

Abstract
Several studies have documented a strong association between smoking and depression. Because cigarette smoke has been reported to inhibit monoamine oxidase (MAO) A in vitro and in animals and because MAO A inhibitors are effective antidepressants, we tested the hypothesis that MAO A would be reduced in the brain of cigarette smokers. We compared brain MAO A in 15 nonsmokers and 16 current smokers with [11C]clorgyline and positron emission tomography (PET). Four of the nonsmokers were also treated with the antidepressant MAO inhibitor drug, tranylcypromine (10 mg/day for 3 days) after the baseline PET scan and then rescanned to assess the sensitivity of [11C]clorgyline binding to MAO inhibition. MAO A levels were quantified by using the model term λk3which is a function of brain MAO A concentration. Smokers had significantly lower brain MAO A than nonsmokers in all brain regions examined (average reduction, 28%). The mean λk3 values for the whole brain were 0.18 ± 0.04 and 0.13 ± 0.03 ccbrain (mlplasma)−1 min−1 for nonsmokers and smokers, respectively; P < 0.0003). Tranylcypromine treatment reduced λk3 by an average of 58% for the different brain regions. Our results show that tobacco smoke exposure is associated with a marked reduction in brain MAO A, and this reduction is about half of that produced by a brief treatment with tranylcypromine. This suggests that MAO A inhibition needs to be considered as a potential contributing variable in the high rate of smoking in depression and in the development of more effective strategies for smoking cessation.
There are approximately 1 billion cigarette smokers in the world today and about 3 million die each year from smoking-associated illnesses (1). This places a sense of urgency on understanding the neuropharmacological properties of tobacco smoke and their relationship to smoking behavior and epidemiology. For example, it is not understood why smoking is more prevalent in depression and why smoking cessation is less successful in depressed patients (2, 3). Though it is unlikely that any one factor accounts for the strong association between smoking and depression, it is possible that tobacco smoke may have antidepressant properties. One of the molecular targets proposed to link smoking and depression is monoamine oxidase (MAO) (4, 5), an enzyme which was first associated with mood over 40 years ago when it was discovered that MAO inhibitors had antidepressant properties (6, 7).
MAO exists in two subtypes (MAO A and B) that are different gene products (8, 9). In the brain, MAO A oxidizes serotonin and norepinephrine and is found primarily in catecholaminergic neurons, whereas MAO B oxidizes benzylamine and phenethylamine and is localized in serotonergic neurons and in glial cells (10). Both forms oxidize dopamine (11). The antidepressant effects of the nonselective MAO inhibitors are generally attributed to the inhibition of MAO A (12).
We recently reported that smokers have reduced brain MAO B relative to nonsmokers and former smokers (13). Others have found that both MAO A and MAO B are reduced in animals exposed to tobacco smoke (14) and in vitro (15, 16), and that heavy smokers have reduced peripheral measures of both MAO A and B (5). It has also been demonstrated that nicotine is not responsible for MAO A inhibition (14, 16). To test the hypothesis that tobacco smoke exposure inhibits MAO A in the human brain, we compared a group of nonsmokers and a group of smokers using positron-emission tomography (PET) and [11C]clorgyline, a radiotracer which binds irreversibly to brain MAO A (17, 18). We also rescanned four of the nonsmokers after they had received a low dose (10 mg per day) of the MAO inhibitor drug tranylcypromine for 3 days to assess the sensitivity of [11C]clorgyline to MAO A inhibition.
SUBJECTS AND METHODS
Participants.
These studies followed the guidelines of the Human Subjects Research Committee at Brookhaven National Laboratory, and informed consent was obtained from each subject after the procedures had been explained. Subjects were recruited by newspaper advertisements. Subjects were screened for a lack of history of current or past psychiatric or neurological disease, as well as for lack of history of drug or alcohol abuse (excluding caffeine for all subjects and nicotine for the smokers). Exclusion criteria were (i) current or past psychiatric disease other than nicotine dependence, (ii) neurological signs and/or history of neurological disease, (iii) history of head trauma, (iv) history of cardiovascular or endocrinological disease, (v) current medical illness, and (vi) dependence on any substance other than nicotine (for the smokers) or caffeine. Inclusion criteria for nonsmokers was that they had never smoked. Inclusion criteria for smokers was to be a current smoker and to have smoked at least 10 cigarettes per day for the preceding 1 year. Except for three of the female subjects who were on hormone replacement therapy, none of the subjects were taking medications at the time of the study, and any previous medications had been discontinued 8 days before the PET scan. Screens for drug use were performed prior to each PET scan. Smokers refrained from smoking during the entire PET study and were scanned 1.5–9.5 h (average time interval, 2.7 h) after the last cigarette. Three of the nonsmokers received a second PET scan on the same day to assess reproducibility of repeated PET measures. Four of the nonsmokers were scanned a second time after receiving tranylcypromine (30 mg total; 10 mg per day for 2 days prior and 10 mg on the day of the second PET scan) to assess the effect of MAO inhibition on [11C]clorgyline binding. The time interval between the baseline scan and the tranylcypromine scan varied from 2 weeks to 6 months.
