

Abstract
A large body of research has established the importance of costimulatory signals and proliferation for the generation of productive T cell immune responses. While costimulation and cell cycle progression are each individually necessary for CD4+ effector T cell differentiation, it has become clear that neither of these processes alone are sufficient to avoid anergy. This review outlines the links between T cell differentiation, tolerance, and the cell cycle, and highlights recent work that has implicated cyclin-dependent kinases as important regulators and potential targets for modulation of T cell immunity and tolerance.
1. Introduction
T lymphocytes form the basis of immunity to foreign pathogens, but also precipitate autoimmune disease and mediate the rejection of organ transplants. For instance, current estimates suggest that as many as 1 in 20 peripheral T cells are alloreactive, and these cells may expand 20- to 50-fold during an alloimmune response in vivo [1]. The proliferation of T lymphocytes from a few clones into a population of antigen-specific effector cells represents a critical and highly-regulated phase of a productive immune response, and the majority of immunosuppressive drugs currently used to ameliorate transplant rejection or autoimmune pathology in experimental models and in the clinic result in inhibition of T cell proliferation.
Growth factors such as IL-2 drive T cell division, clonal expansion, differentiation and oppose tolerance, but the basis for the association between these processes is not known. Does growth factor signaling directly oppose tolerance induction independently of its effects on cell cycle progression, or is tolerance a state that must be ‘escaped’ by cell division? Likewise, do genes induced by growth factors instruct naïve T cells to develop effector function, or does cell cycle progression influence the expression of genes associated with T helper differentiation by a more indirect mechanism? Specifically how mitogenic signals contribute to the avoidance of tolerance and development of effector and memory T cells has been unclear, and a further understanding of the nature of this relationship may allow T cell responses to be modulated with greater specificity and efficacy.
Recent studies have implicated cyclin-dependent kinases (CDK) as major regulators of T cell immunity and tolerance (reviewed in [2]), and point to these molecules as a potential link between the cell cycle and T cell function. These studies have added to our understanding of T cell anergy at the molecular level, but in addition, they have implicated CDK as novel potential targets for therapy in autoimmune disease and organ transplantation. At least four CDK inhibitory drugs are currently at various stages of clinical trials in the treatment of cancer [3], but the activity or efficacy of these drugs as immunosuppressive agents in the context of autoimmunity or transplantation has not been explored.
2. T cell differentiation and anergy are linked to the cell cycle
2.1. Association between the cell cycle and T cell differentiation
Naïve T cells are unable to efficiently produce cytokines such as IFNγ and IL-4, but gain this function several days following their initial activation [4]. A significant step forward in our understanding of T cell differentiation was achieved when several studies demonstrated that the delay in this gain of effector function is not time-dependent, but rather cell division-dependent. Thus, in populations of differentiating Th1 or Th2 cells, only those cells that have undergone several rounds of cell division are able to express the cytokine genes characteristic of these polarized responses [5–7]. This link between cell cycle progression and T cell effector function has been observed in a myriad of studies in a multitude of immunological models, and represents an important clue to the nature of T cell differentiation.
2.2. Association between the cell cycle and T cell clonal anergy
T cells activated in the absence of inflammatory signals fail to produce IL-2 [8], and are rendered hypo-responsive to further stimuli [9,10]. This state, called T cell anergy (reviewed in [11]), is an important mechanism of peripheral tolerance that is associated with altered recruitment of TCR-associated tyrosine kinases, impaired PLCγ-1 activation, defective activation of the ERK and JNK MAP kinase cascades, and results in defective assembly of active transcription factor complexes at the IL2 promoter [12–19]. In addition, anergic T cells suffer from active, dominant repression of IL2 gene transcription [20–22] that may involve epigenetic mechanisms [23–25]. Under physiologic conditions, anergy is induced when T cells receive antigenic stimulation in the absence of CD28 costimulation, and appears to involve signaling through Ca2+/calcineurin to the transcription factor NFAT [26] (Fig. 1 A). These signals induce the expression and/or activity of anergic, negative regulatory factors, which include E3 ubiquitin ligases and transcriptional repressors (reviewed in [27,28]).
Fig. 1.
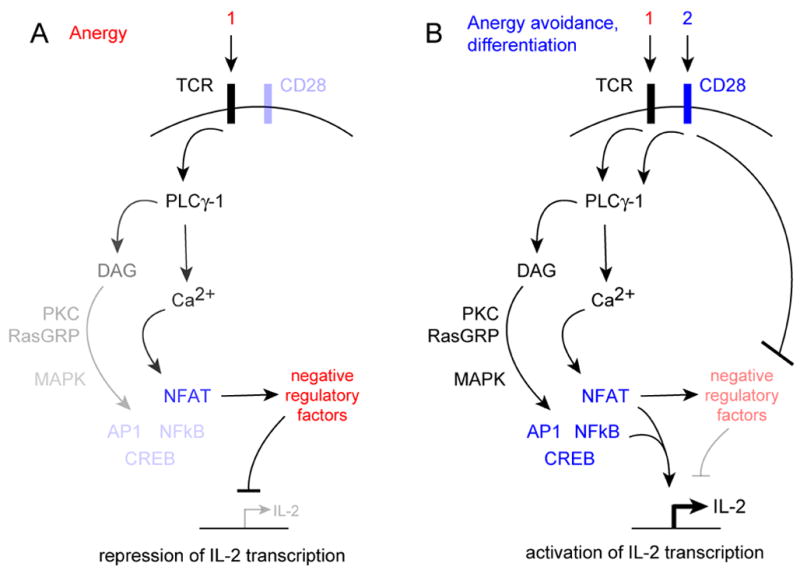
Current molecular view of T cell anergy (A) and anergy avoidance (B).
