, MAP kinases, neuron–glial interaction
INTRODUCTION
Chronic pain, caused by disease and injury, is a major health problem worldwide. Chronic pain includes, but is not limited to, neural damage associated neuropathic pain (Woolf and Mannion, 1999), arthritis associated inflammatory pain (Dubner and Ruda, 1992), and tumor growth associated cancer pain (Mantyh et al., 2002). These chronic conditions can outlive the initial injuries and damage, and are considered disorders in their own right. Analgesic drugs that are useful for treating acute pain are only partially effective for chronic pain. The reasons for this lack of success in pain treatment include a complex etiology of pain and an incomplete understanding of the mechanisms underlying the induction and maintenance of chronic pain. However, it is generally believed that chronic pain results from neural plasticity in the pain pathway (Dubner and Ruda, 1992; Ji and Woolf, 2001).
Previously, it was thought that only neurons and neural circuits mediated pain, and glial cells served only as a structural support for neurons. However, recent evidence indicates that glial cells are active and that they respond to environmental changes and interact with neurons. This neural–glial interaction is bidirectional. On the one hand, glia express different types of neurotransmitter receptors, which enables them to respond to neural signals. On the other hand, glial cells produce numerous mediators (e.g. proinflammatory cytokines and growth factors) that are neuroactive. There are three types of glial cells in the CNS: microglia, astrocytes and oligodendrocytes. The microglia are derived from bone marrow precursors and were believed to be quiescent under normal conditions. However, a recent study shows that resting microglia are highly dynamic surveillants of brain function (Nimmerjahn et al., 2005). Oligodendrocytes and astrocytes are derived from ectodermal precursors and occur in close apposition to neurons. The oligodendrocytes produce myelin, which ensheathes neuronal axons and allows fast nerve conduction. The astrocytes form networks with themselves and are closely associated with neurons and blood vessels. Their activity is generally thought to mirror the metabolic activity of neurons (Haydon and Carmignoto, 2006). In contrast to the rapid propagation of nerve impulses on the order of m sec−1, glial cell signaling is much slower with rates of propagation measured in μm second−1.
Early evidence for the involvement of spinal glial cells in pain regulation came from studies using metabolic inhibitors specific to glial cells such as fluorocitrate (Meller et al., 1994; Watkins et al., 1997). Also, it was believed that proinflammatory cytokines such as interleukin 1β (IL-1β), IL-6 and tumor necrosis factor α (TNF-α) are produced in glial cells. These cytokines are upregulated in the spinal cord in chronic pain conditions. Functional inhibition of these cytokines can attenuate persistent pain and enhance opioid analgesia (Sweitzer et al., 2001; Watkins et al., 2001; Milligan et al., 2003; Watkins et al., 2005). However, the distinct roles of specific glial subtypes in pain sensitization were not assessed in these studies.
Recently, evidence has accumulated that supports a role of spinal microglia in chronic pain, especially the facilitation of neuropathic pain. Nerve injury induces the expression of microglial markers (e.g. CD11b, TLR4 and CD14) within several hours (DeLeo et al., 2004). Initially, microglia appear to be activated, which activates astrocytes (Aldskogius and Kozlova, 1998; DeLeo et al., 2004). Specifically, nerve injury upregulates several receptors, such as the chemokine receptor CX3CR1 and ATP receptor P2X4 in spinal microglia. Either blocking or deleting these receptors results in decreased neuropathic pain (Tsuda et al., 2003; Milligan et al., 2004; Verge et al., 2004; Zhuang et al., 2006b). Intrathecal injection of ATP-activated microglia induces mechanical allodynia (a nociceptive response to normally innocuous mechanical stimulation) that requires microglial production of BDNF (Tsuda et al., 2003; Coull et al., 2005). A non-specific microglial inhibitor minocycline prevents/delays pain development (Raghavendra et al., 2003; Ledeboer et al., 2005; Hua et al., 2005). Notably, studies from different laboratories have demonstrated that p38 mitogen-activated protein kinase (MAPK) is activated in spinal microglia under different chronic pain conditions, and that blocking this kinase attenuates pain hypersensitivity (Table 1 (Jin et al., 2003; Schafers et al., 2003; Tsuda et al., 2004; Boyle et al., 2006; Hains and Waxman, 2006). A recent study shows that nerve injury-induced cleavage of the chemokine fractalkine results in activation of p38 in spinal microglia via CX3CR1 receptors (Zhuang et al., 2006b).
Table I.
Activation of MAPKs in spinal glial cells after SNLa
| MAPKs | Early phase (3 days) | Mid phase (10 days) | Late phase (21 days) |
|---|---|---|---|
| ERK | Microglia | Microglia/Astrocytes | Astrocytes |
| p38 | Microglia | Microglia | Microglia |
| JNK | Astrocytes | Astrocytes | Astrocytes |
Neuropathic pain is induced within 3 days (early phase), fully established at 10 days (mid phase) and maintained at 21 days (late phase).
Compared with the ample evidence for microglial regulation of pain, less is known about the importance of spinal astrocytes in chronic pain, largely because of the lack of specific pharmacological tools. However, there are more astrocytes in the CNS than other cells, and they and have strong structural inter-relationship with neurons by enwrapping synaptic terminals, enabling signaling between glia and neurons. It is becoming evident that astrocytes are involved intimately in neuronal signaling by releasing glutamate and ATP (Haydon, 2001). Astrocytes are thought to signal each other through gap junctions in waves of Ca2+ release (Haydon, 2001). Evidence is accumulating that spinal astrocytes have a role in maintaining chronic pain sensitization.
Involvement of spinal astrocytes in chronic pain
Persistent changes in spinal astrocytes in chronic pain states
Glial fibrillary acidic protein (GFAP), vimentin and S-100β are the markers that are used most often to identify astrocytes (Ridet et al., 1997). Although astrocytes are not as homogenous as previously thought, GFAP appears to label most astrocytes in the spinal cord. The change in staining density of GFAP was analyzed initially in the chronic constriction injury model of neuropathic pain. In this study, increased density of GFAP staining was attributed primarily to hypertrophy of astrocytes rather than either their proliferation or their migration. The magnitude of the increase in GFAP staining appears to correlate with the degree of hyperalgesia (Garrison et al., 1991). A subsequent study shows that the NMDA receptor is, in part, responsible for GFAP expression (Garrison et al., 1994). A correlation between GFAP expression and chronic pain has also been identified in other studies (Colburn et al., 1997; Colburn et al., 1999). However, chronic pain can also been suppressed without inhibition of GFAP expression (Zhuang et al., 2006a). It is unclear whether upregulation of GFAP is required or/and sufficient for chronic pain sensitization, but mounting evidence indicates that persistent activation (e.g. GFAP upregulation) of spinal astrocytes is a unique feature of chronic pain in different animal models following bone cancer (Honore et al., 2000; Mantyh et al., 2002), spinal nerve ligation (SNL) (Tanga et al., 2004; Zhuang et al., 2006a), partial sciatic-nerve ligation (Zhang et al., 2006), spinal cord injury (Nesic et al., 2006) and adjuvant-induced inflammation (Raghavendra et al., 2004). In addition, S100β is also upregulated in the spinal cord after nerve injury. Whereas S100β-deficient mice have reduced mechanical allodynia after nerve injury, allodynia is enhanced in mice that overexpress S100β, supporting a role of this astroglial protein in the pathophysiology of neuropathic pain (Tanga et al., 2006).
Are astrocytes sufficient and required for chronic pain sensitization?
Spinal astrocytes appear to be sufficient to produce persistent pain. Implantation of neural stem cells into the injured spinal cord improves motor recovery and causes allodynia-like hypersensitivity of the forepaws (Hofstetter et al., 2005; Macias et al., 2006). Because most of the stem cells that are implanted in the spinal cord become astrocytes, implantation-induced allodynia is likely to be attributed to the action of astrocytes. This allodynia is associated with aberrant sprouting of pain-mediating calcitonin gene-related peptide (CGRP)-positive fibers in the dorsal horn due to the release of growth factors (e.g. nerve growth factor) from astrocytes (Hofstetter et al., 2005). In contrast, sprouting and allodynia are prevented if neural stem cells are transfected with neurogenin-2 before transplantation to suppress the generation of astrocytes (Hofstetter et al., 2005).
