

Abstract
Objective
Several recent studies have demonstrated that thioredoxin (Trx) is an important anti-apoptotic/cytoprotective molecule. The present study was designed to determine whether Trx activity is altered in the aging heart in a way that may contribute to increased susceptibility to myocardial ischemia/reperfusion (MI/R).
Methods and Results
Compared to young animals, MI/R-induced cardiomyocyte apoptosis and infarct size were increased in aging animals (P<0.01). Trx activity was decreased in the aging heart before MI/R, and this difference was further amplified after MI/R. Trx expression was moderately increased and Trx nitration, a post-translational modification that inhibits Trx activity, was increased in the aging heart. Moreover, Trx-ASK1 complex formation was reduced and activity of p38 MAPK was increased. Treatment with FP15 (a peroxynitrite decomposition catalyst) reduced Trx nitration, increased Trx activity, restored Trx-ASK1 interaction, reduced P38 MAPK activity, attenuated caspase 3 activation and reduced infarct size in aging animals (p<0.01).
Conclusions
Our results demonstrated that Trx activity is decreased in the aging heart by post-translational nitrative modification. Interventions that restore Trx activity in the aging heart may be novel therapies to attenuate MI/R injury in aging patients.
Keywords: Apoptosis, Thioredoxin, Aging, Protein Nitration, Ischemia/Reperfusion
1. Introduction
It is well recognized that age is a major independent risk factor for morbidity and mortality in ischemic heart disease[1]. However, the mechanisms responsible for this age-related pathologic change remain elusive. Accumulating evidence indicates that myocardial apoptosis, a gene regulated programmed cell death, markedly increases with aging[2,3]. The molecular mechanisms and signaling transduction pathways which are responsible for increased susceptibility of cardiomyocytes to apoptosis with aging remain largely unidentified.
Thioredoxin (Trx) is a 12-kDa protein ubiquitously expressed in all living cells that fulfills a variety of biological functions related to cell proliferation and apoptosis[4]. It is not only involved in cytoprotective functions against oxidative stress, but also in the regulation of cellular proliferation and the aging process[5]. Clinical and experimental results have demonstrated that inhibition of Trx promotes apoptosis[6]. Recent in vitro studies demonstrate that Trx interacts directly with, and inhibits, the activity of apoptosis-regulating kinase-1 (ASK1), a mitogen-activated protein (MAP) kinase that activates two pro-apoptotic kinases, p38 MAP kinase (MAPK) and c-Jun N-terminal kinase (JNK)[7]. In aged mouse livers, the ratio of ASK1/Trx-ASK1 (free ASK1/Trx-binding ASK1) increases and this correlates with the increased basal activity of the p38 MAPK pathway[8]. These results suggest that Trx may play a critical role in cell proliferation and cell death in aging. However, whether Trx activity is reduced in the aged heart and whether this alteration may contribute to increased cardiomyocyte apoptosis in the aged heart has never been previously investigated.
Reactive oxygen species (ROS) have long been recognized to cause oxidative protein modification and to act as major mediators of the aging process[9,10]. However, the increased susceptibility of the aging heart to ischemia and reperfusion injury and increased post-ischemic cardiomyocyte apoptosis in aging animals cannot be exclusively explained by ROS production, and many fundamental questions remain unresolved[11]. Recent data have identified nitric oxide–derived reactive nitrogen species (RNS), such as peroxynitrite (ONOO−), as critical contributors of protein modification and cell injury, providing potential targets for therapeutic interventions[12,13]. However, whether increased RNS production may increase cardiomyocyte apoptosis via nitrative modification of a specific protein remains unknown.
Therefore, the aims of the present study were 1) to determine whether Trx activity is reduced in the aging heart; 2) to identify the mechanism responsible for aging-induced Trx alteration; 3) to determine the signaling mechanism by which reduced Trx activity leads to apoptotic cardiomyocyte death in the aging heart; and 4) to investigate whether treatment with FP15 (a peroxynitrite decomposition catalyst) attenuates apoptotic cardiomyocyte death in the aging heart.
2. Material and Methods
2.1. Experimental Protocols
The experiments were performed in adherence with the National Institutes of Health Guidelines on the Use of Laboratory Animals and were approved by the Thomas Jefferson University Committee on Animal Care. Male adult (3 month old) and aging (20 month old) C57/B16 mice were anesthetized with 2% isoflurane. MI was produced by temporarily exteriorizing the heart through a left thoracic incision and placing a 6–0 silk suture slipknot at the distal 1/3 of the left anterior descending artery. After 30 min of MI, the slipknot was released, and the myocardium was reperfused for 3 h or 24 h. Ten minutes before reperfusion, mice were randomized to receive either vehicle (PBS, pH 7.5), or FP15 (a peroxynitrite decomposition catalyst, 5 mg/kg) by i.p. injection. Sham operated control mice (sham MI/R) underwent the same surgical procedures except that the suture placed under the left coronary artery was not tied. At the end of the reperfusion period, the ligature around the coronary artery was retied, and 2% Evans Blue dye was injected into the left ventricular cavity. The heart was quickly excised, and the ischemic/reperfused cardiac tissue was isolated and processed according to the procedures described below.
2.2. Trx Activity Assay
Trx activity was determined by using the insulin disulfide reduction assay[14–16]. In brief, 40 μg of cellular protein extracts were pre-incubated at 37°C for 15 minutes with 2 μl activation buffer (100 mM HEPES, 2 mM EDTA, 1 mg/ml BSA, and 2 mM DTT) to reduce thioredoxin. After addition of 20 μl reaction buffer (100 mM HEPES, 2.0 mM EDTA, 0.2 mM NADPH, and 140 μM insulin), the reaction was started by addition of mammalian Trx reductase (1 μl, 15 mU, Sigma) or water to controls and samples incubated for 30 min at 37 °C. The reaction was terminated by adding 125 μl of stopping solution (0.2 m Tris-CL, 10 M guanidine-HCl and 1.7 mM 3-carboxy-4-nitrophenyl disulfide, DTNB) followed by absorption measurement at 412 nm.
