

Abstract
Four enantiomerically pure (S)-4-alkylthioamphetamine derivatives were evaluated as monoamine oxidase (MAO) inhibitors using the human and rat isoforms of the enzyme. Molecular dockings were performed in order to gain insights regarding the binding mode of these inhibitors. All compounds were potent and selective MAO-A inhibitors although different rank orders of potencies were observed against the enzymes from different species. This behavior can be rationalized on the basis of different binding modes to each enzyme, as determined in silico. These findings further support the concept that MAO inhibitory activity of novel compounds, determined with enzymes from diverse mammalian species, should be considered with caution if human MAO is the final target to be addressed.
Keywords: Monoamine oxidase, MAO inhibitors, amphetamine derivatives, molecular dynamics simulation
1. Introduction
Compounds that inhibit monoamine oxidase (MAO, E.C. 1.4.3.4) have shown therapeutic value in a variety of conditions including affective disorders and neurodegenerative diseases.1 In mammals, two kinds of MAOs (MAO-A and MAO-B) are present in most tissues. Crystal structures of both isoforms of human,2–5 and of rat MAO-A (rMAO-A),6 complexed with pharmacologically relevant inhibitors, have been recently described. The similarities and differences between human MAO-A (hMAO-A) and that from the rat have provided new insights that may be important in drug development.1,7 Although hMAO-A and rMAO-A exhibit ~ 90% sequence identity, both enzymes show important differences in their quaternary structures, as revealed by X-ray diffraction data. Thus, hMAO-A crystallizes and behaves hydrodynamically in solution as a monomer,5 whereas structural data suggest that rMAO-A forms a dimeric structure in vivo.6 In addition, the volume of the active site cavity of rMAO-A (~450Å3) is smaller than that found in hMAO-A (~550Å3), which is due to a different conformation of the cavity-shaping loop (residues 210–216) present in both enzymes.1,5 Even though it has been consistently pointed out,8 the aforementioned data lend further support to the notion that results obtained with non-human forms of MAO (e.g. the evaluation of the inhibitory properties of a given compound) cannot be unambiguously extrapolated to the situation in humans.
Several phenylisopropylamine derivatives (often referred to in the literature as “substituted amphetamines”) have been shown by us and others to be potent MAO inhibitors.9–11 Structure-activity relationship (SAR) studies, including quantitative analyses (QSAR),10–12 have shown that the presence of electron-donating, unbranched, alkylthio substituents at the para position of the aromatic ring of the amphetamine scaffold generate potent and selective MAO-A inhibitors. In addition, a few studies have shown that the (S)-isomers of substituted amphetamines (which are always dextrorotatory) are the eutomers.13–15 However, little is known about the binding mode of this class of compounds, in spite of their close structural similarity to endogenous monoaminergic neurotransmitters which are the main physiological substrates of MAOs. In this sense, molecular docking is a useful approach since protein-ligand configurations produced by the docking programs allow a visual analysis of protein-ligand interactions and facilitate an intuitive interpretation and understanding of the binding process at the protein binding site.16 Furthermore, recent studies have shown that molecular modeling yields good correlations between the calculated and experimental Ki values for a series of known and new MAO inhibitors.17–18
In the present study, four enantiomerically pure (S)-4-alkylthiophenylisopropylamine derivatives were prepared and evaluated as MAO inhibitors using the human and rat enzyme isoforms. In addition, using the crystal structures of the proteins, molecular docking was performed in order to gain insights regarding the binding modes of these inhibitors and also to explain observed differences in their inhibitory properties with MAO from different species.
2. Results and discussion
2.1 Chemistry
The synthesis of the (S)-4-alkylthiophenylisopropylamine derivatives 1–4 (Table 1) was carried out by a reaction sequence involving initial Friedel-Crafts acylation of the corresponding alkyl-phenyl thioethers with (S)-N-trifluoroacetylalanyl chloride, followed by reduction of the (S)-2-trifluoroacetamido-1-arylpropan-1-ones with triethylsilane, and deprotection of the amino group by acid hydrolysis, according to a previously reported procedure.19,20 Hydrochloride salts were obtained by adding gaseous HCl (Scheme 1).
Table 1.
Yields and Physical Properties of (S)-2-Amino-1-arylpropane Derivatives 1–4
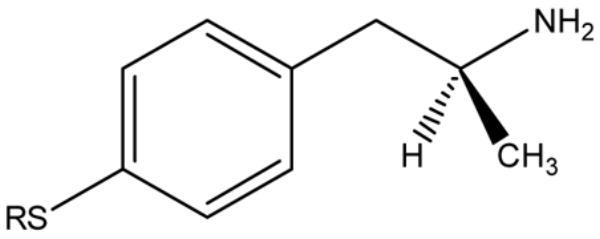
| Compound | R | mp (ºC) | Yield (%) | [α]20Da | m/z (free base) |
|---|---|---|---|---|---|
| 1 | CH3 | 224–225 | 47 | + 7.9 | 181.09 |
| 2 | CH3CH2 | 211–213 | 58 | + 6.4 | 195.11 |
| 3 | CH3CH2CH2 | 135–137 | 50 | + 5.9 | 209.12 |
| 4 | CH3CH2CH2CH2 | 141–143 | 38 | + 6.8 | 223.13 |
(c = 1, MeOH)
Scheme 1a.
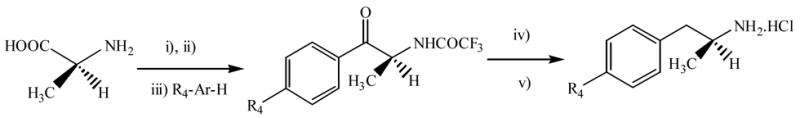
a Reaction conditions: i) 1,1,3,3-tetramethylguanidine/CF3CO2Et/MeOH; ii) (CO)2Cl2/Py/CH2Cl2; iii) AlCl3; iv) (Et3)3SiH / CF3COOH, Reflux; v) 12 M HCl/2-propanol, 70 ºC.
