

Abstract
The AML1 gene, located on chromosome 21, is involved in several distinct chromosomal translocations in human leukemia. In t(8;21) acute myelogenous leukemia (AML), the AML1 gene is juxtaposed to the ETO gene located on chromosome 8, generating an AML1/ETO fusion protein. Both AML1/ETO and the AML1 proteins recognize the same consensus DNA-binding motif (TGT/CGGT), which is found in the promoters of several genes involved in hematopoiesis. We found that two myeloid leukemia cell lines with the t(8;21) translocation, Kasumi and SKNO-1, have elevated levels of BCL-2 protein compared with other myeloid cell lines. In addition, we identified a consensus AML1 binding site in the BCL-2 promoter. Thus far, AML1/ETO has been shown to dominantly repress its target genes; however, we found that AML1/ETO activates transcription of the BCL-2 gene in U937 cells. This activation requires the presence of both the runt homology domain (rhd) and the C-terminal portion of AML1/ETO. We demonstrated sequence specific binding of both AML1A and AML1/ETO to the TGTGGT sequence in the BCL-2 promoter and showed that the AML1 binding site is required for responsiveness to AML1/ETO. Interestingly, AML1A and AML1B do not modulate the activity of the BCL-2 promoter. The elevated levels of BCL-2 in cells that express AML1/ETO may prolong their life span and contribute to the development of t(8;21) leukemia.
Chromosomal translocations found in human leukemia frequently involve genes that code for transcription factors (1). In acute myeloid leukemia (AML) with the t(8;21) chromosomal translocation, which occurs in ≈40% cases of AML with the M2 French–American–British subtype, coding sequences of the AML1 gene (on chromosome 21) are juxtaposed to coding sequences of the ETO gene (on chromosome 8) generating an AML1/ETO fusion protein (2, 3). The AML1 family of transcription factors recognize the binding sequence 5′-TGT/CGGT-3′ (2) through an 117-amino acid region that is highly homologous to the Drosophila segmentation gene runt (2, 3), and has been called the runt homology domain (rhd). This domain is necessary for DNA binding, as well as for protein–protein interactions (3). At least three forms of AML1 protein are produced by alternative splicing (4). The AML1-B isoform (479 amino acids) contains the rhd and a putative C-terminal transcriptional activation domain; the AML1-A isoform (250 amino acids) contains the DNA binding domain, but lacks the potential transcriptional activation domain. AML1-B, but not AML-1A, can transactivate the human granulocyte/macrophage colony-stimulating factor (GM-CSF) promoter (5) and the T cell receptor β enhancer (6), whereas both isoforms can transactivate the human interleukin 3 (IL-3) gene (H.U., S.Z., and S.D.N., unpublished work).
The genes encoding AML1 or its dimerization partner CBFβ, have been shown to be involved in several other translocations in human acute leukemia (7). The AML1 gene is fused to the TEL gene in t(12;21) acute lymphoblastic leukemia (8). In the t(3;21) translocation, seen in the blast crisis phase of chronic myelogenous leukemia and in therapy-related myelodysplastic syndromes or acute leukemia, the AML1 gene is fused to the EVI-1 gene (9) or to the EAP or MDS1 genes (10). The cytogenetic abnormality inversion 16, found in the M4Eo subtype of AML, generates CBFβ/MYH11 fusion transcripts (11). These alterations point to a critical role of AML1 in hematopoiesis, and recently, the targeted disruption of the AML1 gene demonstrated that AML1 is essential for fetal liver hematopoiesis in mice (12, 13). The ETO gene encodes a protein with two putative zinc fingers, and several proline-serine-threonine-rich regions at its C terminus (14); it has been presumed to function as a transcription factor. The chimeric AML1/ETO protein retains the DNA binding specificity of AML1, but lacks the transcriptional activation domain present in AML1B; its transcriptional activity differs from both the wild-type AML1 (5, 6) and ETO proteins (L.K. and S.D.N., unpublished data). AML1/ETO has been shown to repress transcription of the GM-CSF and IL-3 promoters and the T-cell antigen receptor β enhancer (refs. 5 and 6 and unpublished data). However, it is difficult to envision how AML1/ETO exerts its leukemogenic potential by inhibiting the expression of these genes.
To understand the role of the AML1/ETO protein in the development of human leukemia, we are studying its effects on the expression of potential target genes that contain AML1 consensus binding sites. We are mainly interested in studying the effect of the AML1/ETO protein on genes that can affect cell proliferation and those that regulate programmed cell death, such as BCL-2. The BCL-2 gene has been originally discovered at the junction of the t(14;18) chromosomal translocation found in most cases of follicular B-cell lymphomas (15). The t(14;18) chromosomal translocation places the BCL-2 gene under the control of a strong Ig heavy-chain enhancer, which results in overexpression of the BCL-2 protein. BCL-2 can prevent or delay apoptosis in many cell types (16). Although BCL-2 gene rearrangements have not been found in myeloid leukemias, ≈20% of AMLs and 70% of chronic myelogenous leukemias have elevated levels of BCL-2 protein, suggesting that mechanisms other than chromosomal translocation control BCL-2 gene expression.