But if anergy is an active and integral consequence of antigen receptor signaling, then how do T cells avoid this fate to induce immunity? The predominant view is that signals from the CD28 costimulatory receptor oppose the expression or activity of negative regulatory factors that are induced by antigen receptor signals (Fig. 1 B), but a molecular basis for this hypothesis has not been well-defined. One idea is that AP-1 activated by CD28 partners with NFAT, diverting NFAT from a program of anergy-associated genes to genes associated with T cell differentiation [26]. However, CD28 costimulation also leads to synthesis of IL-2 and its receptor, and results in cell cycle progression and clonal expansion (Fig. 2). So, an alternative hypothesis is that costimulation promotes anergy avoidance indirectly by driving proliferation, and that processes associated with cell cycle progression then directly oppose the negative regulatory factors induced by the TCR (Fig. 2). The fact that IL-2 is required for the avoidance of anergy [29], and growth factor signals can replace CD28 costimulation in this process [30,31], argues for this scenario.
Fig. 2.
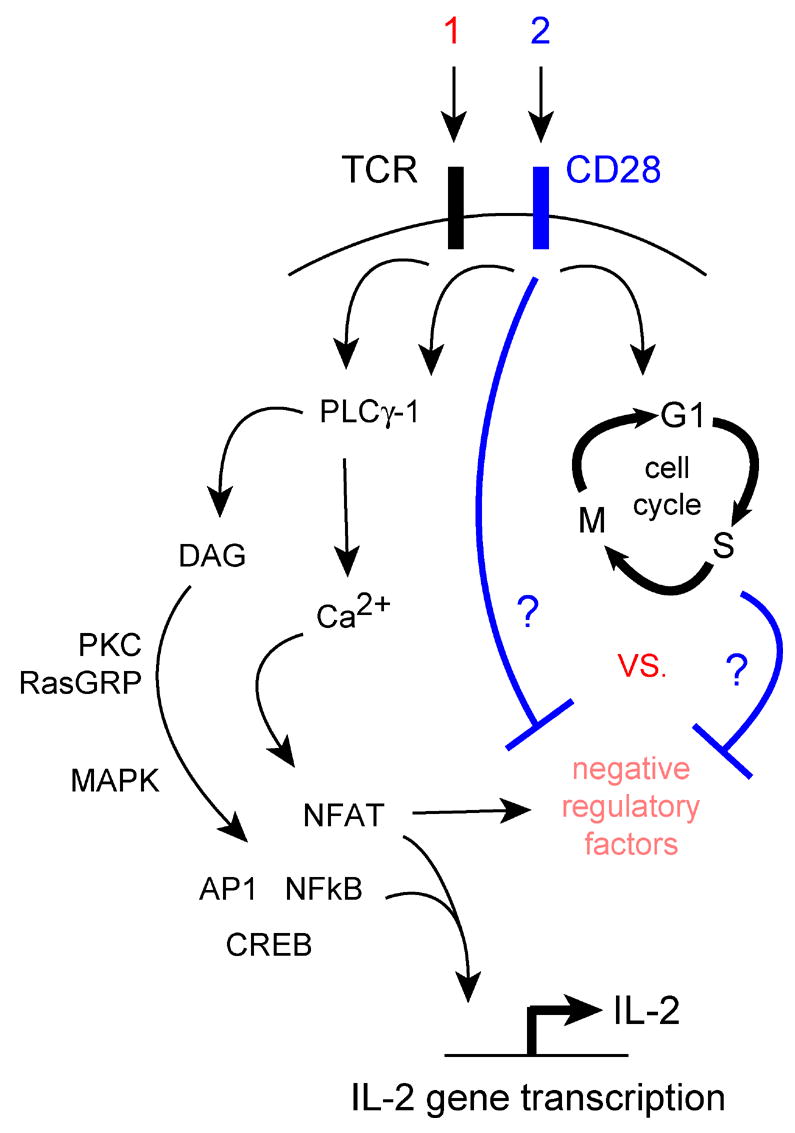
Cell cycle-dependent vs. cell cycle-independent anergy avoidance mediated by CD28 costimulation.
Primary T cells that receive optimal antigen and costimulatory receptor stimulation proceed synchronously through early activation and G1 phase progression, but go on to exhibit marked heterogeneity and asynchrony in mitotic behavior [32]. Those T cells that progress through multiple (i.e., 5–8) cell divisions during the primary response exhibit strong IL-2 production, IFNγ production, and proliferation upon restimulation [5–7,18,33]. Conversely, those cells that did not divide during the primary response fail to produce IL-2 and exhibit growth arrest upon restimulation, despite the fact that these cells were primed in the presence of full TCR and CD28 costimulation [18,33]. These studies have provided support for the idea that CD28 costimulation is not sufficient, while cell cycle progression is necessary, for anergy avoidance. Additional support for the importance of cell cycle progression has come from studies demonstrating that pharmacological inhibition of G1 progression with butyrate [34,35] or IL-2R signaling with rapamycin [36,37] results in induction of anergy in CD4+ T cells despite the presence of full TCR and CD28 costimulation. Cell cycle arrest within the G1 phase appears to be important for anergy induction, because a pharmacologic block within S phase using hydroxyurea failed to induce anergy in T cell clones stimulated through the TCR and CD28 [36]. This implies that biochemical events occurring before the G1 to S phase transition are critical for anergy avoidance.