Spinal astrocytes might also be required for the maintenance of chronic pain. L-alpha-aminoadipate (L-α-AA) is a cytotoxin that is relatively specific for astrocytes. Ultrastructural studies indicate that cell degeneration is confined to astrocytes following the injection of this toxin into the striatum (Huck et al., 1984; Khurgel et al., 1996; Rodriguez et al., 2004). Intrathecal injection of L-α-AA to spinal nerve-ligated rats produces a dose-dependent attenuation of mechanical allodynia, which is a major symptom of chronic pain (Fig. 1). In agreement with previous studies (Khurgel et al., 1996; Rodriguez et al., 2004), L-α-AA produces a marked reduction of GFAP-positive astrocytes in the dorsal horn but has no effect on NeuN-positive spinal neurons (Zhuang et al., 2006a). The anti-allodynic effect of the toxin is reversible, recovering after 3 days (Fig. 1). This reversible effect might be attributed to migration of neighboring astrocytes. Furthermore, intrathecal injection of this astroglial toxin does not alter the basal sensitivity to pain, which indicates that astrocytes might not be essential for mediating basal pain sensitivity (Zhuang et al., 2006a).
Fig. 1. Spinal infusion of astroglial toxin alpha-aminoadipate (L-α-AA) blocks neuropathic pain.
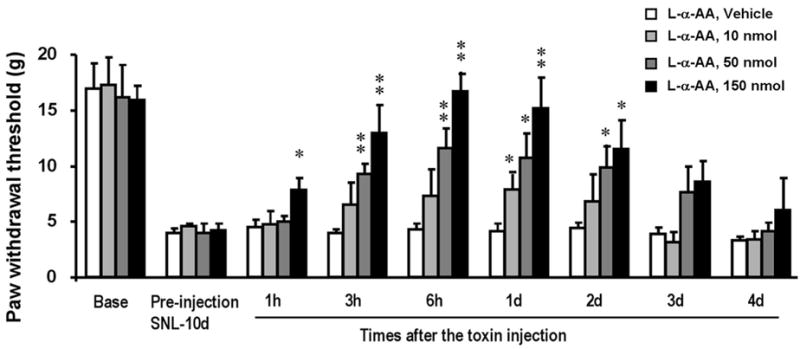
L-α-AA (10, 50, 150 nmol) was injected intrathecally 10 days after SNL. Mechanical allodynia, a major feature of chronic pain, is dose-dependently suppressed by the toxin. *, P<0.05; **, P<0.01, compared to vehicle (saline) control; ANOVA; n=6. Mechanical allodynia was tested using von Frey hairs. Reproduced, with permission, from (Zhuang et al., 2006a).
Gap junctions and astroglial networks
Astrocytes are characterized by the formation of astroglial networks (gap junctions) (Giaume and McCarthy, 1996; Giaume and Venance, 1998; Nagy et al., 2004). Gap junctions exist at apposing plasma membranes of many cell types, and contribute to local metabolic homeostasis and synchronization of cellular activities by allowing direct intercellular movements of ions, metabolites and second messenger molecules up to 1000 Daltons. These junctions are composed of hemi-channels called connexons. Each connexon is composed of six gap-junction proteins, termed connexins (Giaume and Venance, 1998; Nagy et al., 2004). In the adult dorsal horn, gap junctions form predominantly between astrocytes. Connexin-43 (Cx43) is regarded as the main functional connexin in astrocytes (Giaume and Venance, 1998; Nagy et al., 2004). Facial nerve lesion induces a rapid upregulation of Cx43 in the facial nucleus (Rohlmann et al., 1993). Cx43 is also upregulated in the spinal cord after spinal cord injury (Lee et al., 2005). It is noteworthy that the gap-junction blocker carbenoxolone suppresses the spread of pain (Spataro et al., 2004). In a model of nerve inflammation, high concentrations of zymosan delivered to the sciatic nerve produces a ‘mirror pain’ in the contralateral paw (mechanical allodynia), which is suppressed by intrathecal carbenoxolone (Spataro et al., 2004).
Signaling molecules in spinal astrocytes and their roles in pain regulation
Astrocytes express the glutamate transporters GLT-1 and GLAST. These transporters are believed to provide principal route for glutamate removal from synaptic clefts and the extracellular space (Huang and Bergles, 2004; Tawfik et al. 2006). Nerve injury produces a persistent downregulation of the transporters, after an initial rise. Downregulation might result in a decrease in glutamate uptake and a subsequent increase in excitatory synaptic transmission. Thus, neuropathic pain is attenuated by riluzole, a glutamate-transporter activator given intrathecally (Sung et al. 2003). However, GLT-1 appears to have an opposite role in acute pain conditions. For example, intrathecal injection of a GLT-1 inhibitor inhibits rather than enhance long-term potentiation (LTP) of spinal neurons following tetanic stimulation of the sciatic nerve (Wang et al., 2006). Spinal LTP has been implicated in pain sensitization (Sandkuhler, 2000). Also GLT-1 is upregulated transiently in the spinal cord after injection of formalin into the hindpaw, and either inhibition or knockdown of GLT-1 suppresses formalin-induced pain behavior (Niederberger et al., 2003). These studies indicate that glutamate transporters might have different roles in acute and chronic pain conditions.
Spinal cord injury induces an immediate increase in plasma endothelin (ET) levels and a sustained increase in tissue ET levels. ET-1 also induces hypertrophy of astrocytes (Rogers et al., 2003). Interestingly, ET receptor-B (ETB) is induced in spinal astrocytes after spinal cord injury (Table 2) (Peters et al., 2003). Thus, strategies that block ET receptors following spinal cord injury might reduce ischemia and also suppress astrogliosis and chronic pain.
Table 2. Signaling molecules in spinal astrocytes.
Most of them are regulated in chronic pain conditions and have a role in modulating chronic pain.
| Signaling molecule | Change in expression in chronic pain | Role in chronic pain | Refs |
|---|---|---|---|
| pERK | Upregulation | Maintains neuropathic pain | Zhuang et al., 2005 |
| pJNK | Upregulation | Maintains neuropathic pain | Zhuang et al., 2006a |
| p-c-Jun | Upregulation | Not tested | Zhuang et al., 2006a |
| JNK1 | Not tested | Not tested | Zhuang et al., 2006a |
| Neurokinin-2 receptor | Not tested | Maintains neuropathic pain | Garry et al., 2005 |
| Interlukin-1β | Upregulation | Maintains neuropathic pain | Zhang et al., 2005 |
| Glutamate transporter-I | Downregulation | Suppresses neuropathic pain | Sung et al., 2003 |
| Endothelin receptor-B | Upregulation | Not tested | Peters et al., 2003 |
| Connexin-43 | Upregulation | Not tested | Lee et al., 2005 |
| bFGF | Upregulation | Maintains neuropathic pain | Madiai et al., 2005 |
Although cells normally produce L-type amino acids, astrocytes can produce D-serine. Astrocytes also contain the major enzyme for the biosynthesis of D-serine, serine racemase. D-serine increases the sensitivity of NMDA receptors in hippocampal neurons by binding to the glycine site of the receptor and facilitating the induction of LTP (Wolosker et al., 2006). Intrathecal injection of D-serine facilitates pain via NMDA receptors (Kolhekar et al., 1994). Conversely, injection of an inhibitor of the enzyme that degrades D-serine into the anterior cingulated cortex attenuates affective pain (Ren et al. 2006).
MAPKs are a family of protein kinases that play important role in intracellular signal transduction. This family includes three major members: extracellular signal-regulated kinase (ERK), p38 and c-Jun-N-terminal kinase (JNK). As mentioned above, p38 is activated in spinal microglia (Jin et al., 2003), and therefore is not the focus of this review. ERK is the most studied member of the MAPK family. In acute and inflammatory pain conditions, ERK is activated in dorsal horn neurons, which contributes to the induction and maintenance of dorsal horn neuron sensitization and pain hypersensitivity (Ji et al., 1999; Ji et al., 2002). Notably, ERK is activated in spinal glial cells after nerve injury. Phosphorylated ERK (pERK), which is the active form, is induced in spinal astrocytes at late times after SNL (Zhuang et al. 2005). pERK was found in microglia on day 2, in both microglia and astrocytes on day 10, but in astrocytes only on day 21 (Table 1) (Zhuang et al., 2005). pERK is also present in spinal astrocytes 3 weeks after partial sciatic nerve injury (Ma and Quirion, 2002). Spinal inhibition of this late-phase activation of ERK by intrathecal MEK inhibitor reverses mechanical allodynia, supporting a role of astrocytic ERK in the maintenance of neuropathic pain (Zhuang et al., 2005).