We identified a consensus DNA binding sequence for AML1 (TGTGGT) in the 5′ regulatory region of the BCL-2 gene and demonstrated that both AML1 and AML1/ETO proteins can bind to this site. We show that AML1/ETO, but not AML1A, AML1B, or ETO activates BCL-2 gene expression; this activation requires the rhd DNA binding region of AML1/ETO as well as its C terminus. Activation of the BCL-2 promoter is the first demonstration of transcriptional activation by AML1/ETO, in contrast to its previously identified repressor activity. Regulation of the BCL-2 promoter by AML1/ETO, but not by the normal AML1 proteins, points to a unique biological activity of the fusion protein. t(8;21)-containing leukemic cells have high levels of BCL-2 protein compared with other myeloid leukemia cells but similar levels of MCL-1 and BCL-x. This suggests that expression of AML1/ETO may prolong the survival of t(8;21) cells by stimulating BCL-2 expression, and this may represent a key step in the development of t(8;21) leukemia.
MATERIALS AND METHODS
Cell Lines and Electroporation.
The U937, K562, and TF-1 cell lines were grown in RPMI medium 1640, supplemented with 10% fetal bovine serum, and the KG-1 cell line was grown in Iscove’s modified Dulbecco’s medium with 10% fetal bovine serum. Kasumi and SKNO-1 cells, which contain the t(8;21) chromosomal translocation, were grown in RPMI medium 1640 with 10% fetal bovine serum. All media were supplemented with 1% glutamine, penicillin, and streptomycin.
Electroporation was performed with a Bio-Rad Gene Pulser (250V, 960 μF). Cells were cotransfected with 10 μg of a luciferase or chloramphenicol acetyltransferase reporter gene plasmid (as indicated) and 10 μg of either an empty expression vector (pCMV5) or the same vector expressing AML1A, AML1B, or AML1/ETO. Cells were harvested 24–48 h after transfection and luciferase or chloramphenicol acetyltransferase activity was determined using standard techniques. To normalize for transfection efficiency, cells were cotransfected with a human-growth hormone expression plasmid (pXGH5, Nichols Institute, San Juan Capistrano, CA). Conditioned media, obtained at the time of cell harvest was assayed for growth hormone levels according to the manufacturer’s instructions.
Plasmid Construction.
The expression vectors for AML1A (CMV5-AML1A), AML1B (CMV5-AML1B), and AML1/ETO (CMV5-AML1/ETO) have been described (6). The AML1/ETO 406 deletion mutant was created by subcloning a XbaI/StuI fragment of AML1/ETO into the pCMV5 plasmid. The CMV5-ETO expression plasmid was created by deleting the AML1 portion of the AML1/ETO fusion cDNA using PCR-based deletion mutagenesis. An internal deletion of 84 nucleotides within the rhd (immediately upstream of the unique HindIII site in the rhd) was created using a PCR-directed mutagenesis strategy. The BCL-2 promoter-luciferase reporter gene plasmid (pGL2-BCL2) was generated by cloning the KpnI/HindIII fragment of the BCL-2 5′untranslated region (17) into pGL2-basic (Promega). The pTE2 plasmid was described previously (18). The pTE2-SacI plasmid was created by cloning the SacI fragment of the BCL-2 promoter (−1299 to −756)(see Fig. 2) upstream of the herpes simplex virus thymidine kinase promoter in the pTE2 plasmid. A SacI fragment with a mutated AML1 binding site (TGTGGT was changed to cagacT) was generated using the Clontech Site-Directed Mutagenesis kit according to the manufacturer’s instructions. This fragment was subcloned in the pTE2 plasmid to generate the pTE2/SacIm plasmid. All mutations were confirmed by DNA sequencing using Sequenase II (United States Biochemical). AML1/ETO c-DNA was subcloned into the pCIneo vector (Promega) in either sense or antisense orientation, generating the pCI-AML1/ETO sense or pCI-AML1/ETO antisense plasmids.
Figure 2.
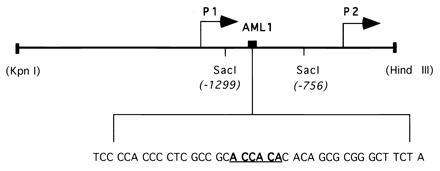
Schematic representation of the BCL-2 5′untranslated region. The location of the P1 and P2 promoters, SacI fragment, and the potential AML1 binding site (shown in bold and underlined) are indicated. The sequence shown is contained in the wild-type oligonucleotide used for the EMSA.
Electrophoretic Mobility-Shift Assay (EMSA).
AML1A and AML1/ETO proteins used in EMSAs were expressed in Escherichia coli (BL21) using the PET vector system (Novagen). The following synthetic oligonucleotides containing wild-type (shown in boldface) or mutant AML1 binding sites from the BCL-2 promoter were used in the electrophoretic mobility-shifts assays: BCL-2 WT: TCC CCA CCC CTC GCC GCA CCA CAC ACA GCG CGG GCT TCT A; and BCL-2 AML-1 M: TCC CCA CCC CTC GCC GCA GTC TGC ACA GCG CGG GCT TCT A.