This synthetic route proceeded with complete retention of chirality, supporting the versatility of using chiral alanine derivatives,20 previously exploited in the synthesis of amphetamine21 and cathinone analogs.22
Enantiomeric excesses were determined to be > 98 % in all cases, as evaluated by the comparison of the 1H NMR signals of the enantiomer with those of the corresponding racemic mixture, both complexed with europium tris-[3-(heptafluoropropylhydroxymethylene)-(+)-camphorate] (Eu(hfc)3) used as chiral shift reagent.
2.2 Biochemistry
Table 2 summarizes the effects of compounds 1–4 upon MAO-A and -B from human and rat. As previously shown for 1,15 and racemic 2,10 all (S)-4-alkylthioamphetamines selectively inhibited MAO-A from both species with Ki values in the nanomolar range. This inhibition was competitive in all cases, as shown for (S)-4-ethylthioamphetamine 2 in Figure 1. Inhibition of both hMAO-B and rMAO-B was observed only in the case of the derivatives bearing a propylthio (3) or butylthio (4) substituent, but the potency was at least 20-fold lower than that observed for the corresponding MAO-A.
Table 2.
MAO Inhibitory Activity of Phenylisopropylamine Derivatives 1–4
| Compound | Rat | Human | ||
|---|---|---|---|---|
| Ki (μM)a | Ki (μM) | |||
| MAO-A | MAO-B | MAO-A | MAO-B | |
| 1 | 0.245 ± 0.030 | >100 | 0.133 ±0.020 | NE |
| 2 | 0.054 ± 0.009 | >100 | 0.075 ± 0.015 | >100 |
| 3 | 0.115 ± 0.016 | 15.000 ± 1.430 | 0.030 ± 0.001 | 14.03 ± 1.082 |
| 4 | 0.392 ± 0.033 | 8.925 ± 1.575 | 0.022 ± 0.001 | 4.58 ± 0.850 |
Ki values for MAOs from rat brain were determined from the IC50 values using the Cheng-Prusoff equation (see experimental section). NE: no effect at the highest concentration tested (100 μM).
Figure 1.
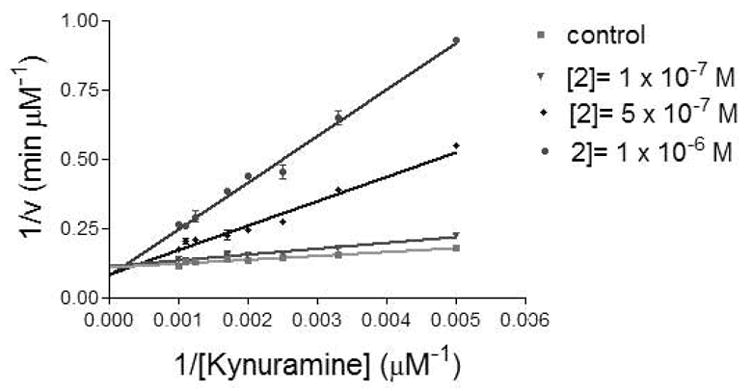
Lineweaver-Burk plot for the inhibition of hMAO-A by 2.
Regarding the MAO-A inhibitory potency, it was similar and followed the same trend for both species in the case of 1 and 2, i.e. lengthening of the substituent from a methylthio to an ethylthio group produced a decrease of the Ki, which agrees with results obtained with racemic mixtures10 and other alkylthio-substituted amphetamine derivatives,11 using MAO from rat brain. However, a clear difference between hMAO-A and rMAO-A was observed when the inhibitory properties of compounds 3 and 4 were compared. This different behavior is demonstrated in the plots in Figure 2 where log Ki is plotted versus the van der Waals volume of the alkyl substituent. A linear correlation is observed with hMAO-A while a strong deviation from linearity is observed for the binding properties of compounds 3 and 4 with rMAO-A.
Figure 2.
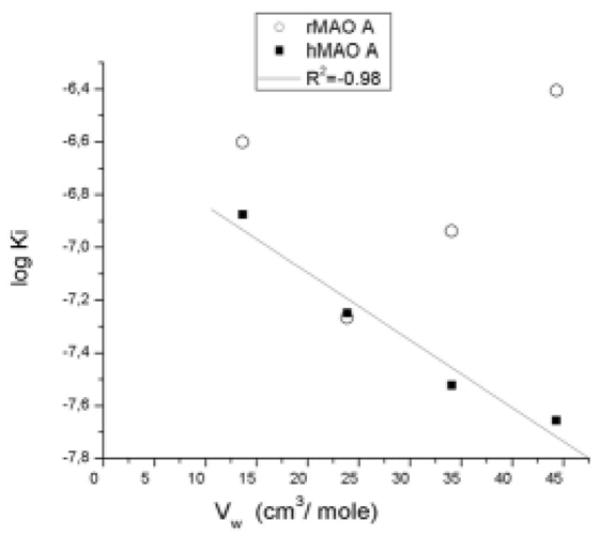
Plot of log Ki versus van der Waals volume of para-alkyl substituents for the inhibition of human and rat MAO-A’s by (S)-4-alkylthioamphetamine analogues. Vw values for the various alkyl substituents were calculated according to Bondi.23
Thus, while potency against hMAO-A increased for the compounds with longer alkyl chains attached to the sulfur atom at the para position (with the four-carbon derivative, i.e. 4, being the most potent), propyl- and butylthio derivatives 3 and 4 were less potent rMAO-A inhibitors than the ethylthio analog 2. These results are in the same line as a recent study by Novaroli et al.,24 which showed that some coumarin and 5H-indeno[1,2-c]pyridazin-5-one derivatives exhibited striking divergences in their inhibitory potency when evaluated against human or rat MAO-B. As stressed by these authors, species-related differences on inhibitor activity such as that of derivative 4 (more than one order of magnitude between rMAO-A and hMAO-A), not only support the idea that results might greatly vary depending on the enzyme source used, but also have important implications for the selection of hit compounds for lead generation, which is an interesting finding considering that both enzymes are ~90% identical in sequence. Accordingly, the different rank orders of potency shown by the compounds tested here strongly suggest that, even though both hMAO-A and rMAO-A are able to accommodate alkylthio-substituted amphetamines in their active sites, the interactions that account for the binding properties in each enzyme, at the least for compounds 3 and 4, are probably different.