3. Cyclin-dependent kinases as a molecular link between the cell cycle and T cell function
3.1. Anergy is accompanied by dysregulated mitogenic signaling
What aspect of cell cycle progression promote anergy avoidance? Two crucial events that occur before the G1 to S phase transition are the activation of the D-type cyclin-dependent kinases CDK4 and 6, and the E-type cyclin-dependent kinase CDK2. Anergic T cells can induce cyclin D2 and CDK6 expression [38,39], but lack cyclin E expression and CDK1 and 2 activity [39]. One consequence of CDK activity is the induction and activation of the E2F family of transcription factors [40]. Anergic T cells exhibit impaired expression and nuclear localization of the pro-mitogenic family members E2F1 and E2F3, and express moderately elevated levels of E2F4, a family member that inhibits the transcriptional activity of the pro-mitogenic members (our unpublished observations). While all the E2F DNA binding activity in TCR/CD28-activated effector cells exists as free E2F1–4 complexes that are capable of transcriptional activation, at least one-third of the E2F DNA binding activity in anergic T cells is assoicated with the retinoblastoma (Rb) tumor-suppressor protein in a complex that represses gene expression and helps to keep cells in a quiescent state (our unpublished observations). Another important consequence of CDK activity is the phosphorylation and targeted degradation p27kip1, a critical inhibitor of CDK activity [41]. In several models, anergic cells exhibit elevated expression of p27kip1, and are unable to downregulate this protein in response to restimulation [18,33,39,42]. These studies together suggest that CDK activity is impaired in anergic T cells, and implicate cyclin-dependent kinases as factors that may promote anergy avoidance in T cells.
3.1. CDK1/2 activity opposes anergy induction
The elevated expression of p27kip1 by anergic T cells has led to the hypothesis that p27kip1, and the regulation of CDK1/2 activity by this protein, is necessary for the induction and/or maintenance of T cell clonal anergy. p27kip1 positively regulates the first G0 to G1 to S phase transition during the activation naïve, quiescent CD4+ T cells [42]. This is because p27kip1 has been shown in other tissues to serve as a scaffold which facilitates the assembly of cyclin D-CDK4/6 complexes during G1 progression, protecting these complexes from inhibition by the ink4 family members [43]. However, after the first cell division (when cells no longer need to undergo a G0 to G1 transition), p27kip1 acts in T cells to oppose clonal expansion by setting the threshold for the amount of CD28 costimulation and growth factor signaling is required to initiate cell cycle progression (reviewed in [2]). In the absence of p27kip1, CD4+ T cells can enter the cell cycle and expand in the absence of CD28 costimulation and in response to small amounts of IL-2, demonstrating that p27kip1 is a crucial intracellular biochemical sensor that ‘tells’ T cells when they have received the appropriate signals to undergo an immune response. This role for p27kip1 is so crucial that, in the absence of this protein, CD4+ T cells activated without CD28 costimulation fail to undergo the silencing of the IL2 gene that is the integral hallmark of anergy, and engage in secondary clonal expansion upon re-encounter with antigen [42,44]. This activity is not shared by all CDK inhibitory proteins, because p18ink4c, which inhibits CDK4 and 6 but not CDK1 or 2, is not required for anergy (our unpublished observations). This implies a unique role for p27kip1 and CDK1/2 in anergy avoidance and effector differentiation.
Anergy induction with CTLA-4Ig is associated with an abortive proliferative response [7,32,33], and genetic elimination of the cell cycle inhibitor p27kip1 renders CD4+ T cells resistant to anergy induced in the absence of CD28 costimulation [2]. The simplest explanation for these results is that the enhanced proliferation afforded by the absence of p27kip1 allows T cells to escape anergy. However, we have found that anergy avoidance by p27kip1−/− T cells can occur in the first 24 hours after stimulation with CTLA-4Ig (our unpublished observations), and therefore does not depend upon cell division. This implies that p27kip1 functions independently of cell cycle regulation to promote anergy in CD4+ T cells, presumably through it’s opposition of CDK activity. CDK might directly regulate IL2 transcription, and several factors involved in IL2 gene expression have been shown in other models to be regulated by these kinases. These include NFκB [45], Sp1 [46], the co-activator and histone acetyltransferase p300/CBP [47], and subunits of the RNA polymerase holoenzyme complex TFIIB and RNApolα [46]. CDK activity could also promote IL-2 production by inhibiting the activity of negative regulatory factors. This idea is supported by a recent study showing that CDK-mediated inactivation of the transcriptional repressor Smad3 [48] occurs in primed, but not anergic, T cells, and that dysregulated Smad3 activity in p27kip1-deficient T cells is an important component of their anergy-resistant phenotype [44]. Therefore, a revised hypothesis would state that cyclin-dependent kinase (most likely CDK2) activity, and not cell cycle progression per se, is crucial for anergy avoidance (Fig. 3). Further molecular studies will be needed to solidify this idea.
Fig. 3.