Radiolabeled oligonucleotides were incubated with 5 μl of bacterially expressed AML1 or AML1/ETO at room temperature in 20 μl reaction buffer containing 20 mM Hepes (pH 7.9), 0.5 mM EDTA, 2 mM DTT, 10% glycerol, 50 mM KCl, and 2 μg poly (dI:dC). Supershift experiments were performed using a polyclonal antiserum raised against the 17 N-terminal amino acids of AML1 (kindly provided by Scott Hiebert, St. Jude Children’s Research Hospital, Memphis, TN).
Western Blot Analysis.
Kasumi, U-937, KG-1, K562, TF-1, or SKNO-1 cells (1 × 106) were resuspended in SDS—lysis buffer and samples were electrophoretically separated on a SDS—12% polyacrylamide gel. After transfer to a nitrocellulose membrane, the gel was stained with Coomassie blue to demonstrate equal loading of proteins in all lanes. The presence and amount of BCL-2 protein were detected using a monoclonal antibody that specifically recognizes BCL-2 (Oncogene Science). The membrane was stripped of bound antibody and reprobed with a polyclonal anti-MCL-1 antibody (PharMingen). BCL-x protein was detected using polyclonal anti-BCL-x antibody (Santa Cruz Biotechnology, Santa Cruz, CA). Western blots were developed using the Amersham Enhanced Chemiluminescence kit and exposed to Kodak XAR film for 30 sec (for BCL-2 and BCL-x detection) or 3 min (for MCL-1 detection).
RESULTS
Cells Expressing AML1/ETO Have Elevated Levels of BCL-2 Protein.
The Kasumi-1 and the SKNO-1 cell lines contain the t(8;21) chromosomal translocation and express AML1/ETO protein (19, 20). We compared the levels of BCL-2, MCL-1, and BCL-x proteins in these cells and in myeloid leukemia cells without the t(8;21) translocation (U937, KG-1, K-562, and TF-1) by Western blot analysis. As shown in Fig. 1A, Kasumi and SKNO-1 cells have significantly higher levels of BCL-2 protein, compared with other myeloid cell lines. In contrast, the amount of MCL-1, a recently described protein that also has anti-apoptotic activity (21, 22, 23) is roughly comparable in all the myeloid cell lines tested (although K562 cells do not express detectable levels of MCL-1 protein). Likewise, the levels of BCL-x protein are comparable in all cell lines tested (Fig. 1B), further suggesting the specific upregulation of the BCL-2 protein in t(8;21) cell lines. To define a possible mechanism leading to the elevated levels of BCL-2 protein in t(8;21) cells, we examined the BCL-2 promoter sequences (24) and identified a potential AML1 consensus binding site at position −1085 to −1091. Thus, we decided to test if AML1, or the AML1/ETO fusion protein, regulates the transcription of the BCL-2 gene.
Figure 1.
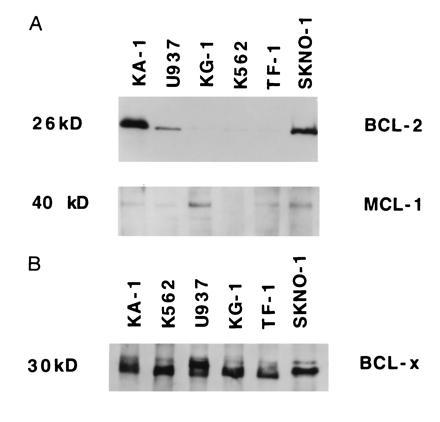
Expression of BCL-2, MCL-1 and BCL-x proteins in myeloid leukemia cell lines. (A) Whole cell lysates from Kasumi, U937, KG-1, K562, TF-1 and SKNO-1 cells were separated on a 12% SDS/PAGE. The gel was stained with Coomassie blue after transfer to demonstrate equal loading of proteins (not shown). The levels of BCL-2 were detected by Western blot analysis, using antibodies specific for BCL-2 proteins. The membrane was stripped of bound antibody and reprobed with an antibody specific for MCL-1. (B) The cell lysates used in Fig. 1A were tested for the presence of the BCL-x protein by Western blot analysis, using antibodies that specifically recognize BCL-x protein.
The BCL-2 Promoter Contains an AML1 Binding Site That Can Bind the AML1 and AML1/ETO Proteins.