2.3. Western Blotting Assay for Trx Expression
Cardiac tissue was homogenized with lysis buffer and centrifuged; the supernatant was used to assay Trx expression. Equal amounts of protein (80 μg protein/lane) were electrophoresed on a 14% SDS-polyacrylamide gel, and then electrophoretically transferred to a poly (vinylidene difluoride) membrane (Millipore). After blocking with 5% skim milk in TBS containing 0.05% Tween 20 at room temperature for 1 hour, the membrane was incubated with a monoclonal anti-murine Trx antibody (Redox Bioscience, Japan), and then with the HRP-linked anti-mouse lgG (Cell Signaling). The blot was developed with a Supersignal chemiluminescent detection kit (Pierce) and visualized with a Kodak Image Station 400. The blot densities were analyzed with Kodak 1D software (version 3.6)[4].
2.4. Quantitation of Tissue Nitrotyrosine Content
Cardiac nitrotyrosine, a footprint of in vivo RNS (e.g., ONOO−) formation, was determined by using an ELISA procedure that has been used by numerous investigators [17,18]. In brief, the free wall of the left ventricle was separated and homogenized in ice-cold PBS (pH 7.0). The homogenates were centrifuged, the supernatants collected, and protein concentrations were determined by the BCA method. A nitrated protein solution was prepared and diluted for use as a standard. These standard samples, along with tissue samples from hearts (100 μl/well), were applied to a sterile ELISA plate pre-coated with a monoclonal anti-nitrotyrosine antibody (Upstate, Charlottesville, VA). After 1 hour incubation at 37°C and three times washing with PBS, a rabbit polyclonal anti-nitrotyrosine antibody (1:200, Upstate) was added. Samples were incubated for another 1 hour at 37°C followed by three times washing with PBS. The secondary antibody (HRP-conjugated anti-rabbit IgG, Upstate, 1:500) was then added (1 hour at 37°C), and the peroxidase reaction product was generated by using O-phenylenediamine dihydrochloride (OPD) solution. The optical density was measured at 490 nm with a SpectraMax-Plus microplate spectrophotometer. The nitrotyrosine content in tissue samples was calculated by using standard curves generated from nitrated BSA containing known amounts of nitrotyrosine and expressed as nanomoles per gram protein.
To verify the specificity of the assay system, samples were pre-treated with sodium hydrosulfite as described by Ischiropoulos et al. [19] to convert 3-nitrotyrosine to aminotyrosine[20–22]. In brief, ischemic/reperfused heart tissue (n=3 rats) was homogenized and the supernatant pH was adjusted to 9.0 by addition of sodium hydroxide. Supernatant from the same heart was aliquated into 4 tubes. Half of the samples were treated with sodium hydrosulfide (final concentration: 0.5M) and another half were treated with vehicle (50 mM sodium bicarbonate pH 9.0). Ten minutes after incubation, nitrotyrosine content was determined as described above. Consistent with previously reported results using solid phase immunoradiochemical assay[19], pre-incubation with sodium hydrosulfite remarkably reduced nitrotyrosine readings determined with this well-accepted ELISA method (0.26±0.03 without sodium hydrosulfide pre-treatment vs. 0.03±0.01 pmole/mg protein with sodium hydrosulfide pre-treatment). This result demonstrated that the ELISA method used in the present study is reliable and specific for nitrotyrosine detection.
2.5. Total NO Assay
Nitric oxide has a very short half-life and is oxidized to form NO2 and NO3 in vivo. Measurement of NOx (NO + NO2 +NO3) concentration has been demonstrated to reflect total NO formation in vivo[23]. Cardiac tissue was homogenized with lysis buffer, and the supernatant was incubated with nitrate reductase for 2 hours to reduce NO3 to NO2. Samples containing NO2 were then injected into the reducing solution (120 mM potassium iodide in glacial acetic acid), and NO was detected by chemiluminescence with a nitric oxide analyzer (NOA 280I; Sievers, Boulder, CO).
2.6. Detection of Trx Nitration and Trx/ASK1 Interaction
Cardiac tissue was homogenized with lysis buffer. Immunoprecipitation and immunoblotting were performed by using a procedure described by Ischiropoulos and colleagues[24]. In brief, endogenous Trx-1 was immunoprecipitated with a monoclonal anti-murine Trx-1 antibody (Redox Bioscience, Japan). After sample separation, Trx-1 nitration was detected with a monoclonal antibody (Upstate, Charlottesville, VA) against nitrotyrosine, and the Trx-1/ASK1 interaction was determined by using a monoclonal antibody against ASK1 (Upstate). The blot was developed with Supersignal-Western reagent (Pierce) and visualized with a Kodak Image Station 400. The blot densities were analyzed with Kodak 1D software (version 3.6).
2.7. p38 MAP kinase activity assay
The p38 MAP kinase activity assay was performed by using a p38 MAPK assay kit (Cell Signaling Technology) with ATF-2 as a substrate by following the manufacture’s instruction [25]. In brief, heart tissue (20–25 mg) was homogenized in 0.5 ml ice-cold cell lysis buffer. The lysates were then sonicated on ice and centrifuged at 15,000×g for 10 min at 4°C. Immunoprecipitation was performed by adding 20 μl of resuspended immobilized monoclonal antibody against phospho-p38 MAP kinase (Thr180/Tyr182) to 100 μl cell lysate containing 250 μg proteins. The mixture was incubated with gentle rocking overnight at 4°C. After centrifuging at 10,000×g at 4°C for 2 min, the pellets were washed twice with lysis buffer and twice with kinase buffer. The kinase reactions were carried out in the presence of 200 μM ATP and 2 μg ATF-2 fusion protein at 30°C for 30 min. After incubation, the samples were separated by SDS–PAGE, and ATF-2 phosphorylation was measured by Western immunoblotting using a monoclonal antibody against phosphorylated ATF-2 followed by an enhanced chemiluminescent detection.
2.8. Measurement of Infarct Size
Twenty-four hours after reperfusion, mice were anesthetized, and the hearts were excised. Myocardial infarct size was determined by using 2,3,5-triphenyltetrazolium chloride (TTC) staining as described (n=8–12 per group)[25].