PET Imaging.
PET scans were run on a whole-body, high-resolution positron emission tomograph [6.5 × 6.5 × 6.5 mm, full-width half maximum, 15 slices; Computer Technologies (Knoxville, TN; model CTI 931] which provided 15 slices of the brain. Subjects were prepared for scanning as described (19). Brain MAO A was measured using [11C]clorgyline, which was prepared as described (20). Each subject received [11C]clorgyline (4–16 mCi; 6–50 μg; 1 Ci = 37 GBq). Time-activity data for all brain regions was accumulated for 75 min following this sequence: ten 1 min frames, four 5 min frames, and six 7.5 min frames. An arterial plasma input function for [11C]clorgyline was measured. Arterial blood samples were withdrawn every 2.5 sec for the first 2 min [Ole Dich (Hvidovre, Denmark) automatic blood sampler], then every minute from 2–6 min, then at 8, 10, 15, 20, 30, 45, 60, and 75 min. Each arterial blood sample was centrifuged and plasma pipetted and counted. Plasma samples at 1, 5, 10, 20, 30, 45, 60, and 75 min were analyzed for [11C]clorgyline using the same solid phase extraction procedure described previously for [11C]l-deprenyl-D2 (19). An arterial input function was calculated from the total carbon-11 in plasma corrected for the fraction present as [11C]clorgyline.
Image Analysis.
Regions of interest were drawn directly on PET scans. For the purpose of region identification, we added the images obtained from 30 to 75 min after tracer injection. A template that used as a reference the brain atlas of Matsui and Hirano (21) was projected into the “averaged” PET image and manually fitted for appropriate neuroanatomical location. The regions were then projected to the dynamic scans to obtain time-activity data. Regions of interest for the following brain areas were obtained: frontal cortex, parietal cortex, occipital cortex, temporal cortex, cingulate gyrus, thalamus, basal ganglia, and cerebellum. Regions were identified in at least two contiguous slices, and the weighted average was obtained for each region. For the thalamus, basal ganglia, frontal, parietal, and temporal cortices, the right and left regions were averaged. For the global region eight central planes were averaged.
Data Analysis.
For each subject, PET time-activity data from different brain regions along with time-activity data for [11C]clorgyline in arterial plasma were used to calculate the model term Ki, a kinetic constant which determines the rate of trapping of [11C]clorgyline (which is a function of both the concentration of MAO A and blood flow), and K1 the blood to tissue transport constant. An approximate blood volume correction (4%) was subtracted from the PET data prior to parameter optimization. K1 is related to blood flow (F) through the following equation (see ref. 22):
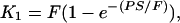 |
1 |
where PS is the permeability-surface area product. Assuming a three-compartment model which allows for both the transport and trapping of ligand, Ki can be written as
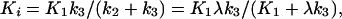 |
2 |
where k2 is the tissue to plasma efflux constant and k3 is proportional to the concentration of MAO A. Ki is also written in terms of the product λk3, which is independent of blood flow (λ = K1/k2) (23) but is a more robust parameter than k3 (24). λk3 can be calculated from Eq. 2 if Ki and K1 are known.
Ki was obtained graphically from a transformation of plasma and tissue time-activity data as described by Patlak et al. (25). Ki was taken as the average of slopes from 6 to 45 min and from 6 to 55 min. The initial time was taken as the time from which linearity was observed. K1 was calculated by using a rearrangement of the method of Blomqvist (26). In this method the tissue radioactivity, region of interest (ROI) and plasma radioactivity (Cp) are related to model parameters as given by
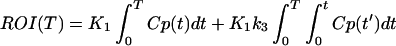 |
3 |
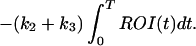 |
Using Ki from Eq. 2, this can be rearranged to give
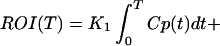 |
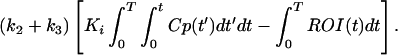 |
4 |
This is a bilinear regression with coefficients K1 and (k2 + k3) computed using standard methods (27).
All data is presented as the mean ± standard deviation of the mean. Model terms K1 and λk3 were compared for nonsmokers and smokers using an unpaired t test (two tail) and for nonsmokers at baseline and after treatment with tranylcypromine using a paired t test. To assess whether K1 and λk3 were affected by gender, a two-factor (smoking status and gender) ANOVA was performed.