Transcription of the BCL-2 gene is regulated by two promoters, P1 and P2 (24). A large region between these two promoters has been shown to have negative regulatory function (25) and we found that it contains an AML1 consensus binding sequence (TGTGGT) (Fig. 2). We performed EMSAs to determine if AML1 proteins can bind to this putative AML1 binding site. Incubation of bacterially expressed AML1A protein with the radiolabeled wild-type BCL-2 oligonucleotide generated a specific gel shift complex (see Fig. 3A, lane 2). This binding was competed by the addition of unlabeled wild-type oligonucleotide (lane 3), but not by an oligonucleotide containing a mutated AML1 binding site (lane 4). Similarly, an oligonucleotide containing the AML1 binding site from the GM-CSF promoter (5) efficiently competed for the binding of AML1A (lane 5), whereas the GM-CSF promoter oligonucleotide with a mutated AML1 binding sequence did not compete (lane 6). Supershift experiments confirmed the presence of AML1A protein in the complex; the anti-AML1 antiserum supershifted the complex (lane 7), whereas the preimmune serum did not affect the formation of the complex (lane 8).
Figure 3.
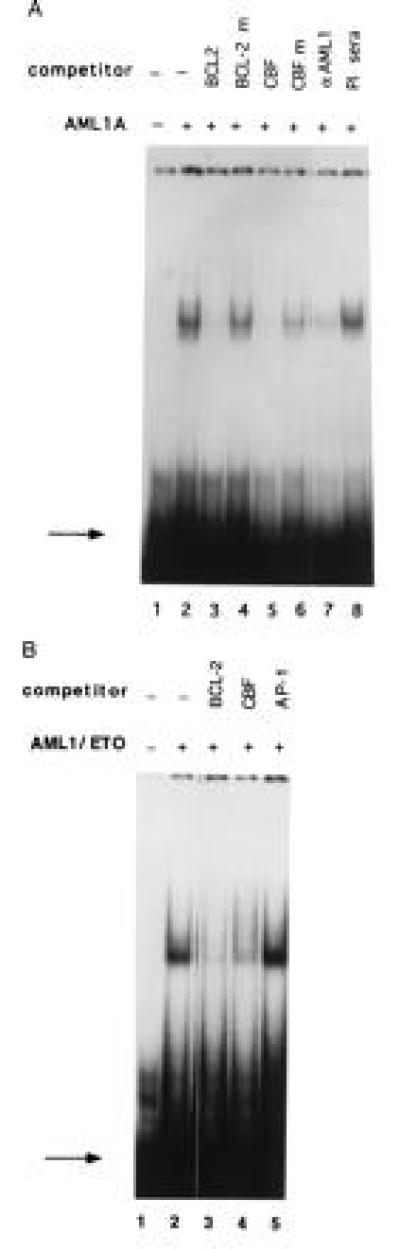
AML1A and AML1/ETO bind to the BCL-2 promoter. (A) A 32P-labeled oligonucleotide containing the AML1 binding site from the BCL-2 promoter was run alone (lane 1), or incubated with 5 μl of bacterially expressed AML1A protein (lane 2). The reaction mixture was preincubated with a 100-fold molar excess of unlabeled wild-type oligonucleotide (lane 3) or an oligonucleotide with a mutated AML1-binding site (lane 4), an oligonucleotide containing the AML1 site from the IL-3 promoter (lane 5) or the IL-3 promoter oligonucleotide with a mutated AML1 site (lane 6). In lane 7, the AML1A protein was preincubated with 2 μl of AML1 antiserum, and in lane 8 with 2 μl of preimmune serum. The binding of AML1A to the BCL-2 promoter was analyzed by EMSA. The position of the free probe is indicated by the arrow. (B) The 32P-labeled 40-mer oligonucleotide (shown in Fig. 2) was incubated with 5 μl of bacterially expressed AML1/ETO alone (lane 2), or with a 100-fold molar excess of unlabeled BCL-2 oligonucleotide (lane 3), a 100-fold excess of unlabeled oligonucleotide containing the AML1 binding site from the IL-3 promoter (lane 4) or a 100-fold excess of unlabeled oligonucleotide containing a consensus AP-1 site (lane 5). No protein was added in lane 1. The position of the free probe is indicated by the arrow.
We further tested whether the AML1/ETO fusion protein can bind to the AML1 binding site in the BCL-2 5′ region. Incubation of bacterially expressed AML1/ETO protein with the 32P-radiolabeled BCL-2 promoter oligonucleotide resulted in sequence specific DNA binding (Fig. 3B, lane 2), which was eliminated by addition of a 100-fold molar excess of unlabeled self-competitor (lane 3), or an oligonucleotide containing the AML1 binding sequence from the IL-3 promoter (lane 4). An unrelated oligonucleotide (containing the AP-1 binding site from the IL-3 promoter) did not compete for binding (lane 5). Supershift assays using the anti-AML1 N-terminal antiserum (and preimmune sera as the negative control) confirmed that this complex contains AML1/ETO (data not shown).
AML1/ETO, but Not AML1A or AML1B, Transactivates the BCL-2 Promoter.