Zerari et al. show that the neurokinin-2 receptor, which is activated by extrasynaptic neurokinin A, is expressed exclusively in spinal astrocytes (Zerari et al., 1998). An NK2 receptor agonist activates ERK and causes behavioral sensitization of pain that is prevented by a MEK inhibitor. Conversely, an NK2 receptor antagonist suppresses ERK activation and neuropathic pain following nerve injury (Garry et al., 2005).
IL-1β is upregulated in the spinal cord in different chronic pain conditions and plays an important role in pain facilitation (Sweitzer et al., 2001; Milligan et al., 2003). In a recent model of bone cancer pain in rats, which results from inoculation of the tibia with prostate cancer cells, IL-1β is induced in spinal astrocytes at certain times (Zhang et al., 2005). However, IL-1β is also found in neurons in the spinal cord (DeLeo et al., 1997; Fu et al., 2006). It is likely that IL-1β is induced in different types of spinal cells either at different times of pain development or in different chronic pain models.
Activation of the JNK cascade in spinal astrocytes and neuropathic pain
JNK is the signaling molecule that is the focus of this review. JNK is the least studied member of the MAPK family. Although all three MAPKs are activated in spinal glial cells after nerve injury, they have different patterns: ERK is activated sequentially in microglia and astrocytes, p38 is activated in microglia, whereas JNK is activated persistently in astrocytes (Table 1) (Jin et al., 2003; Zhuang et al., 2005; Zhuang et al., 2006a). Thus, SNL induces a marked increase in pJNK-immunoreactive (IR) cells in the dorsal horn of the injured side (Fig. 2a,b). Furthermore, although pJNK colocalizes with the astroglial marker GFAP (Fig. 2c,d), pJNK is only expressed in a portion of spinal astrocytes (Zhuang et al., 2006a). JNK activation also occurs in spinal astrocytes 3 weeks after partial sciatic nerve injury (Ma and Quirion, 2002) and in amyotrophic lateral sclerosis (Migheli et al., 1997). JNK is also activated in spinal astrocytes in another chronic pain condition following adjuvant-induced inflammation (Zhuang and Ji, unpublished observation). However, it is noteworthy that pJNK occurs in spinal neurons after traumatic spinal cord injury, a condition that produces robust neuronal apoptosis in the spinal cord. It is known that JNK has a role in stress-induced apoptosis in the nervous system (Borsello et al., 2003). However, following peripheral nerve injury, neuronal apoptosis in the spinal cord is not prominent (Polgar et al., 2005; Scholz et al., 2005). There is no evidence to indicate that the pJNK-positive spinal astrocytes undergo apoptosis after nerve injury.
Fig. 2. SNL induces persistent JNK activation in spinal astroglia.
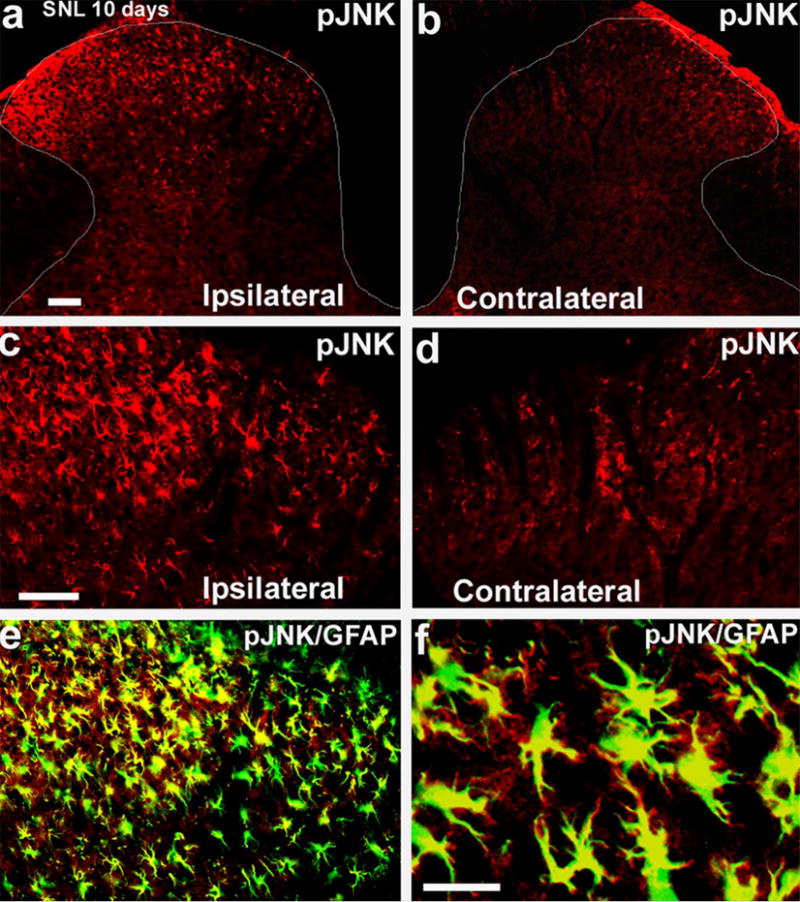
(a,b) Immunohistochemistry reveals an increase in pJNK in the ipsilateral spinal dorsal horn (L5) 10 days after SNL. White lines indicate the border of the dorsal horn. Scale, 50 μm. (c,d) High-magnification images of (a) and (b), respectively, showing pJNK staining in the medial superficial dorsal horn. Scale, 50 μm. (e) Double immunofluorescence shows that pJNK (red) colocalizes with the astroglial marker GFAP (green) in the medial superficial dorsal horn. Two single-stained images are merged. c, d, e have the same magnification. (f) High-magnification image of (e) demonstrates colocalization of pJNK and GFAP. Note that some fine processes of astrocytes are labeled by pJNK but not by GFAP antibody. Scale bar, 25 μm. Reproduced, with permission, from (Zhuang et al., 2006a).
The transcription factor c-Jun is the best-known substrate of JNK. JNK activates c-Jun by phosphorylation to form p-c-Jun. SNL also upregulates p-c-Jun in the ipsilateral spinal cord. P-c-Jun also localizes to GFAP-expressing astrocytes, predominantly in the nucleus (Zhuang et al., 2006a). Of the three isoforms of JNK (JNK1, JNK2 and JNK3), JNK3 is expressed in neurons and JNK1 is expressed in non-neuronal cells (e.g. immune cells) (Ip and Davis 1998; Borsello et al., 2003; Kuan et al., 2003). JNK1 is also expressed in spinal astrocytes and only this isoform is hyperphosphorylated in the spinal cord after SNL (Zhuang et al., 2006a). Thus, the whole JNK cascade is localized preferentially in spinal astrocytes after nerve injury (Table 2).
Is activation of JNK in spinal astrocytes after nerve injury essential for chronic pain sensitization? Answering this question requires a potent, specific inhibition of JNK. Recently, a peptide inhibitor of JNK, which is derived from the JNK-binding domain of JNK-interacting protein-1 (JIP-1), was designed to block selectively the access of JNK to c-Jun and other substrates by a competitive mechanism (Borsello and Bonny, 2004). A TAT sequence (transporter sequence) is linked to the peptide to render it membrane permeable. A convert to D-form amino acids further makes the peptide proteinase-resistant. This highly-specific peptide inhibitor, called D-JNKI-1 is a potent neuroprotectant against excitotoxicity of cortical neurons (Borsello et al., 2003; Borsello and Bonny, 2004). Spinal infusion of this inhibitor intrathecally does not change basal pain thresholds, but it prevents mechanical allodynia, a major neuropathic pain symptom, for >10 days (Zhuang et al., 2006a). Because JNK is also activated transiently (<3 days) in primary sensory neurons, the preventive effect of D-JNKI-1 in the first several days might be mediated by JNK in dorsal root ganglia (DRG). However, the maintenance of neuropathic pain (reversal) is predominantly, if not exclusively, mediated by spinal JNK (Zhuang et al., 2006a).