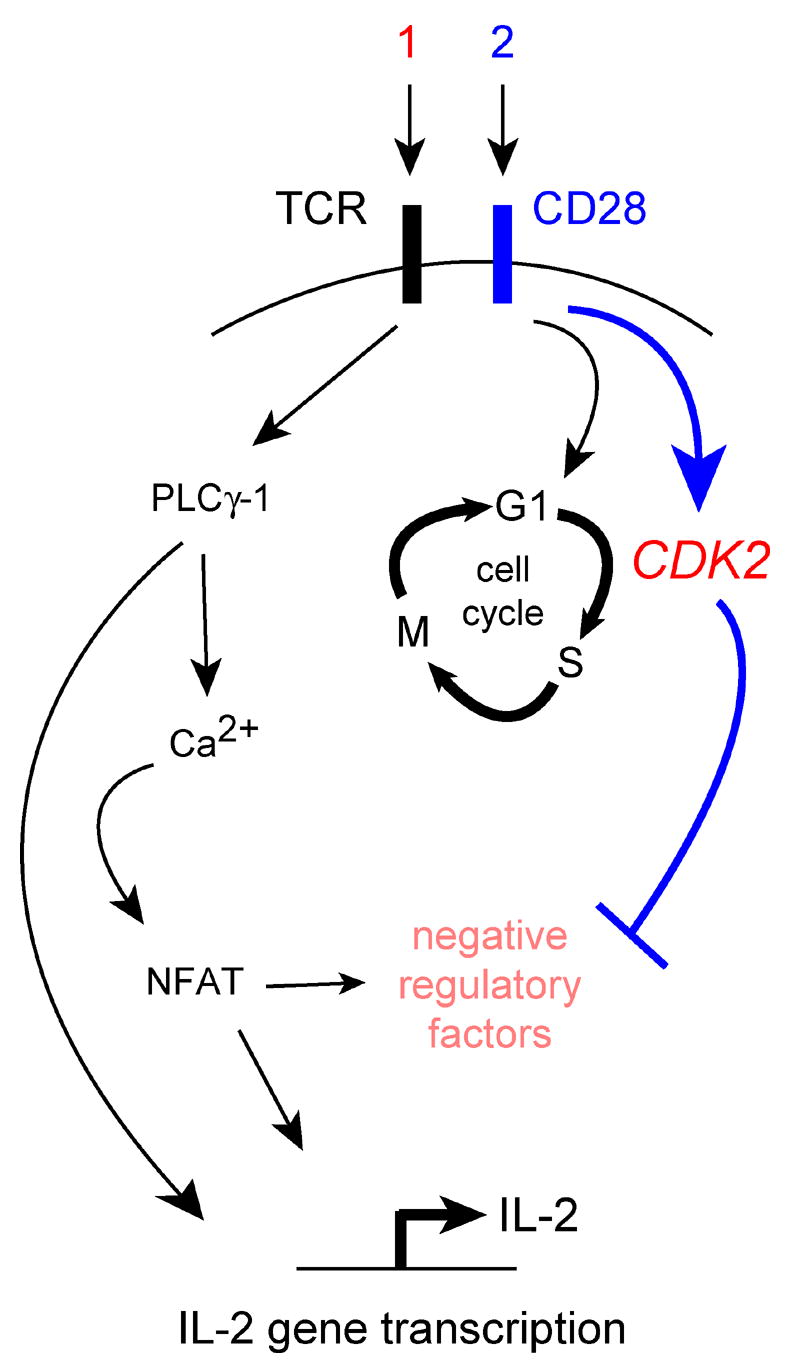
CDK-dependent anergy avoidance.
3. Cyclin-dependent kinases regulate autoimmune and alloimmune tolerance
3.1. CDK activity can influence self tolerance
While the studies described above demonstrate a very significant role for p27kip1 in limiting effector differentiation and promoting anergy induced either in vitro [42] or in vivo [44], these studies do not prove whether p27kip1 or cyclin-dependent kinases are important in regulating the more complex array of T cell functions required for functional immunity or tolerance in vivo. Mice deficient for the CDK inhibitory proteins p27kip1, p18ink4c and p15ink4b exhibit peripheral lymphoid hyperplasia, however, fail to develop signs of autoimmunity (reviewed in [2]). This indicates that these molecules are not required for self-tolerance. p21cip1 inhibits CDK1 and CDK2, and is involved in p53-mediated cell cycle arrest in response to DNA damage. In two studies, aged p21cip1−/− mice were found to have increased frequencies of activated CD4+ T cells, hypergammaglobulinemia, and anti-dsDNA antibodies reminiscent of a lupus-like syndrome [49,50]. Conversely, deletion of p21cip1 on the lupus-prone BXSB background ameliorated disease, apparently due to enhanced apoptosis of activated autoreactive T cells [51]. Thus, p21cip1 may regulate both the activation and survival of autoreactive T cells, and may therefore either promote or oppose self-tolerance depending on underlying genetic predisposition. CDK2 itself also appears to display this duality in life/death function (reviewed in [2]). While this kinase is clearly important for the proliferation and differentiation of mature T cells, mice genetically deficient in CDK2 exhibit defective negative selection of autoreactive T cells in the thymus. This function of CDK2 is apparently mediated through its ability to promote pro-apoptotic signaling in T cells.
3.2. CDK activity influences acquired alloimmune tolerance
The studies above suggest a role for certain cyclin-dependent kinases in central tolerance, and recent work has also demonstrated that CDK activity can strongly influence the acquisition of peripheral tolerance. This was tested recently in a cardiac allograft model using mice with deregulated CDK1/2 activity due to loss of p27kip1 [52], or deregulated CDK4/6 activity due to loss of p18ink4c (our unpublished data). Mice lacking either p27kip1 or p18ink4c reject fully MHC-mismatched cardiac allografts with the same kinetics as wild-type mice, however, pathological findings in the grafts of p27kip1-deficient recipients suggested stronger mononuclear cell infiltration and the involvement of acute humoral alloimmune responses. Allograft rejection requires the CD28 and CD40 costimulatory pathways (reviewed in [53]), and blockade of both of these receptors in wild-type mice leads to long-term allograft survival and donor-specific tolerance [54]. However, blockade of CD28 and CD40 costimulation in p27kip1-deficient mice results in acute, costimulation-independent cellular rejection associated with massive lymphocyte expansion, a 5-fold increase in the frequency of allospecific IFNγ-producing cells in the periphery, and increased infiltration and proliferation of CD4+ T cells in the cardiac grafts [52]. These results show that E-type cyclin-dependent kinase activity strongly promotes T cell differentiation during a physiologic immune response in vivo, and that inhibition of CDK2 and/or CDK1 activity by p27kip1 is required for the development of transplantation tolerance in the absence of CD28/CD40 costimulation.