Having demonstrated sequence specific binding of AML1A and AML1/ETO to the BCL-2 promoter, we examined the functional effect of the AML1A, AML1B, and AML1/ETO proteins on the activity of the BCL-2 promoter in transient transfection assays. We cotransfected the BCL-2 promoter-luciferase reporter gene construct (pGL2- BCL2) with cytomegalovirus promoter-based expression vectors for AML1A, AML1B or AML-1/ETO. Neither AML1A nor AML1B activated the BCL-2 promoter, however, the AML1/ETO fusion protein transactivated the BCL-2 promoter nearly seven-fold in U937 cells (Fig. 4). The activity of the promoterless plasmid (pGL-2-basic) was not modulated by the expression of AML1A, AML1B, or the AML1/ETO fusion protein.
Figure 4.
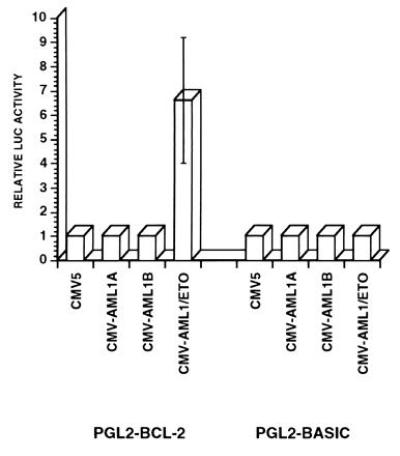
AML1/ETO transactivates the BCL-2 promoter. U937 cells were cotransfected with pGL2-basic or pGL2-BCL-2 reporter gene constructs and with pCMV-AML1A, pCMV-AML1B or pCMV-AML1/ETO expression vectors (together with pGHX5 to normalize transfection efficiency). Promoter activity was measured as the ratio of the luciferase activity and the concentration of growth hormone in the conditioned media. The promoter activity in the presence of the empty CMV5 plasmid was defined as 1 and the promoter activity in the presence of expression vectors for AML1A, AML1B, and AML1/ETO was defined relative to that value. The results shown are the mean of four different experiments (SEs are shown when measurable).
The AML1 Binding Site in the BCL-2 Promoter Is Sufficient and Necessary to Confer Responsiveness to AML1/ETO.
To confirm the importance of the AML1 site and to localize the region of the BCL-2 promoter responsive to transactivation by AML1/ETO, we subcloned a SacI fragment (−1299 to −756) containing the AML1 binding site in front of a heterologous promoter (the herpes simplex virus thymidine kinase promoter) in the pTE2 plasmid. We also created a mutant version of this plasmid (pTE2/SacIm) containing the SacI fragment with mutated AML1 binding site (TGTGGT was changed to cagacT). We cotransfected the wild-type plasmid (pTE2/SacI) or the mutant plasmid (pTE2/SacIm) with either a sense or an antisense AML1/ETO expression vector (pCI-AML1/ETO sense and pCI-AML1/ETO antisense) into U937 cells. The wild-type SacI fragment was able to confer responsiveness to the AML1/ETO protein, whereas the SacI fragment with a mutated AML1 binding site was not responsive to AML1/ETO (Fig. 5). The activity of the pTE2/SacI construct was not modulated by the expression vector containing AML1/ETO in an antisense orientation and the activity of the empty pTE2 plasmid was not modulated by the expression of AML1/ETO (Fig. 5). These data indicate that the AML1 binding site in the SacI region is necessary to confer AML1/ETO-responsiveness to a heterologous promoter. Although the SacI fragment is located in the promoter region previously found to inhibit BCL-2 transcription (25), it appears to exert basal enhancer activity on the thymidine kinase promoter that is abrogated by mutation of the AML1 binding site (Fig. 5).
Figure 5.
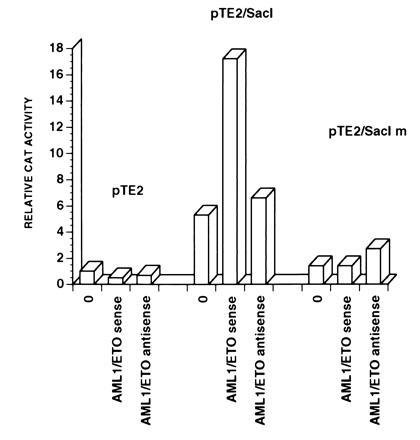
The AML1 binding site in the SacI fragment confers AML1/ETO responsiveness to a heterologous promoter. U937 cells were cotransfected with the pTE2, pTE2/SacI or pTE2/SacIm plasmids and with AML1/ETO expression vector (AML1/ETO sense), with its antisense derivative (AML1/ETO antisense) or with an empty pCIneo vector (0). The activity of the pTE2 plasmid in the presence of an empty vector was assigned a value of one. Results from one of three independent experiments are shown.
The rhd and C Terminus of AML1/ETO Are Necessary for the Transactivation of the BCL-2 Promoter.
Previously, all examined target genes containing an AML1 binding site have been shown to be activated by AML1B and inhibited by AML1/ETO. We, and others, have defined regions of the fusion protein required for its transcriptional repression (ref. 26 and unpublished work). To determine the region of AML1/ETO necessary for the transcriptional upregulation of the BCL-2 promoter, we prepared and analyzed several deletion mutants of the AML1/ETO protein (shown in Fig. 6A). All the deletion mutant plasmids made the appropriate sized proteins when expressed in COS cells (data not shown).