2.3 Molecular docking
In order to test the aforementioned hypothesis, compounds 1–4 were docked into the active sites of rMAO-A and hMAO-A, using the Autodock 3.05 suite,25 and the available crystal structures of both enzymes (PDB: 1O5W or 2BXS respectively). Results are summarized in Figures 3 (rat) and 4 (human), which show the most stable conformations for each drug-enzyme complex.
Figure 3.

Binding modes of phenylisopropylamine derivatives 1–4 to rMAO-A (PDB: 1O5W).
Figure 4.
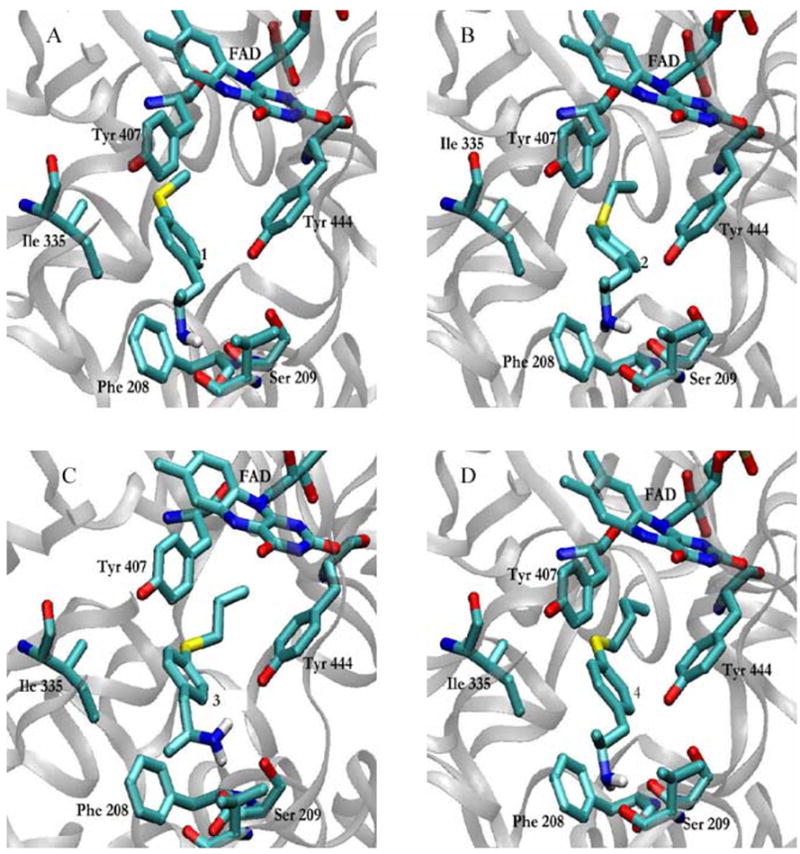
Binding modes of phenylisopropylamine derivatives 1–4 to hMAO-A (PDB: 2BXS).
In rMAO-A, compounds 1 and 2 exhibit binding modes where the amino group points away from the flavin ring forming a hydrogen bond with the carbonyl group of Phe208, whereas the alkylthio chain is located between Tyr407 and Tyr444, a position that would favor interactions between the highly polarizable sulfur atom and either or both aromatic moieties or the hydroxyl groups of the tyrosines (Figs. 3A and B).
In these conformations, the aromatic rings of 1 and 2 seem to be almost identically positioned between the side chains of Gln215 and Ile180, and perpendicular to the flavin ring. A similar binding mode has been recently described for the naturally occurring eugenol (4-allyl-2-methoxyphenol), which has clear structural similarities with phenylisopropylamines, although in that case the aromatic ring of the ligand was located somewhat closer to the isoalloxazine ring.26 As previously discussed for similar results obtained with heteroarylisopropylamine derivatives,27 this binding mode provides a rationale for the observed inhibitory activity, since while blocking the access of any substrate to the active site, these phenylisopropylamines could avoid deamination by adopting a conformation where the amino group is remote from the influence of the flavin ring. In contrast, docking of compounds 3 and 4 yielded binding modes where the inhibitor molecules adopted an almost opposite orientation to derivatives 1 and 2. Thus, the most energetically favorable conformations of 3 and 4 were those where the aminopropyl chain was positioned between Tyr407 and Tyr444 and the alkylthio chain appeared folded into a space surrounded by Ile335, Phe208, Val210 and Thr336, instead of being extended into the hydrophobic channel pointing to the binding site entrance (Figs. 3C and D). The aromatic rings of 3 and 4 were almost in the same position and orientation as those of derivatives 1 and 2. Although different from the foregoing, this binding mode is also consistent with the rMAO-A inhibitory activity of compounds 3 and 4. In spite of an intense debate about the mechanism underlying MAO-catalyzed amineoxidation,28 the two most accepted schemes, i.e. the single electron transfer29 and the polar nucleophilic mechanisms,30 involve the abstraction of the pro-R α-proton of the amine by the N5 atom of the flavin ring, which would act as a base. As can be seen in Figures 3C and D, a possible interaction between the sole α-proton of phenylisopropylamine derivatives 3 and 4 and the FAD N5, is made less likely by the presence of the α-methyl group, which prefers to face the re side of the isoalloxazine ring, leaving the pro-R α-proton far from the isoalloxazine N5. Beyond these considerations, the striking differences in the binding modes of derivatives 1–2 and 3–4 to rMAO-A generated by computational simulations can explain the trend of their inhibitory potencies. Thus, no hydrogen bonds between the inhibitor and active site residues were apparent for the best binding conformations of 3 and 4, which agree with a less stable interaction and higher Ki values.
In the case of hMAO-A, the binding modes obtained for compounds 1–4 from docking simulations were similar in all cases and resemble those observed in rMAO-A for 1 and 2, i.e. the amino group was oriented away from the flavin ring and the alkylthio chain was positioned in the so-called aromatic cage, between Tyr407 and Tyr444 (Fig. 4).
The aromatic rings of the inhibitors occupy a position close to that found in rMAO-A, but they are located in a clearly less perpendicular orientation to the flavin as compared with that observed in the rat enzyme. This positioning might favor stacking interactions with the aromatic rings of Phe208 and Tyr69 (both about 5 Å from the inhibitors’ aromatic moieties), as shown in Figure 5.