It is important to investigate whether inhibition of JNK also reverses established neuropathic pain, a treatment mode that is more relevant to clinical situation. Infusing D-JNKI-1 intrathecally via osmotic pump effectively reverses SNL-induced allodynia for several days (Fig. 3a). A single bolus injection of D-JNKI-1 inhibitors also effectively reverses mechanical allodynia for >12 hours (Fig. 3b). D-JNKI-1 is more potent than the small molecule inhibitor SP600125, which is used currently, with an ED50 that is 50-times less (Zhuang et al., 2006a). D-JNKI-1 also suppresses nerve injury-induced activation of c-Jun in astrocytes, which is a major downstream target of JNK (Zhuang et al., 2006a).
Fig. 3. Spinal infusion of the JNK inhibitors reverses neuropathic pain after SNL.
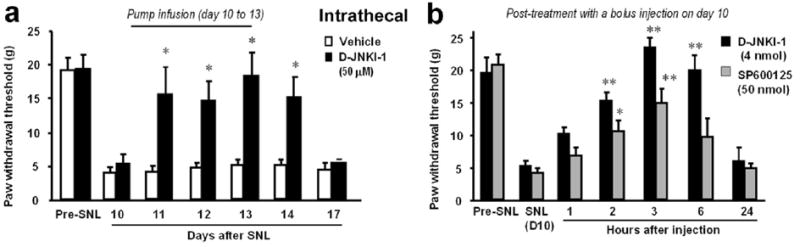
(a) Reversal of SNL-induced mechanical allodynia by intrathecal infusion of the peptide inhibitor of JNK D-JNKI-1 (50 μM) via an osmotic pump (0.5 μl hr−1 for 3 days) starting 10 days after SNL. *, P<0.05, compared to corresponding saline controls; t-test; n=5. (b) Reversal of SNL-induced mechanical allodynia by a bolus intrathecal injection of D-JNKI-1 (4 nmol) and SP600125 (50 nmol) 10 days after SNL. *, P<0.05; **, P<0.01, compared to corresponding pre-injection baseline; ANOVA; n=6. Note that the peptide inhibitor D-JNKI-1 is more potent than the small molecule inhibitor SP600125 in reversing allodynia. Reproduced, with permission, from (Zhuang et al., 2006a).
bFGF/JNK pathway in astrocytes and persistent pain
bFGF (OR FGF-2) is a well-known activator of astrocytes. bFGF is produced by astrocytes and strongly induces their mitosis, growth, differentiation and gliosis (Ferrara et al., 1988; Eclancher et al., 1990). bFGF is induced in the CNS in many injury conditions. For example, bFGF is induced in injured brain regions (mainly in astrocytes) after trauma and in disease pathology such as Alzheimer's where astrogliosis is very active (Gomez-Pinilla, et al., 1990). After spinal cord injury, bFGF is upregulated in the spinal cord, which promotes functional recovery (Koshinaga et al., 1993; Lee et al., 1999; Rabchevsky et al., 2000). bFGF is also upregulated in the spinal cord after sciatic cryoneurolysis but not after chronic constriction injury (DeLeo et al., 1997), which indicates that upregulation might be associated with the severity of the injury. In particular, after SNL, Madiai et al. show that bFGF immunoreactivity increases in reactive astrocytes in the ipsilateral dorsal horn at either 1 week or 3 weeks after nerve ligation (Table 2) (Madiai et al., 2003). The release of bFGF from astrocytes might act in an autocrine manner to further augment astroglial activation (e.g. astrogliosis and proliferation).
As a pleiotropic cytokine, bFGF is synthesized and secreted by both astrocytes and neurons. bFGF is expressed in primary sensory neurons in the DRG. Normally, mRNA that encodes bFGF is expressed in 5% of small neurons in DRG. Nerve injury (e.g. transection of the sciatic nerve and axotomy) induces a dramatic, rapid upregulation of bFGF mRNA; almost all injured DRG neurons contain bFGF mRNA 3 days after axotomy (Fig. 4). An increase in bFGF protein is also evident in DRG neurons after nerve injury (Ji et al., 1995). Although release of bFGF from central terminals of primary sensory neurons after nerve injury has not been shown, bFGF immunoreactivity is present in vesicle-like structures in the cytoplasm, which indicates the possibility that bFGF is released after nerve injury (Ji et al., 1995). bFGF upregulation in the DRG has also been shown in the SNL model (Madiai et al., 2003).
Fig. 4. Nerve injury induces expression of bFGF mRNA in the dorsal root ganglion (DRG).
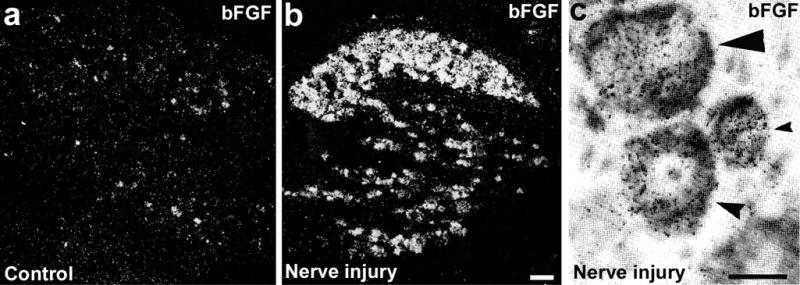
(a,b) Dark-field images following in situ hybridization show expression of bFGF mRNA in the control, noninjured DRG (a) and 3-day axotomized DRG (b). There are much fewer bFGF-positive cells in the control DRG than in the DRG after nerve injury. Scale bar, 100 μm. (c) Bright-field image folowing in situ hybridization shows expression of bFGF mRNA in the injured DRG neurons. The section is counterstained with toluidine blue. Small, medium and large arrowheads indicate small, medium and large neurons, respectively. bFGF-positive cells are labeled with silver grains. The oligodeoxynucleotide probe for bFGF mRNA is labeled with 35S-dATP. Scale bar, 25 μm. Reproduced, with permission, from (Ji et al., 1995).
As discussed above, after peripheral nerve injury, bFGF is produced both in spinal astrocytes and DRG primary sensory neurons. Is endogenous bFGF in the spinal cord important for producing chronic pain? An antagonist of the bFGF receptor is not available, so Madiai et al. tested the role of bFGF in neuropathic pain using a neutralizing antibody to bFGF, delivered intrathecally after SNL. This antibody reduces SNL-induced expression of GFAP in the spinal cord and reverses SNL-induced tactile allodynia, which indicates that endogenous bFGF contributes to maintaining neuropathic tactile allodynia (Madiai et al., 2005).
To examine whether exogenous bFGF is sufficient to induce pain hypersensitivity, we infused bFGF into spinal cord for 1 week through an osmotic pump. Mechanical allodynia develops slowly after infusion of bFGF: mechanical thresholds to Von Frey hair stimuli do not decrease until 4 days after the infusion. Interestingly, allodynia is maintained even after the termination of the infusion (Fig. 5). This slow, persistent development of allodynia is indicative of a role of spinal astrocytes. Infusion of bFGF also increases expression of GFAP in the spinal cord. Notably, spinal injection of adenovirus that encodes bFGF causes overexpression of bFGF in dorsal horn astrocytes and produces persistent hyperalgesia (Romero et al., 2000). Together, these results indicate that bFGF is both sufficient and required for producing chronic pain.
Fig. 5. Spinal infusion of bFGF induces delayed but persistent mechanical allodynia.