2.9. Determination of Ischemic/Reperfusion-Induced Cardiomyocyte Apoptosis
Myocardial apoptosis was analyzed by terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL) staining (n=8–10 hearts per group) and caspase-3 activity assay (n=10–12 per group) as described[26].
2.10. Statistical analysis
All values in the text and figures are presented as means ± SEM of n independent experiments. All data (except Western blot density) were subjected to ANOVA followed by Bonferoni correction for post-hoc t test. Western blot densities were analyzed with the Kruskal-Wallis test followed by Dunn’s post-hoc test. Probabilities of 0.05 or less were considered to be statistically significant.
3. Results
3.1. Aging enhanced myocardial ischemia/reperfusion injury
Consistent with previously published results by other investigators in aging rats[27], MI/R-induced cardiac injury was enhanced in aging mice as evidenced by increased apoptosis (TUNEL: 5.0±1.8 % vs. 8.8±0.9%, caspase 3 activity: 18.0±0.9 vs. 27.4±1.9 nmol/h/mg protein, P<0.01) and enlarged infarct size (38.5±2.7% vs. 52.0±1.8%, P<0.01).
3.2. Trx activity is reduced but its expression is increased in the aging heart
Emerging evidence indicates that Trx is a critical cell survival molecule that exerts strong cytoprotective effects by its anti-oxidant and anti-apoptotic properties. Having demonstrated that the aging heart is more susceptible to ischemia/reperfusion injury, we thought to test a hypothesis that Trx activity/expression might be reduced in the aging heart, thus tilting the death/survival balance toward cell death and promoting ischemia/reperfusion injury. Compared with young animals, cardiac Trx activity was decreased in the aging heart before MI/R (Figure 2A), and this difference was further amplified after MI/R (data not shown). Surprisingly, Trx expression was slightly increased, rather than decreased in aging hearts (Figure 2B). These results indicate that reduced Trx activity in the aging heart is caused by post-translational Trx modification, rather than reduced protein expression.
Figure 2.
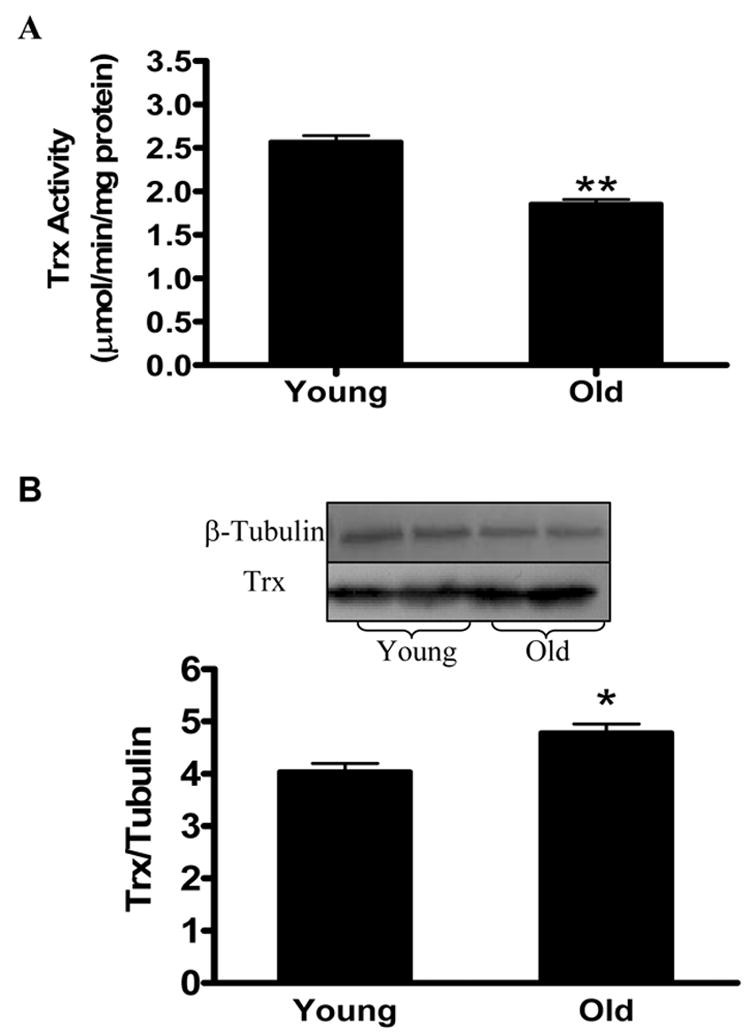
Trx activity is significantly reduced (A) but Trx protein expression is significantly increased (B) in the aging heart before it was subjected to MI/R. N=6–8 mice/group. *P<0.05, **P<0.01 vs. young animals.
3.3. Production of reactive nitrogen species and Trx nitration are increased, Trx/ASK1 interaction is inhibited, and p38 MAPK activity is increased in the aging heart
In a recent study, we have demonstrated that Trx is susceptible to nitrative modification by peroxynitrite, and its activity is irreversibly inhibited by this novel post-translational modification[28]. To determine whether reduced Trx activity observed in the aging heart is caused by increased nitrative stress and subsequent Trx nitration, two additional experiments were performed. In the first series of experiments, total NO production and cardiac nitrotyrosine, the foot-print of in vivo RNS (e.g., peroxynitrite) formation, were determined. As summarized in Figure 3, a greater than 1.5-fold increase in both NOx and nitrotyrosine contents were observed in cardiac tissues obtained from aging animals even before the hearts were subjected to ischemia/reperfusion. Moreover, ischemia/reperfusion-induced overproduction of peroxynitrite was further amplified in the aging heart (0.26±0.01 vs. 0.20±0.01 pmole/mg protein, P<0.05). In the second series of experiments, Trx nitration in young and aging hearts was detected by immunoprecipitation (anti-Trx) followed by immunoblotting (anti-nitrotyrosine). As illustrated in Figure 4A, Trx nitration was not detected in cardiac tissue obtained from young animals. In contrast, clear Trx nitration was detected in cardiac tissues from the aging heart before ischemia/reperfusion (Figure 4A), and this aging-induced Trx nitration was further intensified after ischemia/reperfusion (Figure 5).