Figure 5.

Superimposed structures of compounds 1 (grey), 2 (green), 3 (yellow) and 4 (blue), docked into the active site of hMAO-A.
In addition, stabilizing hydrogen bonds between the amino group and the side chain of Ser209, the backbone carbonyl of Val210, or both, were detected for all compounds. The similarities in the binding modes of 1–4 to hMAO-A suggest that the different potencies of these compounds determined in biochemical assays could be correlated with differential interactions at the active site, mainly associated with the different properties of the alkylthio chain attached to the para position. It has been shown that the affinities of 4-substituted benzylamine and phenethylamine analogs for hMAO-A are more favorable when the size of the para substituent increases, and QSAR analyses have shown that this effect depends exclusively on steric parameters of the para- substituent (van der Waals volume and/or Taft steric parameter Es).31,32 The results presented here completely agree with this notion, and an excellent correlation (r2 = 0.97) was found between log Ki and the van der Waals volume23 of the para substituent of compounds 1–4. In the case of rMAO-A, this SAR does not hold for the compounds tested here, since (as shown by docking experiments) the binding mode is not the same for the whole series. Other examples of arylalkylamine derivatives, in which differential binding modes to MAO from different species have been advocated to explain different inhibitory potencies, include phentermine33 and benzylamine analogs.34
3. Conclusions
The results presented here show that some 4-alkylthiophenylisopropylamine derivatives are able to differentially inhibit MAO-A from rat and human, a behavior that can be rationalized on the basis of differing binding modes to each enzyme exhibited by the compounds in docking studies. These results might be related to the differences in the volume and shape of the active sites of rMAO-A and hMAO-A demonstrated by structural studies, but further experiments are necessary to fully elucidate how these differences influence the binding of this class of compounds to MAO-A. In addition, we have described two binding modes, consistent with inhibitory properties, for the interaction of substituted amphetamine derivatives with MAO-A. It should therefore be kept in mind that drugs with similar activities may not necessarily bind to their receptor(s) in the same orientation, and that differences of this sort must be considered when analyzing SAR.
Finally, our results further support the concept that MAO inhibitory activity of novel compounds, determined with enzymes from diverse mammalian species, should be considered with caution if human MAO is the final target to be addressed.
4. Experimental
4.1. Chemistry
All reagents and solvents were commercially available and were used without further purification. Melting points are uncorrected and were determined with an Electrothermal apparatus. NMR spectra were recorded using either a Bruker AMX 300 or a Bruker Advance 400 spectrometer at 300 or 400 (1H) and 75 (13C) MHz, employing tetramethylsilane as an internal standard. Chemical shifts are reported relative to TMS (δ= 0.00) and coupling constants (J) are given in Hz. Europium tris[3-(heptafluoropropylhydroxymethylene)-(+)-camphorate] was purchased from Aldrich. Optical rotation values were obtained with a Perkin–Elmer 341 polarimeter. The elemental analyses for C, H, N and S were performed on a CE Instruments (model EA 1108) analyzer.
4.1.1. Preparation of (S)-N-Trifluoroacetylalanine
(S)-N-Trifluoroacetylalanine was prepared following a procedure described previously from L-alanine (Aldrich), ethyl trifluoroacetate, and 1,1,3,3-tetramethylguanidine.22 The product (86% yield) had a melting point of 62–64°C (lit.19 70–71°C), and was sufficiently pure for all subsequent uses.
4.1.2. General Procedure for the Preparation of (S)-2-Trifluoroacetamido-1-aryl-1-propanones
(S)-Trifluoroacetamido-1-aryl-1-propanones were prepared by Friedel–Crafts acylation of the corresponding arenes with (S)-N-trifluoroacetylalanyl chloride, obtained in situ by reaction of (S)-N-trifluoroacetylalanine and oxalyl chloride, following previously reported procedures.19,22 Final compounds were purified by column chromatography on silica gel 60, eluting with CH2Cl2. (S)-Methylthio and (S)- ethylthio derivatives were recognized by melting points and NMR spectra from the literature,22 and (S)-propylthio and (S)-butylthio derivatives were identified by comparison with the previously reported NMR signals for the same aromatic substitutions in the racemic compounds.35
4.1.3. General Procedure for the Preparation of (S)-2-Trifluoroacetamido-1-arylpropanes
The (S)-trifluoroacetamido-1-arylpropanes were prepared following a previously reported procedure,20 with some modifications. To a stirred solution of (S)-2-trifluoroacetamido-1-aryl-1-propanone (7 mmol) in trifluoroacetic acid (3 mL) was added triethylsilane (0.55 ml, 35 mmol). The reaction mixture was refluxed and stirred for 6 h. Finally, the mixture was cooled to room temperature and made basic with a saturated solution of NaHCO3. The mixture was extracted with CH2Cl2 (2 x 15 mL) and washed with HCl (1 N). The organic layer was dried over anhydrous Na2SO4 and concentrated by rotatory evaporation at 40°C to afford the crude (S)-trifluoroacetamido-1-arylpropane. The product was purified by column chromatography on silica gel 60, eluting with CH2Cl2.
4.1.3.1. (S)-2-Trifluoroacetamido-1-(4-methylthiophenyl)-propane
Yield 90%. 1H NMR (CDCl3) 1.14 (d, 3H, J = Hz, CHCH3), 2.40 (s, 3H, CH3S), 2.66 (dd, 1H, Ja,b = 13.3 Hz, Ja,c = 9.1 Hz, CHaHbCHc), 2.78 (dd, 1H, J,b,a = 13.4 Hz, Jb,c = 5.1 Hz, CHaHbCHc), 4.18 (m, 1H, CHCH3), 6.04 (s, 1H, NH), 7.30 (d, 2H, J = 8.7 Hz, ArH-3,5), 7.16 (d, 2H, J = 8.7 Hz, ArH-2,6). 13C NMR (CDCl3) 15.9 (CHCH3), 19.4 (CH3S), 41.2 (CH2CH), 47.2 (CHCH3), 117.0 (q, COCF3), 127.0 (ArC-3,5), 129.8 (ArC-2,6), 133.5 (ArC-4), 137.1 (ArC-1), 157.0 (q, COCF3).