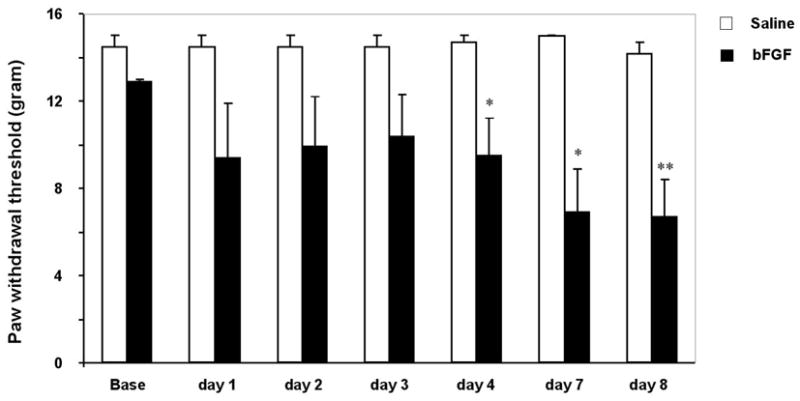
Either bFGF or saline was infused intrathecally via an osmotic pump (10 ng μl−1 h−1) for one week. **, P<0.01 compared to saline control; unpaired t-test; n=6. Mechanical allodynia does not develop until day 4 and is maintained on day 8.
bFGF is a primary ‘activator’ of astrocytes and JNK is an important signaling molecule in spinal astrocytes, so it is reasonable to ask whether bFGF activates JNK in spinal astrocytes. Intrathecal infusion of bFGF induces a marked activation of JNK in the spinal cord (Fig. 6a,b). JNK activation has also been also examined in astroglial cultures. bFGF is a powerful activator of JNK in these cultures (Fig. 6b). However, two additional potential activators of astrocytes, plasminogen (Liu et al., 2000) and ciliary neurotrophic factor (CNTF) (Escartin et al., 2006) do not cause obvious activation of JNK (Fig. 6b). bFGF also induces marked activation of ERK/MAPK in astrocytes, which is evident in the spinal cord at late times of nerve injury (Zhuang et al., 2005). However, activation of p38 MAPK is not evident in astroglial cultures (Fig. 6b), in support of the observation in vivo that p38 is activated in spinal microglia (Jin et al., 2003).
Fig. 6. Activation of JNK by bFGF in astrocytes.
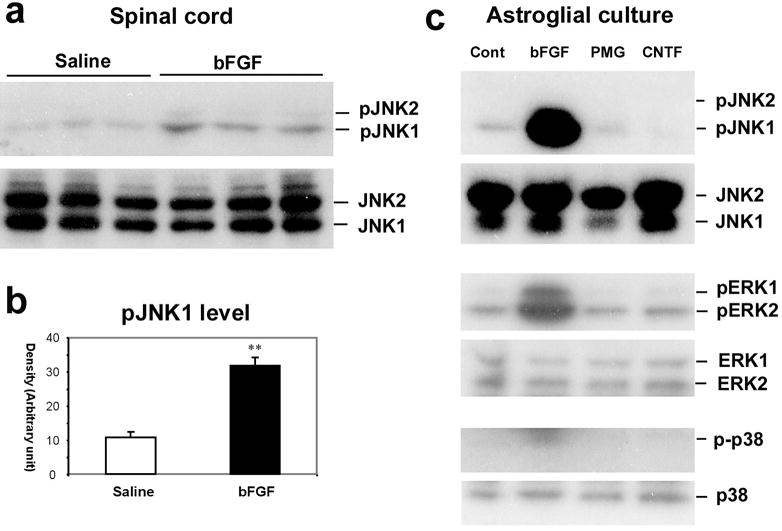
(a) Upper panel: Western blotting in the upper panel reveals an increase in pJNK1 levels in the spinal dorsal horn after bFGF infusion intrathecally via an osmotic pump (10 ng μl−1h−1) for one week. The spinal tissues were collected on day 8 after final behavioral testing (see Fig. 5). Note that only pJNK1 is expressed in the spinal cord. Lower panel: total concentration of JNK does not change after bFGF infusion. (b) Density of pJNK1 bands in (a), normalized to JNK1 loading control. **, P<0.01 compared to saline control. (c) Western blotting shows that bFGF induces pJNK1, pERK1 and pERK2 in astrocytic cultures. By contrast, plasminogen (PMG) and ciliary neurotrophic factor (CNTF) do not activate JNK. p-p38 levels are barely detected in astroglial cultures following all the reagents. Astroglial cultures were prepared from brains of neonatal rats and maintained for 2–3 weeks. Cultures were stimulated with bFGF, PMG and CNTF (each at 100 ng ml−1) for 2 hours.
It is noteworthy that bFGF induces the release of NGF from astrocytes (Fukumoto et al., 1991; Yoshida and Gage, 1991). NGF is a crucial signalling molecule for regulating the phenotype of nociceptive primary sensory neurons. NGF released from spinal astrocytes can be taken up by spinal axonal terminals, causing sprouting of substance P- and CGRP-positive axons and hyperalgesia (Romero et al., 2000). The NGF that is taken up might also be transported retrogradely to DRG neurons, where it induces the expression of pronociceptive genes encoding, for example, the capsaicin receptor transient receptor potential V1 subtype (TRPV1), and the neuropeptides substance P and CGRP.
How doe activation of JNK in spinal astroglia regulate chronic pain? Because inhibition of JNK suppresses SNL-induced phosphorylation of c-Jun in spinal astrocytes (Zhuang et al., 2006a), JNK activation is likely to regulate gene transcription in spinal astrocytes via activation of the transcription factor c-Jun and other transcription factors such as ATF-2. After nerve injury, there is increased synthesis in the spinal cord of inflammatory mediators such as cytokines (IL-1β, TNF-α and IL-6), nitric oxide (NO), which is produced by inducible NO synthase (iNOS), and prostaglandin E2 (PGE2) (produced by either COX-1 or COX-2); all these mediators and enzymes are implicated in pain sensitization (Samad et al., 2001; Watkins et al., 2001; DeLeo et al., 2004; Ji and Strichartz, 2004). Mixed lineage kinases (MLK) are specific JNK kinases. Falsig et al. have shown that MLK inhibitors such as CEP-1347 are potent astrocyte immune modulators (Falsig et al., 2004). In astrocytes in culture, CEP-1347 blocks activation of JNK, expression of COX-2 and iNOS, and release of nitric oxide, PGE2 and IL-6 following challenge with a mixture of cytokines (Falsig et al., 2004). JNK might also regulate chronic pain by modulating the activity of gap junctions that form mainly between astrocytes (Petrich et al., 2004).
Concluding remarks and future directions
Accumulating evidence shows that persistent changes in spinal astrocytes in different chronic pain conditions often outlast microglial changes. This feature of spinal astrocytes implies a role of these cells in the maintenance of chronic pain. Consistent with this, several drugs that affect astroglial function (e.g. JNK inhibitors, MEK inhibitors and propentofylline) reverse persistent, chronic pain. Spinal astrocytes appear to be both sufficient and required for chronic pain sensitization. Although GFAP upregulation and gliosis are generally regarded as the markers of ‘astroglial activation’, studies reviewed here indicate that additional markers are needed that go beyond the vaguely defined state of ‘activation’ and are associated with cellular function related to pain regulation. Signaling molecules such as JNK are not only activated in spinal astrocytes, but also contribute to chronic pain. The bFGF/JNK cascade is an important signaling pathway in spinal astrocytes that promotes chronic pain, especially after nerve and spinal cord injury.
Although current evidence supports a pronociceptive role of spinal astrocytes, presumably by enhancing spinal synaptic transmission via the release of neuroactive substances and inflammatory mediators (e.g. cytokines, NGF and PGE2), studies that establish a causative link between inhibiting spinal cord astrocytic activation and physiological function of spinal neurons are lacking and should be considered in future. Studies in the hippocampus have shown that astroglial activation might have the dual role of enhancing synaptic transmission by releasing glutamate and suppressing synaptic transmission by releasing ATP, which is hydrolyzed to adenosine (Haydon and Carmingnoto, 2006). This discrepancy might be attributable to different roles of astrocytes in physiological and pathological conditions. Recently, 2-photon laser scanning microscopy has been used to image astrocytes in intact brains (Tian et al., 2006). This imaging method will be helpful in demonstrating the activation of astroglial circuits in the intact spinal cord following peripheral noxious and innocuous stimuli. Functional understanding of the cellular and molecular alterations of astroglia-dependent synaptic transmission will help to clarify the role of astrocytes in pain regulation and lead to the identification of novel therapeutic targets for chronic pain.
Acknowledgments
This study is supported by NIH grants DE17794, NS54932, and TW7180 (JRR) and a collaboration grant from International Association for the Study of Pain (JRR and ID).