Figure 3.

Aging enhances nitrative stress in the heart before it was subjected to MI/R as evidenced by increased total NOx production (A) and nitrotyrosine content (B). N=6 mice/group. *P<0.05 vs. young animals.
Figure 4.

Aging enhances Trx nitration (A), decreases Trx/ASK1 binding (B) and increases p38 MAPK activation (C). Inserts: representative Western blots; Bar graphs: Density analysis (5–7 animals/group). *P<0.05 vs. young animals.
Figure 5.

Effect of FP15 treatment on MI/R-induced Trx nitration (A) and Trx inactivation (B) in the aging heart. Inserts: representative Western blots; Bar graphs: Density analysis (N=6–8 mice/group). **P<0.01 vs. aging sham MI/R; ++P<0.01 vs. aging MI/R+V. V=vehicle.
Recent in vitro studies have demonstrated that binding/inhibition of Trx with ASK1 is the primary mechanism by which Trx exerts its anti-apoptotic effect[7,29]. Moreover, the ratio of ASK1/Trx-ASK1 has been shown to be significantly increased in aged mouse livers, and this correlates with the increased basal activity of the p38 MAPK pathway[30]. To determine whether Trx nitration may alter the Trx/ASK1 interaction thus activating the downstream pro-apoptotic kinases, two additional observations were made. As illustrated in Figure 4B, Trx is physically associated with ASK1 (anti-Trx-1 immunoprecipitation and anti-ASK1 immunoblotting) in cardiac tissues isolated from young animals, and this protein/protein interaction was significantly decreased in aging animals. Consequently, activity of p38 MAPK, a pro-apoptotic downstream molecule for ASK1, was significantly enhanced in the aging heart compared with the young heart (Figure 4C).
3.4. Treatment with a peroxynitrite decomposition catalyst blocked Trx nitration and protected the aging heart from ischemia/reperfusion injury
Considerable evidence exists that peroxynitrite is the most pathologically relevant nitrating agent in vivo. To obtain more evidence to support our hypothesis that increased Trx nitration is causatively related to increased susceptibility to myocardial ischemia/reperfusion injury in the aging heart, aging mice were treated with FP-15 (5 mg/kg, 10 minutes before reperfusion), a novel peroxynitrite decomposition catalyst that has recently been shown to attenuate diabetic neuropathy[31,32]. Treatment with FP-15 shortly before reperfusion reduced Trx nitration (Figure 5A), preserved Trx activity (Figure 5B), restored Trx-ASK1 interaction (Figure 6A), reduced P38 MAPK activity (Figure 6B), attenuated caspase 3 activation (Figure 7A) and reduced infarct size (Figure 7B) in aging animals (p<0.01).
Figure 6.
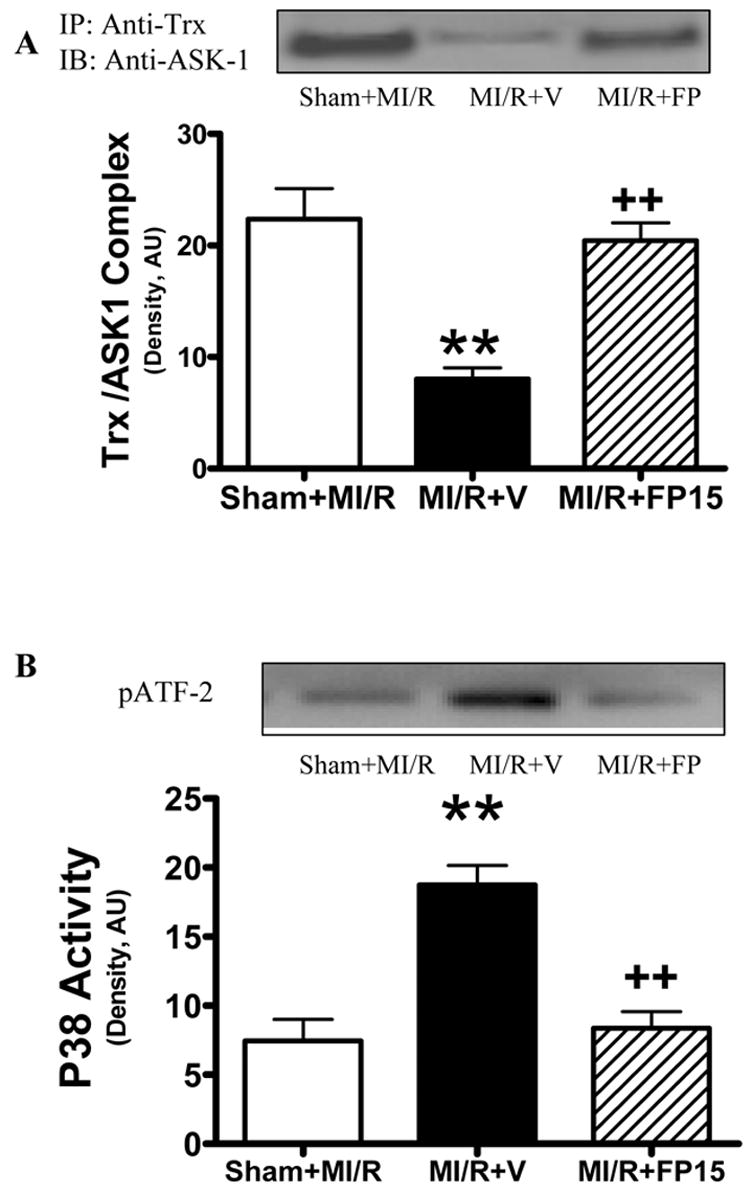
Effect of FP15 treatment on MI/R-induced Trx/ASK1 dissociation (A) and p38MAPK activation (B) in the aging heart. Inserts: Representative Western blots; Bar graphs: Density analysis. N=6–8 mice/group. **P<0.01 vs. aging sham MI/R; ++P<0.01 vs. aging MI/R+V.
Figure 7.