As with p27kip1-deficient T cells [42], T cells with deregulated CDK4 and 6 activity due to p18ink4c deficiency exhibit enhanced cell division and a reduced costimulatory requirement in vitro [55]. However, unlike p27kip1-deficient mice, mice lacking p18ink4c are actually more susceptible to transplantation tolerance than wild-type mice. Blockade of either CD28 or CD40 individually in wild-type mice results in prolonged survival but eventual rejection of allografts. Conversely, blockade of these pathways in p18ink4c-deficient mice results in long-term allograft survival, and p18ink4c−/− T cells exhibit decreased production of proinflammatory cytokines and an increased rate of apoptosis (our unpublished observations). These data suggest that the dominant role for p18ink4c in T cells is to promote survival and differentiation during an immune response. This is also how p18ink4c functions during plasma cell differentiation [56,57], and could explain the disparate roles that p27kip1 and p18ink4c play in the induction of T cell tolerance.
3.3. Do CDK influence regulatory T cell function?
Long-term, donor-specific tolerance to organ transplants requires regulatory T cells [58]. Therefore, while these cyclin-dependent kinase inhibitors clearly influence differentiation and anergy in naïve T cells, these molecules could also modulate allograft tolerance through altering regulatory T cell function. Mice lacking either p27kip1 or p18ink4c contain normal numbers of CD4+CD25+Foxp3+ T cells in the periphery, and these ‘natural’ regulatory T cells function normally in in vitro suppression assays ([52] and our unpublished data). Also, allografts rejecting despite CD28/CD40 blockade in p27kip1−/− animals contain large numbers of Foxp3-positive cells, suggesting that uncontrolled, costimulation-independent effector differentiation is the primary reason for the failure of tolerance in animals that lack p27kip1. However, whether normal or dysregulated CDK activity influences in vivo regulatory T cell function, or the induction of regulatory T cells from naïve T cell precursors, are issues that requires further study.
4. Cyclin-dependent kinases as therapeutic targets for immune modulation
The studies described above suggest that cyclin-dependent kinases and their regulatory partners have discrete and crucial roles in the regulation of T cell immunity vs. tolerance, and imply that CDK may represent novel therapeutic targets for the inhibition of allograft rejection or autoimmunity. Dozens of compounds have been described that inhibit the activity of various cyclin-dependent kinases [59]. Of these, most are ATP analogs that act as competitive inhibitors of ATP binding to the active site of the kinase. For instance, the CDK inhibitory drugs flavopirdol (HMR 1275, L86–8275) and roscovitine (CYC202, seliciclib) are in human trials for the treatment of malignancy [60]. Roscovitine has been used therapeutically in experimental models of cancer [61], stroke [62], glomerulopathy [63], polycystic kidney disease [64], and pleurisy [65], but CDK inhibitory drugs have not been tested in models of autoimmunity or alloimmunity.
Our studies on the effects of CDK de-regulation on allograft rejection and tolerance provide a conceptual framework for novel immuno-modulatory applications of CDK inhibitory drugs. For instance, the fact that p27kip1, a genetically-encoded inhibitor of CDK1 and CDK2, is required for transplantation tolerance suggests that the use of synthetic CDK1/2 inhibitory compounds could promote allograft survival and alloimmune tolerance. Such drugs could potentially ameliorate immunopathologic T cell responses during autoimmune disease, as well. Conversely, de-regulation of CDK4/6 activity appears to have the opposite effect on T cell-mediated immunity in vivo. Therefore, compounds that selectively inhibit CDK4 or CDK6 activity could potentially act as adjuvants to enhance the effects of vaccines or cellular therapies against cancer and infectious diseases. Alternatively, the development of drugs that de-regulate CDK4/6 activity by inhibiting the capacity of ink4 family members to bind to CDK4 or 6 could promote alloreactive or autoreactive T cell apoptosis, thereby forming a therapy for organ transplant rejection and/or autoimmune disease.
5. Summary
Cyclin-dependent kinases are positive regulators of cell cycle progression that are known to act as key regulators of embryonic development and cancer. Recent progress in this field has indicated that these proteins likewise direct cell fate decisions during the development of immune and tolerant T cell responses, and therefore may serve as important targets for immune modulation.