References
- Aldskogius H, Kozlova EN. Central neuron-glial and glial-glial interactions following axon injury. Progress in Neurobology. 1998;55:1–26. doi: 10.1016/s0301-0082(97)00093-2. [DOI] [PubMed] [Google Scholar]
- Borsello T, Bonny C. Use of cell-permeable peptides to prevent neuronal degeneration. Trends in Molecular Medicine. 2004;10:239–244. doi: 10.1016/j.molmed.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF, et al. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Molecular Medicine. 2003;9:1180–1186. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]
- Boyle DL, Jones TL, Hammaker D, Svensson CI, Rosengren S, Albani S, et al. Regulation of Peripheral Inflammation by Spinal p38 MAP Kinase in Rats. PLoS Medicine. 2006;3:e338. doi: 10.1371/journal.pmed.0030338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colburn RW, DeLeo JA, Rickman AJ, Yeager MP, Kwon P, Hickey WF. Dissociation of microglial activation and neuropathic pain behaviors following peripheral nerve injury in the rat. Journal of Neuroimmunology. 1997;79:163–175. doi: 10.1016/s0165-5728(97)00119-7. [DOI] [PubMed] [Google Scholar]
- Colburn RW, Rickman AJ, DeLeo JA. The effect of site and type of nerve injury on spinal glial activation and neuropathic pain behavior. Experimental Neurology. 1999;157:289–304. doi: 10.1006/exnr.1999.7065. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Colburn RW, Rickman AJ. Cytokine and growth factor immunohistochemical spinal profiles in two animal models of mononeuropathy. Brain Research. 1997;759:50–57. doi: 10.1016/s0006-8993(97)00209-6. [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Tanga FY, Tawfik VL. Neuroimmune activation and neuroinflammation in chronic pain and opioid tolerance/hyperalgesia. Neuroscientist. 2004;10:40–52. doi: 10.1177/1073858403259950. [DOI] [PubMed] [Google Scholar]
- Dubner R, Ruda MA. Activity-dependent neuronal plasticity following tissue injury and inflammation. Trends in Neurosciences. 1992;15:96–103. doi: 10.1016/0166-2236(92)90019-5. [DOI] [PubMed] [Google Scholar]
- Eclancher F, Perraud F, Faltin J, Labourdette G, Sensenbrenner M. Reactive astrogliosis after basic fibroblast growth factor (bFGF) injection in injured neonatal rat brain. Glia. 1990;3:502–509. doi: 10.1002/glia.440030609. [DOI] [PubMed] [Google Scholar]
- Escartin C, Brouillet E, Gubellini P, Trioulier Y, Jacquard C, Smadja C, et al. Ciliary neurotrophic factor activates astrocytes, redistributes their glutamate transporters GLAST and GLT-1 to raft microdomains, and improves glutamate handling in vivo. Journal of Neuroscience. 2006;26:5978–5989. doi: 10.1523/JNEUROSCI.0302-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falsig J, Porzgen P, Lotharius J, Leist M. Specific modulation of astrocyte inflammation by inhibition of mixed lineage kinases with CEP-1347. Journal of Immunology. 2004;173:2762–2770. doi: 10.4049/jimmunol.173.4.2762. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Ousley F, Gospodarowicz D. Bovine brain astrocytes express basic fibroblast growth factor, a neurotropic and angiogenic mitogen. Brain Research. 1988;462:223–232. doi: 10.1016/0006-8993(88)90550-1. [DOI] [PubMed] [Google Scholar]
- Fu D, Guo Q, Ai Y, Cai H, Yan J, Dai R. Glial activation and segmental upregulation of interleukin-1beta (IL-1beta) in the rat spinal cord after surgical incision. Neurochemistry Research. 2006;31:333–340. doi: 10.1007/s11064-005-9032-4. [DOI] [PubMed] [Google Scholar]
- Fukumoto H, Kakihana M, Suno M. Recombinant human basic fibroblast growth factor (rhbFGF) induces secretion of nerve growth factor (NGF) in cultured rat astroglial cells. Neuroscience Letters. 1991;122:221–224. doi: 10.1016/0304-3940(91)90863-o. [DOI] [PubMed] [Google Scholar]
- Garrison CJ, Dougherty PM, Carlton SM. GFAP expression in lumbar spinal cord of naive and neuropathic rats treated with MK-801. Experimental Neurology. 1994;129:237–243. doi: 10.1006/exnr.1994.1165. [DOI] [PubMed] [Google Scholar]
- Garrison CJ, Dougherty PM, Kajander KC, Carlton SM. Staining of glial fibrillary acidic protein (GFAP) in lumbar spinal cord increases following a sciatic nerve constriction injury. Brain Research. 1991;565:1–7. doi: 10.1016/0006-8993(91)91729-k. [DOI] [PubMed] [Google Scholar]
- Garry EM, Delaney A, Blackburn-Munro G, Dickinson T, Moss A, Nakalembe I, et al. Activation of p38 and p42/44 MAP kinase in neuropathic pain: involvement of VPAC2 and NK2 receptors and mediation by spinal glia. Molecular and Cellular Neuroscience. 2005;30:523–537. doi: 10.1016/j.mcn.2005.08.016. [DOI] [PubMed] [Google Scholar]
- Giaume C, McCarthy KD. Control of gap-junctional communication in astrocytic networks. Trends in Neurosciences. 1996;19:319–325. doi: 10.1016/0166-2236(96)10046-1. [DOI] [PubMed] [Google Scholar]
- Giaume C, Venance L. Intercellular calcium signaling and gap junctional communication in astrocytes. Glia. 1998;24:50–64. [PubMed] [Google Scholar]
- Gomez-Pinilla F, Cummings BJ, Cotman CW. Induction of basic fibroblast growth factor in Alzheimer's disease pathology. Neuroreport. 1990;1:211–214. doi: 10.1097/00001756-199011000-00009. [DOI] [PubMed] [Google Scholar]
- Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. Journal of Neuroscience. 2006;26:4308–4317. doi: 10.1523/JNEUROSCI.0003-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydon PG. GLIA: listening and talking to the synapse. Nature Reviews Neuroscience. 2001;2:185–193. doi: 10.1038/35058528. [DOI] [PubMed] [Google Scholar]
- Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiological Reviews. 2006;86:1009–1031. doi: 10.1152/physrev.00049.2005. [DOI] [PubMed] [Google Scholar]
- Hofstetter CP, Holmstrom NA, Lilja JA, Schweinhardt P, Hao J, Spenger C, et al. Allodynia limits the usefulness of intraspinal neural stem cell grafts; directed differentiation improves outcome. Nature Neuroscience. 2005;8:346–353. doi: 10.1038/nn1405. [DOI] [PubMed] [Google Scholar]
- Honore P, Rogers SD, Schwei MJ, Salak-Johnson JL, Luger NM, Sabino MC, et al. Murine models of inflammatory, neuropathic and cancer pain each generates a unique set of neurochemical changes in the spinal cord and sensory neurons. Neuroscience. 2000;98:585–598. doi: 10.1016/s0306-4522(00)00110-x. [DOI] [PubMed] [Google Scholar]
- Hua XY, Svensson CI, Matsui T, Fitzsimmons B, Yaksh TL, Webb M. Intrathecal minocycline attenuates peripheral inflammation-induced hyperalgesia by inhibiting p38 MAPK in spinal microglia. Eururopean Journal of Neuroscience. 2005;22:2431–2440. doi: 10.1111/j.1460-9568.2005.04451.x. [DOI] [PubMed] [Google Scholar]
- Huang YH, Bergles DE. Glutamate transporters bring competition to the synapse. Current Opinions in Neurobiology. 2004;14:346–352. doi: 10.1016/j.conb.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Huck S, Grass F, Hortnagl H. The glutamate analogue alpha-aminoadipic acid is taken up by astrocytes before exerting its gliotoxic effect in vitro. Journal of Neuroscience. 1984;4:2650–2657. doi: 10.1523/JNEUROSCI.04-10-02650.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip YT, Davis RJ. Signal transduction by the c-Jun N-terminal kinase (JNK)--from inflammation to development. Current Opinions in Cell Biology. 1998;10:205–219. doi: 10.1016/s0955-0674(98)80143-9. [DOI] [PubMed] [Google Scholar]
- Ji RR, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nature Neuroscience. 1999;2:1114–1119. doi: 10.1038/16040. [DOI] [PubMed] [Google Scholar]
- Ji RR, Befort K, Brenner GJ, Woolf CJ. ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. Journal of Neuroscience. 2002;22:478–485. doi: 10.1523/JNEUROSCI.22-02-00478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Strichartz G. Cell signaling and the genesis of neuropathic pain. Science STKE. 2004:reE14. doi: 10.1126/stke.2522004re14. [DOI] [PubMed] [Google Scholar]