Figure 6.
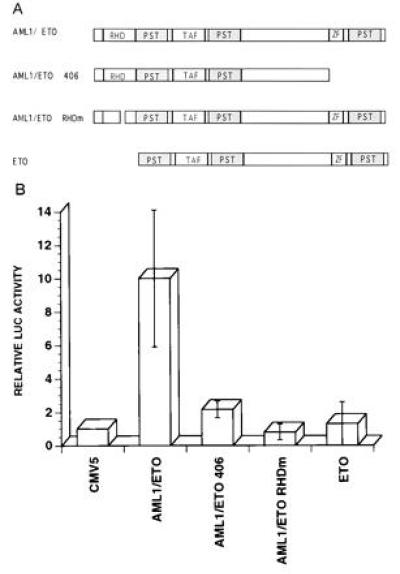
(A) Schematic representation and transcriptional activity of the various AML1/ETO deletion mutants. RHD, rhd; TAF, TATA-binding-protein-associated factor-110 homology domain; PST, proline-serine-threonine-rich region; ZF, zinc finger domain. (B) Transcriptional activity of the wild-type AML1/ETO and mutant AML/ETO proteins. U937 cells were cotransfected with the pGL2-BCL2 plasmid and the wild-type CMV5-AML1/ETO expression plasmid or with its deletion mutants as shown in Fig. 6A. Luciferase activity in the presence of the CMV5 plasmid was assigned a value of 1. Results represent the mean of four independent experiments.
As seen in Fig. 6B, deletion of the rhd portion of the AML1/ETO fusion protein eliminates the stimulatory effect of AML1/ETO on the BCL-2 promoter. This demonstrates that the binding of AML1/ETO to the AML1 DNA binding site and/or protein–protein interactions mediated by the rhd, are necessary for the stimulatory activity of AML1/ETO. The AML1/ETO 406 mutant, which lacks the zinc finger region and PST region of ETO has only minimal stimulatory activity demonstrating that the C-terminal part of the fusion protein is required for the transactivating activity of AML1/ETO as well. This contrasts with the amino acid requirements for repression by AML1/ETO as determined by Lenny et al. (26). Likewise, overexpression of the ETO part of the fusion protein did not modulate the activity of the BCL-2 promoter (Fig. 6B), indicating that fusion of AML1 with ETO is required to generate a transcriptional activator of the BCL-2 gene.
DISCUSSION
The AML1/ETO chimeric protein is invariably found in acute myeloid leukemia cells with an (8;21) chromosomal translocation. Apart from its ability to dominantly interfere with the transcriptional activity of the normal AML1B protein, the function of AML1/ETO within the cell, and its role in leukemogenesis, are not well understood. Antisense oligonucleotides complementary to the junction region of the AML1/ETO fusion mRNA inhibited the growth of t(8;21) myeloid leukemia cell lines and induced partial differentiation, suggesting that AML1/ETO is important for the continuous growth of these cells (27). Likewise, cleavage of AML1/ETO RNA with specific ribozymes inhibits the growth of cells containing the t(8;21) chromosomal translocation (28).
AML1/ETO has been shown to be a transcriptional inhibitor of genes that contain an AML1 binding site, including GM-CSF, T cell receptor β, and IL-3 (refs. 5 and 6 and unpublished work), however AML1/ETO probably does not exert its leukemogenic potential by inhibiting the expression of these genes. Genes that regulate cell death are a potentially important group of targets for leukemia-specific chimeric transcription factors. We identified an AML1 binding site in the 5′ untranslated region of the BCL-2 gene. We demonstrated that AML1/ETO activates transcription of the BCL-2 gene, an activity not shared by the normal AML1A, AML1B, or ETO proteins. The fusion of AML1 with ETO by the t(8;21) translocation thus creates a potent transcriptional activator of the BCL-2 gene exerting unique biological properties. The potential binding site for AML1 in the 5′ flanking region of the BCL-2 gene binds both AML1A and AML1/ETO and a SacI fragment of the BCL-2 promoter (1299 to −756) is sufficient to confer AML1/ETO responsiveness to a heterologous promoter. Moreover, the responsiveness to AML1/ETO is dependent on the presence of an intact AML1 binding site (Fig. 5). Although the AML-1 site is located in a region of the BCL-2 promoter that has been shown to inhibit BCL-2 expression, our results demonstrate that the AML1 site does not act as a negative regulatory element (Fig. 5).