Effect of FP15 treatment on MI/R-induced cardiomyocyte apoptosis (A) and myocardial infarct size (B) in the aging heart (n=10–12/group). **P<0.01 vs. aging sham MI/R; ++P<0.01 vs. aging MI/R+V.
4. Discussion
Myocardial infarction represents a major cause of morbidity and mortality in the elderly. Aging is associated with an increase in myocardial susceptibility to ischemia and a decrease in post-ischemic recovery of cardiac function[33]. Understanding the potential mechanisms of aging could lead to improved medical treatment of this increasing patient population. Our present study provided the first evidence that the activity of Trx, a critical anti-oxidant and anti-apoptotic molecule, is significantly reduced in aging hearts even before they are subjected to myocardial ischemia/reperfusion. Moreover, we have demonstrated that it is the post-translational nitrative modification of Trx, not its expression, which is responsible for this reduced Trx activity in the aging heart. In addition, we have demonstrated that increased nitrative modification of Trx results in a decreased Trx-ASK1 binding in the aging heart before myocardial ischemia/reperfusion. Finally, we have demonstrated that treatment with a novel peroxynitrite decomposition catalyst shortly before reperfusion blocked nitrative Trx inactivation, attenuated ASK1 activation and reduced post-ischemic myocardial apoptosis in the aging heart. Taken together, these results provided strong evidence that nitrative Trx inactivation plays a causative role in increased post-ischemic cardiomyocyte apoptosis in the aging heart.
Trx, a small, ubiquitous protein, is one of the most important regulators of reduction–oxidation (redox) balance and, thus, redox controlled cell functions[34]. Although Trx was discovered 40 years ago in bacteria, the number and diversity of processes that Trx influences in human cells has only recently been appreciated. Processes influenced by Trx include the control of cellular redox balance, the promotion of cell growth, the inhibition of apoptosis and the modulation of inflammation. Not surprisingly, the role of Trx has been discovered in a wide range of human diseases and conditions, including cancer, viral disease, and ischemia/reperfusion injury[35]. Emerging evidence suggests that Trx plays a critical role in promoting cell proliferation/survival and reducing cell death. Trx and Trx reductase (TrxR) are upregulated in cancer tissues, and molecules that inhibit Trx or TrxR promote apoptosis and reduce cancer development[36]. In contrast, Trx activity is reduced in diseased tissues where pathologic apoptosis is increased[37]. Moreover, a very recent study has demonstrated that aging is associated with a TrxR2 reduction in skeletal muscle which thus enhances the susceptibility to free radical-induced apoptosis[38]. Trx is involved not only in cytoprotective functions against oxidative stress, but also in the regulation of cellular proliferation and the aging process[39]. Cumulatively, these data strongly suggest that altered Trx expression and/or activity play critical pathogenic roles in aging, and that Trx may be a novel therapeutic target for age-associated diseases, such as cardiovascular disease, where a physiologic balance between cell death and cell proliferation is disturbed.
Recent studies have demonstrated that besides upregulation or downregulation of Trx expression at the gene level, Trx activity is regulated by post-translational modification. Three forms of post-translational modification of Trx have been previously identified. These include oxidation, glutathionylation and S-nitrosylation. Interestingly, all three forms of modification occur at cysteine residues but affect Trx function differently. Oxidation of the thiol groups of Cys-32 and -35 forms a disulfide bond which results in Trx inactivation. However, previous studies have demonstrated that administration of oxidized Trx-1 exerts significant anti-oxidant and cytoprotective effects unless intracellular Trx reductase is inhibited, indicating that oxidative Trx inhibition is reversible and this form of post-translational modification may not be the major mechanism responsible for Trx inactivation in vivo[40,41]. Glutathionylation occurs at Cys-73, and this post-translational modification significantly inhibits Trx activity[42]. However, whether Trx glutathionylation may occur in vivo in diseased tissues remains completely unknown and the role of this form of post-translational modification in regulating Trx function in vivo remains to be determined. S-nitrosylation has been reported to occur at either Cys-69[4,43] or Cys-73[44]. In contrast to oxidation and glutathionylation, S-nitrosylation increases Trx activity and further enhances its anti-apoptotic effect[4,43,44].
In a recent study, we have demonstrated that besides three previously reported post-translational Trx modifications which all occur at the cysteine residue, Trx can also be modified at the tyrosine residue (protein nitration) in a peroxynitrite-dependent fashion[28]. More interestingly, in contrast to the reversible (by Trx reductase) oxidative Trx inactivation, nitrative modification of Trx results in an irreversible inactivation [28]. Therefore, nitric oxide and its secondary reaction products, particularly peroxynitrite, exert opposite effect on Trx activity. Specifically, nitric oxide itself induces Trx S-nitrosylation and enhances its activity[4,43,44]. In contrast, peroxynitrite results in Trx nitration and causes an irreversible inactivation[28]. Our present study demonstrated for the first time that this nitrative inhibitory post-translational modification of Trx is increased in the aging heart. ASK1 is a member of mitogen activated protein kinases (MAPK’s) and functions as an upstream activator of JNK and p38 MAPK. Under physiologic conditions, ASK1 activity is inhibited by several cellular factors, including Trx, glutaredoxin, and phosphoserine-binding protein 14-3-3[45]. Previous studies have demonstrated that many cellular stresses and apoptotic stimuli activate mitochondrial-dependent apoptotic pathways by facilitating dissociation of ASK1 with its inhibitory proteins[46,47]. Our current study demonstrated that Trx is physically associated with ASK1 in cardiac tissues from young animals. However, Trx/ASK1 binding was reduced in cardiac tissue from aging animals. Therefore, it is likely that increased Trx nitration in aging hearts results in disassociation of Trx from ASK1, thus increasing post-ischemic myocardial apoptosis by increasing p38 MAPK activity. In this connection, we have provided direct evidence in the current study that treatment with a peroxynitrite decomposition catalyst in aging animals significantly reduced Trx nitration, partially restored Trx/ASK1 binding, reduced post-ischemic myocardial apoptosis, and reduced myocardial infarct size in aging hearts. It is worth noticing that treatment with FP15 also reduced myocardial infarct size in young animals (from 37.7±2.8 to 24.3±1.9%, a 36% reduction). However, this treatment reduced infarct size to a much greater extent in the aging heart (from 52±2.2 to 27.9±1.9%, a 46% reduction). These results strongly suggest that increased nitrative Trx modification is a novel pathologic pathway by which aging increases the susceptibility to ischemic/reperfusion injury. In addition, it should be noted that treatment with FP15 failed to completely block Trx nitration (Figure 5A). Two possible explanations exist. Decomposition of peroxynitrite by FP15 leads to formation of nitrite [48] and high concentrations of nitrite has been shown to cause protein nitration in vitro. However, several previous studies [31,49] have demonstrated that treatment with FP15 markedly reduces protein nitration in diseased tissues, indicating that nitrite itself is not a major nitrating agent in vivo. Another more likely explanation is that besides peroxynitrite, peroxidases, particularly myeloperoxidase, may catalyze protein nitration in the presence of nitrite and hydrogen peroxide.