Acknowledgments
ADW is supported by NIH grants AI054643 and AI059881, the Joseph Stokes, Jr. Research Institute, and the Fred & Suzanne Biesecker Pediatric Liver Center at The Children’s Hospital of Philadelphia.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Suchin EJ, Langmuir PB, Palmer E, Sayegh MH, Wells AD, Turka LA. Quantifying the frequency of alloreactive T cells in vivo: new answers to an old question. J Immunol. 2001;166:973–81. doi: 10.4049/jimmunol.166.2.973. [DOI] [PubMed] [Google Scholar]
- 2.Rowell EA, Wells AD. The role of cyclin-dependent kinases in T-cell development, proliferation, and function. Crit Rev Immunol. 2006;26:189–212. doi: 10.1615/critrevimmunol.v26.i3.10. [DOI] [PubMed] [Google Scholar]
- 3.Senderowicz AM. Novel small molecule cyclin-dependent kinases modulators in human clinical trials. Cancer Biol Ther. 2003;2:S84–95. [PubMed] [Google Scholar]
- 4.Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. 2002;2:933–44. doi: 10.1038/nri954. [DOI] [PubMed] [Google Scholar]
- 5.Bird JJ, Brown DR, Mullen AC, Moskowitz NH, Mahowald MA, Sider JR, et al. Helper T cell differentiation is controlled by the cell cycle. Immunity. 1998;9:229–37. doi: 10.1016/s1074-7613(00)80605-6. [DOI] [PubMed] [Google Scholar]
- 6.Gett AV, Hodgkin PD. Cell division regulates the T cell cytokine repertoire, revealing a mechanism underlying immune class regulation. Proc Natl Acad Sci U S A. 1998;95:9488–93. doi: 10.1073/pnas.95.16.9488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gudmundsdottir H, Wells AD, Turka LA. Dynamics and requirements of T cell clonal expansion in vivo at the single-cell level: effector function is linked to proliferative capacity. J Immunol. 1999;162:5212–23. [PubMed] [Google Scholar]
- 8.Linsley PS, Brady W, Grosmaire L, Aruffo A, Damle NK, Ledbetter JA. Binding of the B cell activation antigen B7 to CD28 costimulates T cell proliferation and interleukin 2 mRNA accumulation. J Exp Med. 1991;173:721–30. doi: 10.1084/jem.173.3.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harding FA, McArthur JG, Gross JA, Raulet DH, Allison JP. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature. 1992;356:607–9. doi: 10.1038/356607a0. [DOI] [PubMed] [Google Scholar]
- 10.Gimmi CD, Freeman GJ, Gribben JG, Gray G, Nadler LM. Human T-cell clonal anergy is induced by antigen presentation in the absence of B7 costimulation. Proc Natl Acad Sci USA. 1993;90:6586–90. doi: 10.1073/pnas.90.14.6586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwartz RH. T cell anergy. Annu Rev Immunol. 2003;21:305–34. doi: 10.1146/annurev.immunol.21.120601.141110. [DOI] [PubMed] [Google Scholar]
- 12.Boussiotis VA, Barber DL, Lee BJ, Gribben JG, Freeman GJ, Nadler LM. Differential association of protein tyrosine kinases with the T cell receptor is linked to the induction of anergy and its prevention by B7 family-mediated costimulation. J Exp Med. 1996;184:365–76. doi: 10.1084/jem.184.2.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeSilva DR, Feeser WS, Tancula EJ, Scherle PA. Anergic T cells are defective in both jun NH2-terminal kinase and mitogen-activated protein kinase signaling pathways. J Exp Med. 1996;183:2017–23. doi: 10.1084/jem.183.5.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fields PE, Gajewski TF, Fitch FW. Blocked Ras activation in anergic CD4+ T cells. Science. 1996;271:1276–8. doi: 10.1126/science.271.5253.1276. [DOI] [PubMed] [Google Scholar]
- 15.Li W, Whaley CD, Mondino A, Mueller DL. Blocked signal transduction to the ERK and JNK protein kinases in anergic CD4+ T cells. Science. 1996;271:1272–6. doi: 10.1126/science.271.5253.1272. [DOI] [PubMed] [Google Scholar]
- 16.Sloan-Lancaster J, Steinberg TH, Allen PM. Selective activation of the calcium signaling pathway by altered peptide ligands. J Exp Med. 1996;184:1525–30. doi: 10.1084/jem.184.4.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boussiotis VA, Freeman GJ, Berezovskaya A, Barber DL, Nadler LM. Maintenance of human T cell anergy: blocking of IL-2 gene transcription by activated Rap1. Science. 1997;278:124–8. doi: 10.1126/science.278.5335.124. [DOI] [PubMed] [Google Scholar]
- 18.Wells AD, Walsh MC, Sankaran D, Turka LA. T cell effector function and anergy avoidance are quantitatively linked to cell division. J Immunol. 2000;165:2432–43. doi: 10.4049/jimmunol.165.5.2432. [DOI] [PubMed] [Google Scholar]
- 19.Wells AD, Liu QH, Hondowicz B, Zhang J, Turka LA, Freedman BD. Regulation of T cell activation and tolerance by phospholipase cgamma-1-dependent integrin avidity modulation. J Immunol. 2003;170:4127–33. doi: 10.4049/jimmunol.170.8.4127. [DOI] [PubMed] [Google Scholar]
- 20.Telander DG, Malvey EN, Mueller DL. Evidence for repression of IL-2 gene activation in anergic T cells. J Immunol. 1999;162:1460–5. [PubMed] [Google Scholar]
- 21.Kitagawa-Sakakida S, Schwartz RH. Multifactor cis-dominant negative regulation of IL-2 gene expression in anergized T cells. J Immunol. 1996;157:2328–39. [PubMed] [Google Scholar]
- 22.Powell JD, Lerner CG, Ewoldt GR, Schwartz RH. The -180 site of the IL-2 promoter is the target of CREB/CREM binding in T cell anergy. J Immunol. 1999;163:6631–9. [PubMed] [Google Scholar]