- Ji RR, Woolf CJ. Neuronal plasticity and signal transduction in nociceptive neurons: implications for the initiation and maintenance of pathological pain. Neurobiology of Disease. 2001;8:1–10. doi: 10.1006/nbdi.2000.0360. [DOI] [PubMed] [Google Scholar]
- Ji RR, Zhang Q, Zhang X, Piehl F, Reilly T, Pettersson RF, et al. Prominent expression of bFGF in dorsal root ganglia after axotomy. European Journal of Neuroscience. 1995;7:2458–2468. doi: 10.1111/j.1460-9568.1995.tb01044.x. [DOI] [PubMed] [Google Scholar]
- Jin SX, Zhuang ZY, Woolf CJ, Ji RR. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. Journal of Neuroscience. 2003;23:4017–4022. doi: 10.1523/JNEUROSCI.23-10-04017.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurgel M, Koo AC, Ivy GO. Selective ablation of astrocytes by intracerebral injections of alpha-aminoadipate. Glia. 1996;16:351–358. doi: 10.1002/(SICI)1098-1136(199604)16:4<351::AID-GLIA7>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Kolhekar R, Meller ST, Gebhart GF. N-methyl-D-aspartate receptor-mediated changes in thermal nociception: allosteric modulation at glycine and polyamine recognition sites. Neuroscience. 1994;63:925–936. doi: 10.1016/0306-4522(94)90560-6. [DOI] [PubMed] [Google Scholar]
- Koshinaga M, Sanon HR, Whittemore SR. Altered acidic and basic fibroblast growth factor expression following spinal cord injury. Experimental Neurology. 1993;120:32–48. doi: 10.1006/exnr.1993.1038. [DOI] [PubMed] [Google Scholar]
- Kuan CY, Whitmarsh AJ, Yang DD, Liao G, Schloemer AJ, Dong C, et al. A critical role of neural-specific JNK3 for ischemic apoptosis. Proceedings of the National Academy of Sciences of the USA. 2003;100:15184–15189. doi: 10.1073/pnas.2336254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledeboer A, Sloane EM, Milligan ED, Frank MG, Mahony JH, Maier SF, et al. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005;115:71–83. doi: 10.1016/j.pain.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Lee IH, Lindqvist E, Kiehn O, Widenfalk J, Olson L. Glial and neuronal connexin expression patterns in the rat spinal cord during development and following injury. Journal of Comparative Neurology. 2005;489:1–10. doi: 10.1002/cne.20567. [DOI] [PubMed] [Google Scholar]
- Lee TT, Green BA, Dietrich WD, Yezierski RP. Neuroprotective effects of basic fibroblast growth factor following spinal cord contusion injury in the rat. Journal of Neurotrauma. 1999;16:347–356. doi: 10.1089/neu.1999.16.347. [DOI] [PubMed] [Google Scholar]
- Liu Y, Honda S, Kohsaka S, Nakajima K. Plasminogen enhances the secretion of plasminogen activator inhibitor-1 from cultured rat astrocytes. Neuroscience Letters. 2000;282:137–140. doi: 10.1016/s0304-3940(00)00860-0. [DOI] [PubMed] [Google Scholar]
- Ma W, Quirion R. Partial sciatic nerve ligation induces increase in the phosphorylation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) in astrocytes in the lumbar spinal dorsal horn and the gracile nucleus. Pain. 2002;99:175–184. doi: 10.1016/s0304-3959(02)00097-0. [DOI] [PubMed] [Google Scholar]
- Macias MY, Syring MB, Pizzi MA, Crowe MJ, Alexanian AR, Kurpad SN. Pain with no gain: allodynia following neural stem cell transplantation in spinal cord injury. Experimental Neurology. 2006;201:335–348. doi: 10.1016/j.expneurol.2006.04.035. [DOI] [PubMed] [Google Scholar]
- Madiai F, Goettl VM, Hussain SR, Clairmont AR, Stephens RL, Jr, Hackshaw KV. Anti-fibroblast growth factor-2 antibodies attenuate mechanical allodynia in a rat model of neuropathic pain. Journal of Molecular Neuroscience. 2005;27:315–324. doi: 10.1385/JMN:27:3:315. [DOI] [PubMed] [Google Scholar]
- Madiai F, Hussain SR, Goettl VM, Burry RW, Stephens RL, Jr, Hackshaw KV. Upregulation of FGF-2 in reactive spinal cord astrocytes following unilateral lumbar spinal nerve ligation. Experimental Brain Research. 2003;148:366–376. doi: 10.1007/s00221-002-1286-3. [DOI] [PubMed] [Google Scholar]
- Mantyh PW, Clohisy DR, Koltzenburg M, Hunt SP. Molecular mechanisms of cancer pain. Nature Reviews Cancer. 2002;2:201–209. doi: 10.1038/nrc747. [DOI] [PubMed] [Google Scholar]
- Meller ST, Dykstra C, Grzybycki D, Murphy S, Gebhart GF. The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuropharmacology. 1994;33:1471–1478. doi: 10.1016/0028-3908(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Migheli A, Piva R, Atzori C, Troost D, Schiffer D. c-Jun, JNK/SAPK kinases and transcription factor NF-kappa B are selectively activated in astrocytes, but not motor neurons, in amyotrophic lateral sclerosis. Journal of Neuropathology and Experimental Neurology. 1997;56:1314–1322. doi: 10.1097/00005072-199712000-00006. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Twining C, Chacur M, Biedenkapp J, O'Connor K, Poole S, et al. Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. Journal of Neuroscience. 2003;23:1026–1040. doi: 10.1523/JNEUROSCI.23-03-01026.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Zapata V, Chacur M, Schoeniger D, Biedenkapp J, O'Connor KA, Verge GM, et al. Evidence that exogenous and endogenous fractalkine can induce spinal nociceptive facilitation in rats. European Journal of Neuroscience. 2004;20:2294–2302. doi: 10.1111/j.1460-9568.2004.03709.x. [DOI] [PubMed] [Google Scholar]
- Nagy JI, Dudek FE, Rash JE. Update on connexins and gap junctions in neurons and glia in the mammalian nervous system. Brain Research. Brain Research Review. 2004;47:191–215. doi: 10.1016/j.brainresrev.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Nesic O, Lee J, Johnson KM, Ye Z, Xu GY, Unabia GC, Wood TG, et al. Transcriptional profiling of spinal cord injury-induced central neuropathic pain. Journal of Neurochemistry. 2005;95:998–1014. doi: 10.1111/j.1471-4159.2005.03462.x. [DOI] [PubMed] [Google Scholar]
- Niederberger E, Schmidtko A, Rothstein JD, Geisslinger G, Tegeder I. Modulation of spinal nociceptive processing through the glutamate transporter GLT-1. Neuroscience. 2003;116:81–87. doi: 10.1016/s0306-4522(02)00547-x. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Peters CM, Rogers SD, Pomonis JD, Egnaczyk GF, Keyser CP, Schmidt JA, et al. Endothelin receptor expression in the normal and injured spinal cord: potential involvement in injury-induced ischemia and gliosis. Experimental Neurology. 2003;180:1–13. doi: 10.1016/s0014-4886(02)00023-7. [DOI] [PubMed] [Google Scholar]
- Petrich BG, Eloff BC, Lerner DL, Kovacs A, Saffitz JE, Rosenbaum DS, et al. Targeted activation of c-Jun N-terminal kinase in vivo induces restrictive cardiomyopathy and conduction defects. Journal of Biological Chemistry. 2004;279:15330–15338. doi: 10.1074/jbc.M314142200. [DOI] [PubMed] [Google Scholar]
- Polgar E, Hughes DI, Arham AZ, Todd AJ. Loss of neurons from laminas I–III of the spinal dorsal horn is not required for development of tactile allodynia in the spared nerve injury model of neuropathic pain. Journal of Neuroscience. 2005;25:6658–6666. doi: 10.1523/JNEUROSCI.1490-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabchevsky AG, Fugaccia I, Turner AF, Blades DA, Mattson MP, Scheff SW. Basic fibroblast growth factor (bFGF) enhances functional recovery following severe spinal cord injury to the rat. Experimental Neurology. 2000;164:280–291. doi: 10.1006/exnr.2000.7399. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. Journal of Pharmacology and Experimental Therapeutics. 2003;306:624–630. doi: 10.1124/jpet.103.052407. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga FY, DeLeo JA. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. European Journal of Neuroscience. 2004;20:467–473. doi: 10.1111/j.1460-9568.2004.03514.x. [DOI] [PubMed] [Google Scholar]