In conclusion, our results demonstrated that Trx activity is decreased in the aging heart by post-translational nitrative modification. Blocking ONOO− production and inhibiting Trx inactivation significantly protected the aging heart from MI/R injury. These results suggest that therapeutic interventions that preserve Trx activity in the aging patient may improve outcome after MI.
Figure 1.

MI/R injury is significantly increased in aging hearts. A: Myocardial infarct size; B: Apoptotic index; C: Cardiac caspase 3 activity. N=8 mice/group. **P<0.01 vs. Young+MI/R.
Acknowledgments
This research was supported by the following grants: NIH 2R01HL-63828, the American Diabetes Association 7-05-RA-83, the Commonwealth of Pennsylvania-Department of Health (X.M.), the American Diabetes Association 7-06-JF59 (L.T.), and the American Heart Association Grant-in-Aid 0555447U (to T.C.).
List of Abbreviations
- ASK-1
Apoptosis-regulating kinase-1
- JNK
c-Jun N-terminal kinase
- MAPK
Mitogen activated protein kinase
- MI/R
Myocardial ischemia/reperfusion
- NO
Nitric oxide
- ONOO−
Peroxynitrite
- RNS
Reactive nitrogen species
- ROS
Reactive oxygen species
- Trx
Thioredoxin
- TTC
2,3,5-triphenyltetrazolium chloride
- TUNEL
Terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lakatta EG, Levy D. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part I: Aging Arteries: A “Set Up” for vascular disease. Circulation. 2003;107:139–146. doi: 10.1161/01.cir.0000048892.83521.58. [DOI] [PubMed] [Google Scholar]
- 2.Pollack M, Phaneuf S, Dirks A, Leeuwenburgh C. The role of apoptosis in the normal aging brain, skeletal muscle, and heart. Ann N Y AcadSci. 2002;959:93–107. doi: 10.1111/j.1749-6632.2002.tb02086.x. [DOI] [PubMed] [Google Scholar]
- 3.Kajstura J, Cheng W, Sarangarajan R, Li P, Li B, Nitahara JA, Chapnick S, Reiss K, Olivetti G, Anversa P. Necrotic and apoptotic myocyte cell death in the aging heart of Fischer 344 rats. Am J Physiol. 1996;271:H1215–H1228. doi: 10.1152/ajpheart.1996.271.3.H1215. [DOI] [PubMed] [Google Scholar]
- 4.Tao L, Gao E, Bryan NS, Qu Y, Liu HR, Hu A, Christopher TA, Lopez BL, Yodoi J, Koch WJ, Feelisch M, Ma XL. Cardioprotective effects of thioredoxin in myocardial ischemia and reperfusion: Role of S-nitrosation. Proc Natl Acad Sci U S A. 2004;101:11471–11476. doi: 10.1073/pnas.0402941101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshida T, Oka Si, Masutani H, Nakamura H, Yodoi J. The Role of Thioredoxin in the Aging Process: Involvement of Oxidative Stress. Antioxidants Redox Signaling. 2003;5:563–570. doi: 10.1089/152308603770310211. [DOI] [PubMed] [Google Scholar]
- 6.Lincoln DT, li Emadi EM, Tonissen KF, Clarke FM. The thioredoxin-thioredoxin reductase system: over-expression in human cancer. Anticancer Res. 2003;23:2425–2433. [PubMed] [Google Scholar]
- 7.Liu Y, Min W. Thioredoxin promotes ASK1 ubiquitination and degradation to inhibit ASK1-mediated apoptosis in a redox activity-independent manner. Circ Res. 2002;90:1259–1266. doi: 10.1161/01.res.0000022160.64355.62. [DOI] [PubMed] [Google Scholar]
- 8.Bian JS, Yong QC, Pan TT, Feng ZN, Ali MY, Zhou S, Moore PK. Role of Hydrogen Sulfide in the Cardioprotection Caused by Ischemic Preconditioning in the Rat Heart and Cardiac Myocytes. J Pharmacol Exp Ther. 2006;316:670–678. doi: 10.1124/jpet.105.092023. [DOI] [PubMed] [Google Scholar]
- 9.Harman D. Aging: phenomena and theories. Ann NY Acad Sci. 1998;854:1–7. doi: 10.1111/j.1749-6632.1998.tb09886.x. [DOI] [PubMed] [Google Scholar]
- 10.Sastre J, Pallardo FV, Vina J. Mitochondrial oxidative stress plays a key role in aging and apoptosis. IUBMB Life. 2000;49:427–435. doi: 10.1080/152165400410281. [DOI] [PubMed] [Google Scholar]
- 11.Schoneich C. Reactive oxygen species and biological aging: a mechanistic approach. Exp Gerontol. 1999;34:19–34. doi: 10.1016/s0531-5565(98)00066-7. [DOI] [PubMed] [Google Scholar]
- 12.Ischiropoulos H, Beckman JS. Oxidative stress and nitration in neurodegeneration: Cause, effect, or association? J Clin Invest. 2003;111:163–169. doi: 10.1172/JCI17638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brookes P, Darley-Usmar VM. Hypothesis: the mitochondrial NO(*) signaling pathway, and the transduction of nitrosative to oxidative cell signals: an alternative function for cytochrome C oxidase. Free Radic Biol Med. 2002;32:370–374. doi: 10.1016/s0891-5849(01)00805-x. [DOI] [PubMed] [Google Scholar]