- 23.Bruniquel D, Schwartz RH. Selective, stable demethylation of the interleukin-2 gene enhances transcription by an active process. Nat Immunol. 2003;4:235–40. doi: 10.1038/ni887. [DOI] [PubMed] [Google Scholar]
- 24.Thomas RM, Gao L, Wells AD. Signals from CD28 induce stable epigenetic modification of the IL-2 promoter. J Immunol. 2005;174:4639–46. doi: 10.4049/jimmunol.174.8.4639. [DOI] [PubMed] [Google Scholar]
- 25.Murayama A, Sakura K, Nakama M, Yasuzawa-Tanaka K, Fujita E, Tateishi Y, et al. A specific CpG site demethylation in the human interleukin 2 gene promoter is an epigenetic memory. Embo J. 2006;25:1081–92. doi: 10.1038/sj.emboj.7601012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Macian F, Garcia-Cozar F, Im SH, Horton HF, Byrne MC, Rao A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell. 2002;109:719–31. doi: 10.1016/s0092-8674(02)00767-5. [DOI] [PubMed] [Google Scholar]
- 27.Mueller DL. E3 ubiquitin ligases as T cell anergy factors. Nat Immunol. 2004;5:883–90. doi: 10.1038/ni1106. [DOI] [PubMed] [Google Scholar]
- 28.Powell JD. The induction and maintenance of T cell anergy. Clin Immunol. 2006;120:239–46. doi: 10.1016/j.clim.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 29.DeSilva DR, Urdahl KB, Jenkins MK. Clonal anergy is induced in vitro by T cell receptor occupancy in the absence of proliferation. J Immunol. 1991;147:3261–7. [PubMed] [Google Scholar]
- 30.Boussiotis VA, Barber DL, Nakarai T, Freeman GJ, Gribben JG, Bernstein GM, et al. Prevention of T cell anergy by signaling through the gamma c chain of the IL-2 receptor. Science. 1994;266:1039–42. doi: 10.1126/science.7973657. [DOI] [PubMed] [Google Scholar]
- 31.Beverly B, Kang SM, Lenardo MJ, Schwartz RH. Reversal of in vitro T cell clonal anergy by IL-2 stimulation. Int Immunol. 1992;4:661–71. doi: 10.1093/intimm/4.6.661. [DOI] [PubMed] [Google Scholar]
- 32.Wells AD, Gudmundsdottir H, Turka LA. Following the fate of individual T cells throughout activation and clonal expansion. Signals from T cell receptor and CD28 differentially regulate the induction and duration of a proliferative response. J Clin Invest. 1997;100:3173–83. doi: 10.1172/JCI119873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wells AD, Walsh MC, Bluestone JA, Turka LA. Signaling through CD28 and CTLA-4 controls two distinct forms of T cell anergy. J Clin Invest. 2001;108:895–903. doi: 10.1172/JCI13220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jackson SK, DeLoose A, Gilbert KM. Induction of anergy in Th1 cells associated with increased levels of cyclin-dependent kinase inhibitors p21Cip1 and p27Kip1. J Immunol. 2001;166:952–8. doi: 10.4049/jimmunol.166.2.952. [DOI] [PubMed] [Google Scholar]
- 35.Jackson SK, DeLoose A, Gilbert KM. The ability of antigen, but not interleukin-2, to promote n-butyrate-induced T helper 1 cell anergy is associated with increased expression and altered association patterns of cyclin-dependent kinase inhibitors. Immunology. 2002;106:486–95. doi: 10.1046/j.1365-2567.2002.01457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Powell JD, Lerner CG, Schwartz RH. Inhibition of cell cycle progression by rapamycin induces T cell clonal anergy even in the presence of costimulation. J Immunol. 1999;162:2775–84. [PubMed] [Google Scholar]
- 37.Vanasek TL, Khoruts A, Zell T, Mueller DL. Antagonistic roles for CTLA-4 and the mammalian target of rapamycin in the regulation of clonal anergy: enhanced cell cycle progression promotes recall antigen responsiveness. J Immunol. 2001;167:5636–44. doi: 10.4049/jimmunol.167.10.5636. [DOI] [PubMed] [Google Scholar]
- 38.Verdoodt B, Blazek T, Rauch P, Schuler G, Steinkasserer A, Lutz MB, et al. The cyclin-dependent kinase inhibitors p27Kip1 and p21Cip1 are not essential in T cell anergy. Eur J Immunol. 2003;33:3154–63. doi: 10.1002/eji.200323960. [DOI] [PubMed] [Google Scholar]
- 39.Boussiotis VA, Freeman GJ, Taylor PA, Berezovskaya A, Grass I, Blazar BR, et al. p27kip1 functions as an anergy factor inhibiting interleukin 2 transcription and clonal expansion of alloreactive human and mouse helper T lymphocytes. Nat Med. 2000;6:290–7. doi: 10.1038/73144. [DOI] [PubMed] [Google Scholar]
- 40.Harbour JW, Dean DC. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev. 2000;14:2393–409. doi: 10.1101/gad.813200. [DOI] [PubMed] [Google Scholar]
- 41.Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, et al. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 1994;78:59–66. doi: 10.1016/0092-8674(94)90572-x. [DOI] [PubMed] [Google Scholar]
- 42.Rowell EA, Walsh MC, Wells AD. Opposing roles for the cyclin-dependent kinase inhibitor p27kip1 in the control of CD4+ T cell proliferation and effector function. J Immunol. 2005;174:3359–68. doi: 10.4049/jimmunol.174.6.3359. [DOI] [PubMed] [Google Scholar]
- 43.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–12. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 44.Li L, Iwamoto Y, Berezovskaya A, Boussiotis VA. A pathway regulated by cell cycle inhibitor p27(Kip1) and checkpoint inhibitor Smad3 is involved in the induction of T cell tolerance. Nat Immunol. 2006;7:1157–65. doi: 10.1038/ni1398. [DOI] [PubMed] [Google Scholar]