4.1.3.2. (S)-2-Trifluoroacetamido-1-(4-ethylthiophenyl)-propane
Yield 93%. 1H NMR (CDCl3) 1.14 (d, 3H, J = 6.6 Hz, CHCH3), 1.23 (t, 3H, J = 7.3 Hz, CH3CH2S), 2.71 (dd, 1H, Ja,b = 13.9 Hz, Ja,c = 7.9 Hz, CHaHbCHc), 2.79 (dd, 1H, Jb,a = 13.8 Hz, Jb,c = 7.0 Hz, CHaHbCHc), 2.85 (q, 2H, J = 7.3 Hz, CH3CH2S), 4.18 (m, 1H, CHCH3), 6.00 (s, 1H, NH), 7.10 (d, 2H, J = 8.1 Hz, ArH-3,5), 7.21 (d, 2H, J = 8.6 Hz, ArH-2,6). 13C NMR (CDCl3) 14.4 (CHCH3), 19.4 (CH3CH2S), 27.8 (CH3CH2S), 41.3 (CH2CH), 47.2 (CHCH3), 115.7 (q, COCF3) 129.4 (ArC-3,5), 129.8 (ArC-2,6), 134.3 (ArC-4), 135.3 (ArC-1), 156.3 (q, COCF3).
4.1.3.3. (S)-2-Trifluoroacetamido-1-(4-propylthiophenyl)-propane
Yield 90 %. 1H NMR (CDCl3) 0.95 (t, 3H, J = Hz, CH3CH2CH2S), 1.14 (d, 3H, J = Hz, CHCH3), 1.60 (m, 2H, CH3CH2CH2S), 2.75 (m, 4H, CH2CH, CH3CH2CH2S), 4.17 (m, 1H, CHCH3), 5.98 (s, 1H, NH), 7.02 (d, 2H, J = 8.6 Hz, ArH-3,5), 7.23 (d, 2H, J = 8.6 Hz, ArH-2,6). 13C NMR (CDCl3) 13.4 (CHCH3), 19.4 (CH3CH2CH2S), 22.5 (CH3CH2CH2S), 35.8 (CH3CH2CH2S), 41.3 (CH2CH), 47.2 (CHCH3), 116.0 (q, COCF3) 129.3 (ArC-3,5), 129.8 (ArC-2,6), 134.2 (ArC-4), 135.7 (ArC-1), 156.6 (q, COCF3).
4.1.3.4. (S)-2-Trifluoroacetamido-1-(4-butylthiophenyl)-propane
Yield 95 %. 1H NMR (CDCl3) 0.84 (t, 3H, J = 7.4 Hz, CH3CH2CH2CH2S), 1.13 (d, 3H, J = 6.6 Hz, CHCH3), 1.37 (m, 2H, CH3CH2CH2CH2S), 1.53 (m, 2H, CH3CH2CH2CH2S), 2.68 (dd, 1H, Ja,b = 13.9 Hz, Ja,c = 7.4 Hz, CHaHbCHc), 2.77 (dd, 1H, Jb,a = 13.4 Hz, Jb,c = 7.0 Hz, CHaHbCHc), 2.84 (t, 2H, J = 7.2 Hz, CH3CH2CH2CH2S), 4.18 (m, 1H, CHCH3), 6.00 (s, 1H, NH), 6.99 (d, 2H, J = 8.5 Hz, ArH-3,5), 7.20 (d, 2H, J = 8.5 Hz, ArH-2,6). 13C NMR (CDCl3) 13.6 (CHCH3), 19.4 (CH3CH2CH2CH2S), 22.0 (CH3CH2CH2CH2S), 31.2 (CH3CH2CH2CH2S), 33.4 (CH3CH2CH2CH2S), 41.3 (CH2CH), 47.2 (CHCH3), 115.7 (q, COCF3) 129.2 (ArC-3,5), 129.8 (ArC-2,6), 134.1 (ArC-4), 135.8 (ArC-1), 156.0 (q, COCF3).
4.1.4. General Procedure for the Preparation of (S)-2-Amino-1-arylpropane hydrochlorides, 1–4
The (S)-2-trifluoroacetamido-1-aryl-1-propane derivatives (0.7 mmol) were dissolved in 2-propanol (16 ml) and concentrated HCl (12 ml). The resulting solutions were then stirred at 70°C for 10 h. The mixtures were allowed to cool to room temperature and the solvent was eliminated by rotary evaporation. The solid mixtures were treated with 1 N NaOH (15 ml) and extracted with CH2Cl2 (2 x 15 ml). The organic phases were combined and extracted with 1 N HCl and the aqueous layers were separated and made basic with 2 N NaOH. These solutions were extracted with CH2Cl2 (2 x 15 ml) and the organic phases were dried over anhydrous Na2SO4 and concentrated. The residual amines were dissolved in dry ether (10 mL) and the hydrochlorides precipitated with gaseous HCl.