The deletion mutant analysis of the fusion protein demonstrates that both the rhd and the C-terminal portion of the ETO protein, are critical for the transactivating activity of AML1/ETO. The rhd has also been shown to be important for the inhibitory activity of the AML1/ETO, however, deletion of the C-terminal part of ETO did not affect the repressing activity of the fusion protein (26). The rhd is required for sequence specific binding of AML1 to DNA, as well as its interactions with other proteins, such as CBFβ or ets-1 (26). Thus, it is possible that AML1/ETO must bind to the AML1 binding site to stimulate BCL-2 transcription, with the C-terminal portion of ETO functioning as the transcriptional activation domain. Another possibility is that the rhd of AML1/ETO recruits a potential coactivator, and that this coactivator protein requires the presence of the C-terminal portion of the fusion protein to exert its function. The inability of AML1B (and AML1A) to modulate the activity of the BCL-2 promoter is consistent with this model; however it may merely reflect the absence of adjacent binding sites for transcription factors that can cooperate with AML1 proteins, such as ETS or MYB (29, 30). Our results do not allow us to distinguish between these possibilities. Several other transcription factors have been shown to act as both a transcriptional activator and a transcriptional repressor, such as the glucocorticoid receptor, NF-IL6 and p53 (31, 32, 33). The molecular basis of the dual functions of AML1/ETO remains to be elucidated.
Both of the established cell lines with the t(8;21) translocation, Kasumi and SKNO-1, have elevated levels of BCL-2 protein compared with other myeloid leukemia cell lines. The levels of AML1/ETO fusion protein are higher in Kasumi than in SKNO-1 cells (data not shown), and it is noteworthy that Kasumi cells also have higher levels of BCL-2 protein (Fig. 1A). Although the expression of BCL-2 in myeloid cells is most likely controlled at multiple levels, the AML1/ETO-mediated increase in BCL-2 transcription may result in overexpression of BCL-2 protein in cells that contain the t(8;21) translocation. Analysis of primary clinical samples would be helpful in addressing this issue. We have, however, recently demonstrated that TF-1 cells engineered to express AML1/ETO, unlike the parental TF-1 cell line, maintain the level of BCL-2 protein upon growth factor withdrawal (L.K., J.Z., and S.D.N., unpublished work).
Wild-type p53 has been shown to inhibit BCL-2 promoter activity (17), however, we did not find a correlation between the presence of a p53 and the levels of BCL-2 protein in the cell lines tested (data not shown). Several other fusion proteins found in myeloid leukemias, such as PML-RARα and BCR-ABL, have also been shown to modulate programmed cell death (34, 35).
Inhibition (or delay) of cell death by AML1/ETO may be a key step in development of t(8;21) leukemia. However, cells containing AML1/ETO mRNA can be detected in the bone marrow of patients with t(8;21) AML who have been in remission for years, which suggests that other abnormalities may need to be present for leukemic cell development and proliferation.
Acknowledgments
We thank Wei Zhang for expressing the AML1/ETO protein, Michael Cleary for providing the BCL-2 genomic DNA, and Richard Frank, Hans-Georg Wisniewski, and Anne Altmeyer for reading the manuscript. This work was supported by National Institutes of Health RO1 Grants DK43052 to S.D.N. and CA58842 to A.O.Z. and the DeWitt Wallace Foundation.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: GM-CSF, granulocyte/macrophage colony-stimulating factor; EMSA, electrophoretic mobility-shift assay; IL, interleukin; AML, acute myelogenous leukemia; rhd, runt homology domain.
References
- 1.Nichols J, Nimer S D. Blood. 1992;80:2953–2963. [PubMed] [Google Scholar]
- 2.Erickson P, Gao K S, Chang T, Look T, Whisenant E, Raimondi S, Lasher J, Trujillo J, Rowley J, Drabkin H. Blood. 1992;80:1825–1831. [PubMed] [Google Scholar]
- 3.Meyers S, Downing J R, Hiebert S W. Mol Cell Biol. 1993;23:6336–6345. doi: 10.1128/mcb.13.10.6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bae S C, Ogawa E, Maruyama M, Oka H, Satake M, Shigaseda K, Jenkins N A, Gilbert D J, Copela N G, Ito Y. Mol Cell Biol. 1994;14:3242–3252. doi: 10.1128/mcb.14.5.3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frank R, Zhang J, Hiebert S W, Meyers S, Nimer S D. Oncogene. 1995;11:2667–2674. [PubMed] [Google Scholar]