- 14.Yamamoto M, Yang G, Hong C, Liu J, Holle E, Yu X, Wagner T, Vatner SF, Sadoshima J. Inhibition of endogenous thioredoxin in the heart increases oxidative stress and cardiac hypertrophy. J Clin Invest. 2003;112:1395–1406. doi: 10.1172/JCI17700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holmgren A, Bjornstedt M. Thioredoxin and thioredoxin reductase. Methods Enzymol. 1995;252:199–208. doi: 10.1016/0076-6879(95)52023-6. [DOI] [PubMed] [Google Scholar]
- 16.Das KC, Guo XL, White CW. Induction of thioredoxin and thioredoxin reductase gene expression in lungs of newborn primates by oxygen. Am J Physiol. 1999;276:L530–L539. doi: 10.1152/ajplung.1999.276.3.L530. [DOI] [PubMed] [Google Scholar]
- 17.Thomson L, Christie J, Vadseth C, Lanken PN, Fu X, Hazen SL, Ischiropoulos H. Identification of Immunoglobulins that Recognize 3-Nitrotyrosine in Patients with Acute Lung Injury after Major Trauma. Am J Respir Cell Mol Biol. 2007;36:152–157. doi: 10.1165/rcmb.2006-0288SM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Naviliat M, Gualco G, Cayota A, Radi R. Protein 3-nitrotyrosine formation during Trypanosoma cruzi infection in mice. Braz J Med Biol Res. 2005;38:1825–1834. doi: 10.1590/s0100-879x2005001200011. [DOI] [PubMed] [Google Scholar]
- 19.Ischiropoulos H, Beers MF, Ohnishi ST, Fisher D, Garner SE, Thom SR. Nitric oxide production and perivascular tyrosine nitration in brain after carbon monoxide poisoning in the rat. J Clin Invest. 1996;97:2260–2267. doi: 10.1172/JCI118667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shigenaga MK, Lee HH, Blount BC, Christen S, Shigeno ET, Yip H, Ames BN. Inflammation and NO(X)-induced nitration: assay for 3-nitrotyrosine by HPLC with electrochemical detection. Proc Natl Acad Sci U S A. 1997;94:3211–3216. doi: 10.1073/pnas.94.7.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ikeda K, Yukihiro HB, Iwai H, Matsumoto T, Mineki R, Taka H, Takamori K, Ogawa H, Yamakura F. Detection of 6-nitrotryptophan in proteins by Western blot analysis and its application for peroxynitrite-treated PC12 cells. Nitric Oxide. 2007;16:18–28. doi: 10.1016/j.niox.2006.04.263. [DOI] [PubMed] [Google Scholar]
- 22.Viera L, Ye YZ, Estevez AG, Beckman JS. Immunohistochemical methods to detect nitrotyrosine. Methods Enzymol. 1999;301:373–381. doi: 10.1016/s0076-6879(99)01101-5. [DOI] [PubMed] [Google Scholar]
- 23.Ma XL, Gao F, Nelson AH, Lopez BL, Christopher TA, Yue TL, Barone FC. Oxidative inactivation of nitric oxide and endothelial dysfunction in stroke-prone spontaneous hypertensive rats. J Pharmacol Exp Ther. 2001;298:879–885. [PubMed] [Google Scholar]
- 24.Vadseth C, Souza JM, Thomson L, Seagraves A, Nagaswami C, Scheiner T, Torbet J, Vilaire G, Bennett JS, Murciano JC, Muzykantov V, Penn MS, Hazen SL, Weisel JW, Ischiropoulos H. Pro-thrombotic state induced by post-translational modification of fibrinogen by reactive nitrogen species. J Biol Chem. 2004;279:8820–8826. doi: 10.1074/jbc.M306101200. [DOI] [PubMed] [Google Scholar]
- 25.Gao F, Yue TL, Shi DW, Christopher TA, Lopez BL, Ohlstein EH, Barone FC, Ma XL. p38 MAPK inhibition reduces myocardial reperfusion injury via inhibition of endothelial adhesion molecule expression and blockade of PMN accumulation. Cardiovasc Res. 2002;53:414–422. doi: 10.1016/s0008-6363(01)00488-6. [DOI] [PubMed] [Google Scholar]
- 26.Gao F, Gong B, Christopher TA, Lopez BL, Karasawa A, Ma XL. Anti-apoptotic effect of benidipine, a long-lasting vasodilating calcium antagonist, in ischaemic/reperfused myocardial cells. Br J Pharmacol. 2001;132:869–878. doi: 10.1038/sj.bjp.0703881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu P, Xu B, Cavalieri TA, Hock CE. Age-related difference in myocardial function and inflammation in a rat model of myocardial ischemia-reperfusion. Cardiovasc Res. 2002;56:443–453. doi: 10.1016/s0008-6363(02)00603-x. [DOI] [PubMed] [Google Scholar]
- 28.Tao L, Jiao X, Gao E, Lau WB, Yuan Y, Lopez B, Christopher T, Ramachandrarao SP, Williams W, Southan G, Sharma K, Koch W, Ma XL. Nitrative inactivation of thioredoxin-1 and its role in postischemic myocardial apoptosis. Circulation. 2006;114:1395–1402. doi: 10.1161/CIRCULATIONAHA.106.625061. [DOI] [PubMed] [Google Scholar]
- 29.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsieh CC, Papaconstantinou J. Thioredoxin-ASK1 complex levels regulate ROS-mediated p38 MAPK pathway activity in livers of aged and long-lived Snell dwarf mice. FASEB J. 2006;20:259–268. doi: 10.1096/fj.05-4376com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Obrosova IG, Mabley JG, Zsengeller Z, Charniauskaya T, Abatan OI, Groves JT, Szabo C. Role for nitrosative stress in diabetic neuropathy: evidence from studies with a peroxynitrite decomposition catalyst. FASEB J. 2005;19:401–403. doi: 10.1096/fj.04-1913fje. [DOI] [PubMed] [Google Scholar]