4.1.4.1. (S)-2-Amino-1-(4-methylthiophenyl)-propane hydrochloride (1)
1H NMR (DMSO-d6) 1.11 (d, 3H, J = 6.6 Hz, CHCH3), 2.44 (s, 3H, CH3S), 2.64 (dd, 1H, Ja,b = 13.3 Hz, Ja,c = 9.1 Hz, CHaHbCHc), 3.02 (dd, 1H, J,b,a = 13.4 Hz, Jb,c = 5.1 Hz, CHaHbCHc), 3.31 (br, 1H, CHCH3), 7.18 (d, 2H, J = 8.7 Hz, ArH-3,5), 7.22 (d, 2H, J = 8.7 Hz, ArH-2,6), 8.23 (s, 3H, NH3). 13C NMR (DMSO-d6) 15.2 (CHCH3), 17.9 (CH3S), 39.8 (CH2CH), 48.4 (CHCH3), 126.7 (ArC-3,5), 130.3 (ArC-2,6), 133.9 (ArC- 4), 136.8 (ArC-1). HRMS m/z 181.0926, calculated for C10H15NS (M-HCl)+, 181.0925. Elemental analysis of this compound was reported previously.15
4.1.4.2. (S)-2-Amino-1-(4-ethylthiophenyl)-propane hydrochloride (2)
1H NMR (D2O) 1.26 (t, 3H, J = 7.3 Hz, CH3CH2S), 1.28 (d, 3H, J = 6.6 Hz, CHCH3), 2.88 (dd, 1H, Ja,b = 13.9 Hz, Ja,c = 7.3 Hz, CHaHbCHc), 2.93 (dd, 1H, J,b,a = 13.8 Hz, Jb,c = 7.0 Hz, CHaHbCHc) 2.98 (q, 2H, J = 7.3 Hz, CH3CH2S), 3.60 (m, 1H, CHCH3), 7.25 (d, 2H, J = 8.1 Hz, ArH-3,5), 7.38 (d, 2H, J = 8.6 Hz, ArH-2,6). 13C NMR (D2O) 16.2 (CH CH3), 20.0 (CH3CH2S), 29.5 (CH3CH2S), 42.2 (CH2CH), 51.6 (CHCH3), 131.8 (ArC-3,5), 132.8 (ArC-2,6), 136.7 (ArC-4), 137.0 (ArC-1). HRMS m/z 195.1078, calculated for C11H17NS (M-HCl)+, 195.1082. (C11H18ClNS): C, 57.07%; H, 8.57%; N, 6.05%; S, 15.07% (calcd: C, 57.00%; H, 7.83%; N, 6.04%; S, 13.83%).
4.1.4.3. (S)-2-Amino-1-(4-propylthiophenyl)-propane hydrochloride (3)
1H NMR (D2O) 0.96 (t, 3H, J = 7.3 Hz, CH3CH2CH2S), 1.28 (d, 3H, J = 6.6 Hz, CHCH3), 1.62 (m, 2H, CH3CH2CH2S), 2.94 (t, 2H, J = 7.3 Hz, CH3CH2CH2S), (m, 4H, CH2CH, CH3CH2CH2S) 3.60 (m, 1H, CHCH3), 7.24 (d, 2H, J = 8.6 Hz, ArH-3,5), 7.37 (d, 2H, J = 8.6 Hz, ArH-2,6). 13C NMR (D2O) 15.2 (CHCH3) 20.0 (CH3CH2CH2S) 24.6 (CH3CH2CH2S), 37.4 (CH3CH2CH2S) 42.2 (CH2CH), 51.6 (CHCH3), 131.8 (ArC-3,5) 132.8 (ArC-2,6) 136.6 (ArC-4) 137.2 (ArC-1). HRMS m/z 209.1240, calculated for C12H19NS (M-HCl)+, 209.1238. (C12H20ClNS): C, 57.77%; H, 9.01%; N, 5.56%; S, 14.44% (calcd: C, 58.63%; H, 8.20%; N, 5.70%; S, 13.04%).
4.1.4.4. (S)-2-Amino-1-(4-butylthiophenyl)-propane hydrochloride (4)
1H NMR (D2O) 0.85 (t, 3H, J = 7.4 Hz, CH3CH2CH2CH2S), 1.26 (d, 3H, J = 6.6 Hz, CHCH3), 1.38 (m, 2H, CH3CH2CH2CH2S), 1.56 (m, 2H, CH3CH2CH2CH2S), 2.86 (dd, 1H, Ja,b = 13.9 Hz, Ja,c = 7.4 Hz, CHaHbCHc), 2.91 (dd, 1H, Jb,a = 13.4 Hz, Jb,c = 7.0 Hz, CHaHbCHc), 2.95 (t, 2H, J = 7.2 Hz, CH3CH2CH2CH2S), 3.58 (m, 1H, CHCH3), 7.22 (d, 2H, J = 8.5 Hz, ArH-3,5), 7.36 (d, 2H, J = 8.5 Hz, ArH-2,6). 13C NMR (D2O) 15.5 (CHCH3), 20.0 (CH3CH2CH2CH2S), 23.9 (CH3CH2CH2CH2S), 33.2 (CH3CH2CH2CH2S), 35.0 (CH3CH2CH2CH2S), 42.2 (CH2CH), 51.6 (CHCH3), 131.8 (ArC-3,5), 132.7 (ArC-2,6), 136.6 (ArC-4), 137.3 (ArC-1). HRMS m/z 223.1392, calculated for C13H21NS (M-HCl)+, 223.1395. (C13H22ClNS): C, 59.10%; H, 9.39%; N, 5.26%; S, 14.03% (calcd: C, 60.09%; H, 8.53%; N, 5.39%; S, 12.34%).