- 6.Meyers S, Lenny N, Hiebert S. Mol Cell Biol. 1995;15:1974–1982. doi: 10.1128/mcb.15.4.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nucifora G, Rowley J D. Blood. 1995;86:1–14. [PubMed] [Google Scholar]
- 8.Golub T R, Barker G F, Bohlander S K, Hiebert S W, Ward D C, Bray-Ward P, Morgan E, Raimondi S C, Rowley J D, Gilliland D G. Proc Natl Acad Sci USA. 1995;92:4917–4921. doi: 10.1073/pnas.92.11.4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanaka T, Mitani K, Kurokawa M, Ogawa S, Tanaka K, Nishida J, Yazaki Y, Shibata Y, Hirai H. Mol Cell Biol. 1995;15:2383–2392. doi: 10.1128/mcb.15.5.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zent C S, Mathieu C, Claxton D F, Zhang D E, Tenen D G, Rowley J D, Nucifora G. Proc Natl Acad Sci USA. 1996;6:1044–1048. doi: 10.1073/pnas.93.3.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu P, Tarle S A, Hajra A, Claxton D F, Marlton P, Freedman M, Siciliano M J, Collins F S. Science. 1993;261:1041–1044. doi: 10.1126/science.8351518. [DOI] [PubMed] [Google Scholar]
- 12.Okuda T, Deursen J V, Hiebert S, Grosvelt G, Downing J. Cell. 1996;84:321–330. doi: 10.1016/s0092-8674(00)80986-1. [DOI] [PubMed] [Google Scholar]
- 13.Wang Q, Stacy T, Binder M, Marin-Padilla M, Sharpe A H, Speck N. Proc Natl Acad Sci USA. 1996;93:3444–3449. doi: 10.1073/pnas.93.8.3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Erickson P F, Robinson M, Owens G, Drabkin H A. Cancer Res. 1994;54:1782–1786. [PubMed] [Google Scholar]
- 15.Tsujimoto Y, Gorham J, Cossman J, Jaffe E, Croce C M. Science. 1985;229:1390–1393. doi: 10.1126/science.3929382. [DOI] [PubMed] [Google Scholar]
- 16.Vaux D, Cory S, Adams J. Nature (London) 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 17.Miyashita T, Harigai M, Hanada M, Reed J. Cancer Res. 1994;54:3131–3135. [PubMed] [Google Scholar]
- 18.Nimer S D, Zhang W, Kwan K, Wang Y, Zhang J. Blood. 1996;87:3694–3703. [PubMed] [Google Scholar]
- 19.Asou H, Tashiro S, Hamamoto K, Otsuji A, Kita K, Kamada N. Blood. 1991;77:2031–2036. [PubMed] [Google Scholar]
- 20.Matozaki S, Nakagawa T, Kawaguchi R, Aozaki R, Tsusumi M, Murayama T, Koizumi T, Nishimura R, Isobe T, Chihara K. Br J Haematol. 1995;89:805–811. doi: 10.1111/j.1365-2141.1995.tb08418.x. [DOI] [PubMed] [Google Scholar]
- 21.Kozopas K, Yang T, Buchan H L, Zhou P, Craig R. Proc Natl Acad Sci USA. 1993;90:3516–3520. doi: 10.1073/pnas.90.8.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reynolds J E, Yang T, Quian L, Jenkinson J D, Zhou P, Eastman A, Craig R. Cancer Res. 1994;54:6348–6352. [PubMed] [Google Scholar]
- 23.Lomo J, Smeland B, Krayewski S, Reed C J, Blomhoff H K. Cancer Res. 1996;56:40–43. [PubMed] [Google Scholar]
- 24.Seto M, Jaeger U, Hockett R D, Graninger W, Bennett S, Goldman P, Korsmeyer S J. EMBO J. 1988;7:123–130. doi: 10.1002/j.1460-2075.1988.tb02791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Young L R, Korsmeyer S. Mol Cell Biol. 1993;4:3686–3697. doi: 10.1128/mcb.13.6.3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lenny N, Meyers S, Hiebert S W. Oncogene. 1995;11:1761–1769. [PubMed] [Google Scholar]
- 27.Sakukara C, Yamaguch-Iwai Y, Satake M, Bae S C, Takahashi A, Ogawa F, Hagiwara A, Takahashi T, Murakami A, Makino A, Nakagawa T, Kamada N, Ito Y. Proc Natl Acad Sci USA. 1994;91:11723–11727. doi: 10.1073/pnas.91.24.11723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mitsushita H, Kobayashi H, Mori S, Kizaki M, Ikeda Y. Biochem Biophys Res Commun. 1995;215:431–437. doi: 10.1006/bbrc.1995.2483. [DOI] [PubMed] [Google Scholar]
- 29.Giese K, Kirshner J R, Grosschedl R. Genes Dev. 1995;9:995–1008. doi: 10.1101/gad.9.8.995. [DOI] [PubMed] [Google Scholar]
- 30.Hernandez-Munain C, Krangel M S. Mol Cell Biol. 1994;14:473–483. doi: 10.1128/mcb.14.1.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ko L J, Prives C. Genes Dev. 1996;10:1054–1072. doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- 32.Descombes P, Schibler U. Cell. 1991;67:569–579. doi: 10.1016/0092-8674(91)90531-3. [DOI] [PubMed] [Google Scholar]
- 33.Diamond M I, Milner J I, Yoshinaga S K, Yamamoto K. Science. 1990;249:1266–1272. doi: 10.1126/science.2119054. [DOI] [PubMed] [Google Scholar]
- 34.Fu S, Consoli U, Hanania E G, Zu Z, Claxton D F, Andreef M, Deisseroth A B. Clin Cancer Res. 1995;1:583–587. [PubMed] [Google Scholar]
- 35.Sanchez-Garcia I, Grutz G. Proc Natl Acad Sci USA. 1995;92:5287–5291. doi: 10.1073/pnas.92.12.5287. [DOI] [PMC free article] [PubMed] [Google Scholar]