- Ren K, Hylden JL, Williams GM, Ruda MA, Dubner R. The effects of a non-competitive NMDA receptor antagonist, MK-801, on behavioral hyperalgesia and dorsal horn neuronal activity in rats with unilateral inflammation. Pain. 1992;50:331–344. doi: 10.1016/0304-3959(92)90039-E. [DOI] [PubMed] [Google Scholar]
- Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends in Neurosciences. 1997;20:570–577. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- Rodriguez MJ, Martinez-Sanchez M, Bernal F, Mahy N. Heterogeneity between hippocampal and septal astroglia as a contributing factor to differential in vivo AMPA excitotoxicity. Journal of Neuroscience Research. 2004;77:344–353. doi: 10.1002/jnr.20177. [DOI] [PubMed] [Google Scholar]
- Romero MI, Rangappa N, Li L, Lightfoot E, Garry MG, Smith GM. Extensive sprouting of sensory afferents and hyperalgesia induced by conditional expression of nerve growth factor in the adult spinal cord. Journal of Neuroscience. 2000;20:4435–4445. doi: 10.1523/JNEUROSCI.20-12-04435.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers SD, Peters CM, Pomonis JD, Hagiwara H, Ghilardi JR, Mantyh PW. Endothelin B receptors are expressed by astrocytes and regulate astrocyte hypertrophy in the normal and injured CNS. Glia. 2003;41:180–190. doi: 10.1002/glia.10173. [DOI] [PubMed] [Google Scholar]
- Rohlmann A, Laskawi R, Hofer A, Dobo E, Dermietzel R, Wolff JR. Facial nerve lesions lead to increased immunostaining of the astrocytic gap junction protein (connexin 43) in the corresponding facial nucleus of rats. Neuroscience Letters. 1993;154:206–208. doi: 10.1016/0304-3940(93)90208-3. [DOI] [PubMed] [Google Scholar]
- Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, et al. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–475. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- Sandkuhler J. Learning and memory in pain pathways. Pain. 2000;88:113–118. doi: 10.1016/S0304-3959(00)00424-3. [DOI] [PubMed] [Google Scholar]
- Schafers M, Svensson CI, Sommer C, Sorkin LS. Tumor necrosis factor-alpha induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. Journal of Neuroscience. 2003;23:2517–2521. doi: 10.1523/JNEUROSCI.23-07-02517.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz J, Broom DC, Youn DH, Mills CD, Kohno T, Suter MR, et al. Blocking caspase activity prevents transsynaptic neuronal apoptosis and the loss of spinal inhibition following peripheral nerve injury. Journal of Neuroscience. 2005;25:7317–7323. doi: 10.1523/JNEUROSCI.1526-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spataro LE, Sloane EM, Milligan ED, Wieseler-Frank J, Schoeniger D, Jekich BM, et al. Spinal gap junctions: potential involvement in pain facilitation. Journal of Pain. 2004;5:392–405. doi: 10.1016/j.jpain.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Sung B, Lim G, Mao J. Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. Journal of Neuroscience. 2003;23:2899–2910. doi: 10.1523/JNEUROSCI.23-07-02899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweitzer S, Martin D, DeLeo JA. Intrathecal interleukin-1 receptor antagonist in combination with soluble tumor necrosis factor receptor exhibits an anti-allodynic action in a rat model of neuropathic pain. Neuroscience. 2001;103:529–539. doi: 10.1016/s0306-4522(00)00574-1. [DOI] [PubMed] [Google Scholar]
- Tanga FY, Raghavendra V, DeLeo JA. Quantitative real-time RT-PCR assessment of spinal microglial and astrocytic activation markers in a rat model of neuropathic pain. Neurochemistry International. 2004;45:397–407. doi: 10.1016/j.neuint.2003.06.002. [DOI] [PubMed] [Google Scholar]
- Tanga FY, Raghavendra V, Nutile-McMenemy N, Marks A, DeLeo JA. Role of astrocytic S100beta in behavioral hypersensitivity in rodent models of neuropathic pain. Neuroscience. 2006;140:1003–1010. doi: 10.1016/j.neuroscience.2006.02.070. [DOI] [PubMed] [Google Scholar]
- Tawfik VL, Lacroix-Fralish ML, Bercury KK, Nutile-McMenemy N, Harris BT, Deleo JA. Induction of astrocyte differentiation by propentofylline increases glutamate transporter expression in vitro: heterogeneity of the quiescent phenotype. Glia. 2006;54:193–203. doi: 10.1002/glia.20365. [DOI] [PubMed] [Google Scholar]
- Tian GF, Takano T, Lin JH, Wang X, Bekar L, Nedergaard M. Imaging of cortical astrocytes using 2-photon laser scanning microscopy in the intact mouse brain. Advanced Drug Delivery Reviews. 2006;58:773–787. doi: 10.1016/j.addr.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Mizokoshi A, Shigemoto-Mogami Y, Koizumi S, Inoue K. Activation of p38 mitogen-activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia. 2004;45:89–95. doi: 10.1002/glia.10308. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, et al. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- Verge GM, Milligan ED, Maier SF, Watkins LR, Naeve GS, Foster AC. Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. European Journal of Neuroscience. 2004;20:1150–1160. doi: 10.1111/j.1460-9568.2004.03593.x. [DOI] [PubMed] [Google Scholar]
- Wang ZY, Zhang YQ, Zhao ZQ. Inhibition of tetanically sciatic stimulation-induced LTP of spinal neurons and Fos expression by disrupting glutamate transporter GLT-1. Neuropharmacology. 2006;51:764–772. doi: 10.1016/j.neuropharm.2006.05.024. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Johnston IN, Maier SF. Glia: novel counter-regulators of opioid analgesia. Trends in Neurosciences. 2005;28:661–669. doi: 10.1016/j.tins.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Martin D, Ulrich P, Tracey KJ, Maier SF. Evidence for the involvement of spinal cord glia in subcutaneous formalin induced hyperalgesia in the rat. Pain. 1997;71:225–235. doi: 10.1016/s0304-3959(97)03369-1. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends in Neurosciences. 2001;24:450–455. doi: 10.1016/s0166-2236(00)01854-3. [DOI] [PubMed] [Google Scholar]
- Wolosker H. D-serine regulation of NMDA receptor activity. Science STKE. 2006:e41. doi: 10.1126/stke.3562006pe41. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Mannion RJ. Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet. 1999;353:1959–1964. doi: 10.1016/S0140-6736(99)01307-0. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Gage FH. Fibroblast growth factors stimulate nerve growth factor synthesis and secretion by astrocytes. Brain Research. 1991;538:118–126. doi: 10.1016/0006-8993(91)90385-9. [DOI] [PubMed] [Google Scholar]
- Zerari F, Karpitskiy V, Krause J, Descarries L, Couture R. Astroglial distribution of neurokinin-2 receptor immunoreactivity in the rat spinal cord. Neuroscience. 1998;84:1233–1246. doi: 10.1016/s0306-4522(97)00548-4. [DOI] [PubMed] [Google Scholar]
- Zhang J, De Koninck Y. Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. Journal of Neurochemistry. 2006;97:772–783. doi: 10.1111/j.1471-4159.2006.03746.x. [DOI] [PubMed] [Google Scholar]
- Zhang RX, Liu B, Wang L, Ren K, Qiao JT, Berman BM, Lao L. Spinal glial activation in a new rat model of bone cancer pain produced by prostate cancer cell inoculation of the tibia. Pain. 2005;118:125–136. doi: 10.1016/j.pain.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Kawasaki Y, Tan PH, Wen YR, Huang J, Ji RR. Role of the CX3CR1/p38 MAPK pathway in spinal microglia for the development of neuropathic pain following nerve injury-induced cleavage of fractalkine. Brain Behavior and Immunity. 2006b doi: 10.1016/j.bbi.2006.11.003. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114:149–159. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, et al. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. Journal of Neuroscience. 2006a;26:3551–3560. doi: 10.1523/JNEUROSCI.5290-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]