4.1.5. Determination of the Enantiomeric Excess by 1H NMR Using (Eu(hfc)3) as Chiral Shift Reagent
Trifluoroacetamides, suitable for the 1H NMR analysis in CDCl3, were prepared from the (S)-2-amino-1-(4-alkylthiophenyl)-propane hydrochlorides. Trifluoroacetic anhydride (0.1 mL) was added separately to suspensions of 5 mg (0.022 mmol) of (R,S) and (S)-2-amino-1-(4-alkylthiophenyl)-propane hydrochlorides in CH2Cl2. The reaction mixtures were stirred for 1 hour and rotary evaporated to give solid (R,S) and (S)-2-trifluoroacetamido-1-(4-alkylthiophenyl)- propane, respectively. To each solid, 0.5 eq. of Eu(hfc)3 was added and the complexes were dissolved in CDCl3 for analysis. Inspection of the 1H NMR signals indicated that in the case of the (S)-enantiomers, the methyl group linked to the asymmetric center gave a doublet in the spectrum with a chemical shift relative to TMS close to 1.44 ppm, whereas the racemic mixture give rise to two partially overlapping doublets
4.2. Biochemistry
4.2.1. Rat MAO
All the chemicals used were of the highest grade commercially available. Tetrahydrofuran and acetonitrile were Merck HPLC grade. 5-HT, 5- hydroxyindoleacetic acid, yeast aldehyde dehydrogenase and β-nicotinamide dinucleotide were from Sigma (St. Louis, MO, USA). The effects of the enantiomers on MAO-A or MAO-B activities were studied using a crude rat brain mitochondrial suspension (male Sprague–Dawley rats weighing 180–220 g, sacrificed by decapitation), using 5-HT (100 μM) and 4-dimethylaminophenethylamine (DMAPEA, 5 μM) as selective substrates for MAO-A and -B, respectively, and detecting these compounds and their metabolites by HPLC with electrochemical detection (HPLC-ED) as described previously.10 A C18 reverse phase column (Lichrospher 250 mm x 4.6 mm, 5 μm) and an amperometric detector (Merck-Recipe L3500A), were used to analyze the reaction mixtures. All other conditions were as previously described.10,15 IC50 values (mean ± SD from at least two independent experiments, each in triplicate) were determined using Prism Graph Pad software, from plots of inhibition percentages (calculated in relation to a sample of the enzyme treated under the same conditions without inhibitors) versus -log inhibitor concentration. Ki were determined from the IC50 values using the Cheng-Prusoff equation: Ki = IC50 / (1 + [S]/ Km).36
4.2.2. Human MAO
Human recombinant MAO-A and MAO-B were expressed in Pichia pastoris and purified as described previously.37,38 Both enzymes (1–2 mg) were desalted from glycerol stock solutions using a G-25 (fine) Sephadex column (1 x 20 cm, Sigma) in 50 mM potassium phosphate buffer, pH = 7.5, containing 0.8% (w/v) β-octyl-glucoside before use. Enzymatic activity measurements were carried out as previously described.39 Briefly, hMAO-A and hMAO-B activities were determined spectrophotometrically, in the presence or absence of inhibitors, using kynuramine and benzylamine (purchased from Sigma) as substrates, respectively, in 50 mM HEPES buffer, pH 7.5, containing 0.5% (w/v) reduced Triton X-100, at 25°C. The oxidation of kynuramine catalyzed by MAO-A was monitored at 316 nm (absorption maximum for aldehyde product, extinction coefficient = 11,800 M−1 cm−1), and that of benzylamine by MAO-B was monitored at 250 nm (maximum absorption by benzaldehyde, extinction coefficient = 12,800 M−1 cm−1). All spectral data were obtained using a Perkin-Elmer Lambda 2 UV-VIS double-beam spectrophotometer with thermostatted cuvette holders. Kinetic data were evaluated and plotted using Microcal Origin or Prism Graph Pad software.
4.3. Molecular Simulation
The crystallographic data of rMAO-A or hMAO-A (PDB: 1O5W or 2BXS respectively) were used for all calculations. The Autodock 3.05 suite25 was then used to perform the docking simulations and conditions were as previously reported,27,35 with some modifications. Briefly, the hydrogen atoms of the protein and the FAD molecule were built using Insight II,40 and the structures were relaxed following a minimization protocol using Discover_3 and the ESFF force field. The grid maps were calculated using the autogrid3 option and were centred on the putative ligand-binding site. The volumes chosen for the grid maps were made up of 40 x 40 x 40 points, with a grid-point spacing of 0.375 Å. The partial charges of 4-alkylthiophenylisopropylamine derivatives 1–4 were corrected using RESP methodology.41 The dielectric constant was adjusted either to 10 (hMAO-A) or to 2 (rMAO-A) in the grid parameter file (gpf) of the Autodock suite. This parameter adjustment gave the best correlations between the calculated docking energies and biochemical results. The docked compound complexes were built using the lowest free-energy binding positions.
Acknowledgments
This work was partially funded by FONDECYT grant 1060199 and DICYT grant 020591RPRP. DEE acknowledges research support from the National Institutes of Health (GM29433). The compounds described were synthesized by MOO during a stay in Prof. E. Breitmaier’s laboratory at the University of Bonn as the recipient of a DAAD grant. We also thank Dr. Juan Guerrero for his assistance in the NMR spectroscopy with the chiral shift reagent, Ms. Milagros Aldeco for the preparation of purified recombinant human MAO A and MAO B and Drs. G. Zapata-Torres and C. Mascayano and Prof. M.C. Rezende for their contribution to the computational work. AF was the recipient of CONICYT and MECESUP scholarships.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Youdim MBH, Edmondson D, Tipton KF. Nat Rev Neurosci. 2006;7:295. doi: 10.1038/nrn1883. [DOI] [PubMed] [Google Scholar]