- 32.Pieper GM, Nilakantan V, Chen M, Zhou J, Khanna AK, Henderson JD, Jr, Johnson CP, Roza AM, Szabo C. Protective mechanisms of a metalloporphyrinic peroxynitrite decomposition catalyst, WW85, in rat cardiac transplants. J Pharmacol Exp Ther. 2005;314:53–60. doi: 10.1124/jpet.105.083493. [DOI] [PubMed] [Google Scholar]
- 33.Sumeray MS, Yellon DM. Characterisation and validation of a murine model of global ischaemia-reperfusion injury. Mol Cell Biochem. 1998;186:61–68. [PubMed] [Google Scholar]
- 34.Nakamura H. Thioredoxin as a Key Molecule in Redox Signaling. Antioxidants Redox Signaling. 2004;6:15–17. doi: 10.1089/152308604771978309. [DOI] [PubMed] [Google Scholar]
- 35.Das DK. Thioredoxin Regulation of Ischemic Preconditioning. Antioxidants Redox Signaling. 2004;6:405–412. doi: 10.1089/152308604322899477. [DOI] [PubMed] [Google Scholar]
- 36.Powis G, Mustacich D, Coon A. The role of the redox protein thioredoxin in cell growth and cancer. Free Radic Biol Med. 2000;29:312–322. doi: 10.1016/s0891-5849(00)00313-0. [DOI] [PubMed] [Google Scholar]
- 37.Lovell MA, Xie C, Gabbita SP, Markesbery WR. Decreased thioredoxin and increased thioredoxin reductase levels in Alzheimer’s disease brain. Free Radic Biol Med. 2000;28:418–427. doi: 10.1016/s0891-5849(99)00258-0. [DOI] [PubMed] [Google Scholar]
- 38.Rohrbach S, Gruenler S, Teschner M, Holtz J. The thioredoxin system in aging muscle: key role of mitochondrial thioredoxin reductase in the protective effects of caloric restriction? Am J Physiol Regul Integr Comp Physiol. 2006;291:R927–R935. doi: 10.1152/ajpregu.00890.2005. [DOI] [PubMed] [Google Scholar]
- 39.Shioji K, Nakamura H, Masutani H, Yodoi J. Redox Regulation by Thioredoxin in Cardiovascular Diseases. Antioxidants Redox Signaling. 2003;5:795–802. doi: 10.1089/152308603770380106. [DOI] [PubMed] [Google Scholar]
- 40.Andoh T, Chiueh CC, Chock PB. Cyclic GMP-dependent protein kinase regulates the expression of thioredoxin and thioredoxin peroxidase-1 during hormesis in response to oxidative stress-induced apoptosis. J Biol Chem. 2003;278:885–890. doi: 10.1074/jbc.M209914200. [DOI] [PubMed] [Google Scholar]
- 41.Andoh T, Chock PB, Chiueh CC. The roles of thioredoxin in protection against oxidative stress-induced apoptosis in SH-SY5Y cells. J Biol Chem. 2002;277:9655–9660. doi: 10.1074/jbc.M110701200. [DOI] [PubMed] [Google Scholar]
- 42.Casagrande S, Bonetto V, Fratelli M, Gianazza E, Eberini I, Massignan T, Salmona M, Chang G, Holmgren A, Ghezzi P. Glutathionylation of human thioredoxin: a possible crosstalk between the glutathione and thioredoxin systems. Proc Natl Acad Sci U S A. 2002;99:9745–9749. doi: 10.1073/pnas.152168599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haendeler J, Hoffmann J, Zeiher AM, Dimmeler S. Antioxidant effects of Statins via S-nitrosylation and activation of thioredoxin in endothelial cells. A novel vasculoprotective function of Statins. Circulation. 2004;110:856–861. doi: 10.1161/01.CIR.0000138743.09012.93. [DOI] [PubMed] [Google Scholar]
- 44.Mitchell DA, Marletta MA. Thioredoxin catalyzes the S-nitrosation of the caspase-3 active site cysteine. Nat Chem Biol. 2005;1:154–158. doi: 10.1038/nchembio720. [DOI] [PubMed] [Google Scholar]
- 45.Bishopric NH, Webster KA. Preventing apoptosis with thioredoxin: ASK me how. Circ Res. 2002;90:1237–1239. doi: 10.1161/01.res.0000025101.04065.83. [DOI] [PubMed] [Google Scholar]
- 46.Zhang R, He X, Liu W, Lu M, Hsieh JT, Min W. AIP1 mediates TNF-{alpha}-induced ASK1 activation by facilitating dissociation of ASK1 from its inhibitor 14-3-3. J Clin Invest. 2003;111:1933–1943. doi: 10.1172/JCI17790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamawaki H, Berk BC. Thioredoxin: a multifunctional antioxidant enzyme in kidney, heart and vessels. Curr Opin Nephrol Hypertens. 2005;14:149–153. doi: 10.1097/00041552-200503000-00010. [DOI] [PubMed] [Google Scholar]
- 48.Szabo C, Mabley JG, Moeller SM, Shimanovich R, Pacher P, Virag L, Soriano FG, Van Duzer JH, Williams W, Salzman AL, Groves JT. Part I: pathogenetic role of peroxynitrite in the development of diabetes and diabetic vascular complications: studies with FP15, a novel potent peroxynitrite decomposition catalyst. Mol Med. 2002;8:571–580. [PMC free article] [PubMed] [Google Scholar]
- 49.Pacher P, Liaudet L, Bai P, Mabley JG, Kaminski PM, Virag L, Deb A, Szabo E, Ungvari Z, Wolin MS, Groves JT, Szabo C. Potent metalloporphyrin peroxynitrite decomposition catalyst protects against the development of doxorubicin-induced cardiac dysfunction. Circulation. 2003;107:896–904. doi: 10.1161/01.cir.0000048192.52098.dd. [DOI] [PubMed] [Google Scholar]