- 45.Chen E, Li CC. Association of Cdk2/cyclin E and NF-kappa B complexes at G1/S phase. Biochem Biophys Res Commun. 1998;249:728–34. doi: 10.1006/bbrc.1998.9224. [DOI] [PubMed] [Google Scholar]
- 46.Dynlacht BD. Regulation of transcription by proteins that control the cell cycle. Nature. 1997;389:149–52. doi: 10.1038/38225. [DOI] [PubMed] [Google Scholar]
- 47.Perkins ND, Felzien LK, Betts JC, Leung K, Beach DH, Nabel GJ. Regulation of NF-kappaB by cyclin-dependent kinases associated with the p300 coactivator. Science. 1997;275:523–7. doi: 10.1126/science.275.5299.523. [DOI] [PubMed] [Google Scholar]
- 48.Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature. 2004;430:226–31. doi: 10.1038/nature02650. [DOI] [PubMed] [Google Scholar]
- 49.Balomenos D, Martin-Caballero J, Garcia MI, Prieto I, Flores JM, Serrano M, et al. The cell cycle inhibitor p21 controls T-cell proliferation and sex-linked lupus development. Nat Med. 2000;6:171–6. doi: 10.1038/72272. [DOI] [PubMed] [Google Scholar]
- 50.Santiago-Raber ML, Lawson BR, Dummer W, Barnhouse M, Koundouris S, Wilson CB, et al. Role of cyclin kinase inhibitor p21 in systemic autoimmunity. J Immunol. 2001;167:4067–74. doi: 10.4049/jimmunol.167.7.4067. [DOI] [PubMed] [Google Scholar]
- 51.Lawson BR, Baccala R, Song J, Croft M, Kono DH, Theofilopoulos AN. Deficiency of the cyclin kinase inhibitor p21(WAF-1/CIP-1) promotes apoptosis of activated/memory T cells and inhibits spontaneous systemic autoimmunity. J Exp Med. 2004;199:547–57. doi: 10.1084/jem.20031685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rowell EA, Wang L, Hancock WW, Wells AD. The Cyclin-Dependent Kinase Inhibitor p27kip1 Is Required for Transplantation Tolerance Induced by Costimulatory Blockade. J Immunol. 2006;177:5169–76. doi: 10.4049/jimmunol.177.8.5169. [DOI] [PubMed] [Google Scholar]
- 53.Wells AD. T cell Costimulatory Pathways Relevant to Transplant Rejection and Tolerance. Transplantation Reviews. 2002;16:205. [Google Scholar]
- 54.Larsen CP, Elwood ET, Alexander DZ, Ritchie SC, Hendrix R, Tucker-Burden C, et al. Long-term acceptance of skin and cardiac allografts after blocking CD40 and CD28 pathways. Nature. 1996;381:434–8. doi: 10.1038/381434a0. [DOI] [PubMed] [Google Scholar]
- 55.Kovalev GI, Franklin DS, Coffield VM, Xiong Y, Su L. An important role of CDK inhibitor p18(INK4c) in modulating antigen receptor-mediated T cell proliferation. J Immunol. 2001;167:3285–92. doi: 10.4049/jimmunol.167.6.3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tourigny MR, Ursini-Siegel J, Lee H, Toellner KM, Cunningham AF, Franklin DS, et al. CDK inhibitor p18(INK4c) is required for the generation of functional plasma cells. Immunity. 2002;17:179–89. doi: 10.1016/s1074-7613(02)00364-3. [DOI] [PubMed] [Google Scholar]
- 57.Huang X, Di Liberto M, Cunningham AF, Kang L, Cheng S, Ely S, et al. Homeostatic cell-cycle control by BLyS: Induction of cell-cycle entry but not G1/S transition in opposition to p18INK4c and p27Kip1. Proc Natl Acad Sci U S A. 2004;101:17789–94. doi: 10.1073/pnas.0406111101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wood KJ, Sakaguchi S. Regulatory T cells in transplantation tolerance. Nat Rev Immunol. 2003;3:199–210. doi: 10.1038/nri1027. [DOI] [PubMed] [Google Scholar]
- 59.Knockaert M, Greengard P, Meijer L. Pharmacological inhibitors of cyclin-dependent kinases. Trends Pharmacol Sci. 2002;23:417–25. doi: 10.1016/s0165-6147(02)02071-0. [DOI] [PubMed] [Google Scholar]
- 60.Senderowicz AM. Targeting cell cycle and apoptosis for the treatment of human malignancies. Curr Opin Cell Biol. 2004;16:670–8. doi: 10.1016/j.ceb.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 61.McClue SJ, Blake D, Clarke R, Cowan A, Cummings L, Fischer PM, et al. In vitro and in vivo antitumor properties of the cyclin dependent kinase inhibitor CYC202 (R-roscovitine) Int J Cancer. 2002;102:463–8. doi: 10.1002/ijc.10738. [DOI] [PubMed] [Google Scholar]
- 62.Osuga H, Osuga S, Wang F, Fetni R, Hogan MJ, Slack RS, et al. Cyclin-dependent kinases as a therapeutic target for stroke. Proc Natl Acad Sci U S A. 2000;97:10254–9. doi: 10.1073/pnas.170144197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gherardi D, D’Agati V, Chu TH, Barnett A, Gianella-Borradori A, Gelman IH, et al. Reversal of collapsing glomerulopathy in mice with the cyclin-dependent kinase inhibitor CYC202. J Am Soc Nephrol. 2004;15:1212–22. doi: 10.1097/01.asn.0000124672.41036.f4. [DOI] [PubMed] [Google Scholar]
- 64.Bukanov NO, Smith LA, Klinger KW, Ledbetter SR, Ibraghimov-Beskrovnaya O. Long-lasting arrest of murine polycystic kidney disease with CDK inhibitor roscovitine. Nature. 2006;444:949–52. doi: 10.1038/nature05348. [DOI] [PubMed] [Google Scholar]
- 65.Rossi AG, Sawatzky DA, Walker A, Ward C, Sheldrake TA, Riley NA, et al. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat Med. 2006;12:1056–64. doi: 10.1038/nm1468. [DOI] [PubMed] [Google Scholar]