- 2.Binda C, Newton-Vinson P, Hubálek F, Edmondson DE, Mattevi A. Nat Struct Biol. 2002;9:22. doi: 10.1038/nsb732. [DOI] [PubMed] [Google Scholar]
- 3.Binda C, Li M, Hubálek F, Restelli N, Edmondson DE, Mattevi A. Proc Natl Acad Sci USA. 2003;100:9750. doi: 10.1073/pnas.1633804100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Binda C, Hubálek F, Li M, Herzig Y, Sterling J, Edmondson DE, Mattevi A. J Med Chem. 2005;48:8148. doi: 10.1021/jm0506266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Colibus L, Li M, Binda C, Lustig A, Edmondson DE, Mattevi A. Proc Natl Acad Sci USA. 2005;102:12684. doi: 10.1073/pnas.0505975102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma J, Yoshimura M, Yamashita E, Nakagawa A, Ito A, Tsukihara T. J Mol Biol. 2004;338:103. doi: 10.1016/j.jmb.2004.02.032. [DOI] [PubMed] [Google Scholar]
- 7.Reyes-Parada M, Fierro A, Iturriaga-Vásquez P, Cassels BK. Curr Enz Inhib. 2005;1:85. [Google Scholar]
- 8.Tipton KF, Boyce S, O’Sullivan J, Davey GP, Healy J. Curr Med Chem. 2004;11:1965. doi: 10.2174/0929867043364810. [DOI] [PubMed] [Google Scholar]
- 9.Florvall L, Ask AL, Ögren SO, Ross SB. J Med Chem. 1978;21:56. doi: 10.1021/jm00199a010. [DOI] [PubMed] [Google Scholar]
- 10.Scorza MC, Carrau C, Silveira R, Zapata-Torres G, Cassels BK, Reyes-Parada M. Biochem Pharmacol. 1997;54:1361. doi: 10.1016/s0006-2952(97)00405-x. [DOI] [PubMed] [Google Scholar]
- 11.Gallardo-Godoy A, Fierro A, McLean TH, Castillo M, Cassels BK, Reyes-Parada M, Nichols DE. J Med Chem. 2005;48:2407. doi: 10.1021/jm0493109. [DOI] [PubMed] [Google Scholar]
- 12.Vallejos G, Rezende MC, Cassels BK. J Comput-Aided Mol Des. 2002;16:95. doi: 10.1023/a:1016344030772. [DOI] [PubMed] [Google Scholar]
- 13.Fowler CJ, Oreland L. J Pharm Pharmacol. 1981;33:403. doi: 10.1111/j.2042-7158.1981.tb13819.x. [DOI] [PubMed] [Google Scholar]
- 14.Robinson JB. Biochem Pharmacol. 1985;34:4105. doi: 10.1016/0006-2952(85)90201-1. [DOI] [PubMed] [Google Scholar]
- 15.Hurtado-Guzmán C, Fierro A, Iturriaga-Vásquez P, Sepúlveda-Boza S, Cassels BK, Reyes-Parada M. J Enz Inhib Med Chem. 2003;18:339. doi: 10.1080/1475636031000118437. [DOI] [PubMed] [Google Scholar]
- 16.Evers A, Hessler G, Matter H, Klabunde T. J Med Chem. 2005;48:5448. doi: 10.1021/jm050090o. [DOI] [PubMed] [Google Scholar]
- 17.Toprakçí M, Yelekçi K. Bioorg Med Chem Lett. 2005;15:4438. doi: 10.1016/j.bmcl.2005.07.043. [DOI] [PubMed] [Google Scholar]
- 18.Chimenti F, Bolasco A, Manna F, Secci D, Chimenti P, Granese A, Befani O, Turín P, Alcaro S, Ortuso F. Chem Biol Drug Des. 2006;67:206. doi: 10.1111/j.1747-0285.2006.00367.x. [DOI] [PubMed] [Google Scholar]
- 19.Curphey TJ. J Org Chem. 1979;44:2805–2807. [Google Scholar]
- 20.Nordlander JE, Payne MJ, Njoroge FG, Balk MA, Laicos GD, Vishwanath VM. J Org Chem. 1984;49:4107. [Google Scholar]
- 21.Chambers JJ, Kurrasch-Orbaugh DM, Parker MA, Nichols DE. J Med Chem. 2001;44:1003. doi: 10.1021/jm000491y. [DOI] [PubMed] [Google Scholar]
- 22.Osorio-Olivares M, Rezende MC, Sepúlveda-Boza S, Cassels BK, Baggio RF, Muñoz-Acevedo JC. Tetrahedron: Asymmetry. 2003;14:1473. [Google Scholar]
- 23.Bondi A. J Phys Chem. 1964;68:441. [Google Scholar]
- 24.Novaroli L, Daina A, Favre E, Bravo J, Carotti A, Leonetti F, Catto M, Carrupt PA, Reist M. J Med Chem. 2006;49:6264. doi: 10.1021/jm060441e. [DOI] [PubMed] [Google Scholar]
- 25.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. J Computat Chem. 1998;19:1639. [Google Scholar]
- 26.Tao G, Irie Y, Li DJ, Keung WM. Bioorg Med Chem. 2005;13:4777. doi: 10.1016/j.bmc.2005.04.081. [DOI] [PubMed] [Google Scholar]
- 27.Vallejos G, Fierro A, Rezende MC, Sepúlveda-Boza S, Reyes-Parada M. Bioorg Med Chem. 2005;13:4450. doi: 10.1016/j.bmc.2005.04.045. [DOI] [PubMed] [Google Scholar]
- 28.Scrutton NS. Nat Prod Rep. 2004;21:722. doi: 10.1039/b306788m. [DOI] [PubMed] [Google Scholar]
- 29.Silverman RB. Acc Chem Res. 1995;28:335. [Google Scholar]
- 30.Edmondson DE, Mattevi A, Binda C, Li M, Hubálek F. Curr Med Chem. 2004;11:1983. doi: 10.2174/0929867043364784. [DOI] [PubMed] [Google Scholar]
- 31.Miller JR, Edmondson DE. Biochemistry. 1999;38:13670. doi: 10.1021/bi990920y. [DOI] [PubMed] [Google Scholar]
- 32.Nandigama RK, Edmondson DE. Biochemistry. 2000;39:15258. doi: 10.1021/bi001957h. [DOI] [PubMed] [Google Scholar]
- 33.Nandigama RK, Newton-Vinson P, Edmondson DE. Biochem Pharmacol. 2002;63:865. doi: 10.1016/s0006-2952(02)00840-7. [DOI] [PubMed] [Google Scholar]
- 34.Edmondson DE, Bhattacharrya AK, Xu J. Biochim Biophys Acta. 2000;1479:52. doi: 10.1016/s0167-4838(00)00055-8. [DOI] [PubMed] [Google Scholar]
- 35.Osorio-Olivares M, Caroli-Rezende M, Sepúlveda-Boza S, Cassels BK, Fierro A. Bioorg Med Chem. 2004;12:4055. doi: 10.1016/j.bmc.2004.05.033. [DOI] [PubMed] [Google Scholar]
- 36.Cheng YC, Prusoff WH. Biochem Pharmacol. 1973;22:3099. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 37.Li M, Hubálek F, Newton-Vinson P, Edmondson DE. Protein Expr Purif. 2002;24:152. doi: 10.1006/prep.2001.1546. [DOI] [PubMed] [Google Scholar]
- 38.Newton-Vinson P, Hubálek F, Edmondson DE. Protein Expr Purif. 2000;20:334. doi: 10.1006/prep.2000.1309. [DOI] [PubMed] [Google Scholar]
- 39.Hubálek F, Binda C, Li M, Herzig Y, Sterling J, Youdim MBH, Mattevi A, Edmondson DE. J Med Chem. 2004;47:1760. doi: 10.1021/jm0310885. [DOI] [PubMed] [Google Scholar]
- 40.Insight II and Discover. User Guide. Accelrys; San Diego, CA, USA: 1998. [Google Scholar]
- 41.Bayly CI, Cieplak P, Cornell WD, Kollman PA. J Phys Chem. 1993;97:10269. [Google Scholar]