RESULTS
Subject data is presented in Table 1. The model term λk3 was reduced for smokers relative to nonsmokers (Table 2). An ANOVA analysis showed that the effect of smoking on λk3 was significant (F = 16; P < 0.0003 for the global value), but that there was not a significant effect of gender (F = 3; P = 0.1) and no smoking versus gender interaction (F = 0.3; P = 0.6). The region with highest difference between groups was the occipital cortex (reduced by 38%), and the average reduction for all cortical and subcortical structures was 28 ± 4%. Values for λk3 for the different brain regions are shown in Table 2. Individual data for the thalamus are shown in Fig. 1, and brain images are shown in Fig. 2. We found no relationship between number of cigarettes smoked per day and values for λk3 in this initial study. However, because the variability in smoking behavior in addition to dose would contribute to degree of MAO inhibition, this issue requires further investigation.
Table 1.
Characteristics of subjects
| Nonsmokers | Smokers | |
|---|---|---|
| Number | 15 | 16 |
| Age | ||
| Mean | 34.4 ± 9.6 | 38.4 ± 10.6 |
| Range | 21–58 | 18–60 |
| Males/females | 8/7 | 10/6 |
| Years smoking | ||
| Mean | 20 ± 11 | |
| Range | 8–35 | |
| Smoking dose, pack per day | ||
| Mean | 1.10 ± 0.36 | |
| Range | 0.5–2 | |
| Time between last cigarette and PET scan, h | ||
| Mean | 2.7 ± 2 | |
| Range | 2–9.5 |
Table 2.
Comparison of model terms K1 and λk3 for nonsmokers and smokers for different brain regions
| Brain region |
K1
(ml/cc/min)
|
P value | λk3 [ccbrain
(mlplasma)−1
min−1]
|
P value | |||
|---|---|---|---|---|---|---|---|
| Nonsmokers (n = 15) | Smokers (n = 16) | Nonsmokers (n = 15) | Smokers (n = 16) | % difference | |||
| Global | 0.30 ± 0.07 | 0.28 ± 0.05 | 0.3 | 0.18 ± 0.04 | 0.13 ± 0.03 | −28 | 0.0003 |
| Cingulate | 0.42 ± 0.11 | 0.36 ± 0.08 | 0.1 | 0.25 ± 0.06 | 0.18 ± 0.043 | −28 | 0.0006 |
| Gyrus | |||||||
| Occipital cortex | 0.49 ± 0.11 | 0.46 ± 0.07 | 0.4 | 0.32 ± 0.10 | 0.20 ± 0.06 | −38 | 0.0001 |
| Basal ganglia | 0.42 ± 0.04 | 0.40 ± 0.08 | 0.5 | 0.23 ± 0.04 | 0.18 ± 0.04 | −22 | 0.0014 |
| Thalamus | 0.46 ± 0.12 | 0.42 ± 0.06 | 0.2 | 0.33 ± 0.07 | 0.24 ± 0.05 | −27 | 0.0003 |
| Frontal cortex | 0.36 ± 0.06 | 0.34 ± 0.06 | 0.6 | 0.22 ± 0.05 | 0.16 ± 0.035 | −27 | 0.0008 |
| Parietal cortex | 0.37 ± 0.09 | 0.34 ± 0.07 | 0.3 | 0.21 ± 0.04 | 0.16 ± 0.03 | −24 | 0.0003 |
| Temporal | 0.40 ± 0.10 | 0.35 ± 0.08 | 0.2 | 0.24 ± 0.05 | 0.17 ± 0.04 | −29 | 0.0001 |
| Cortex | |||||||
| Cerebellum | 0.42 ± 0.08 | 0.40 ± 0.06 | 0.4 | 0.14 ± 0.03 | 0.099 ± 0.025 | −29 | 0.0005 |
Figure 1.
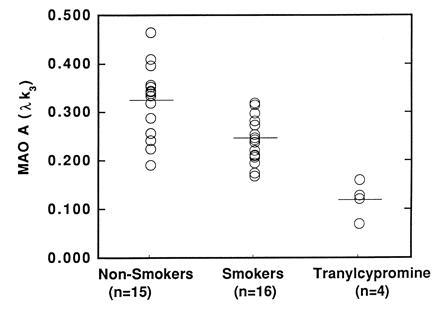
Comparison of MAO A levels in the thalamus (as expressed by the model term λk3) for nonsmokers (n = 15), smokers (n = 16), and nonsmokers who were treated with tranylcypromine (n = 4).
Figure 2.
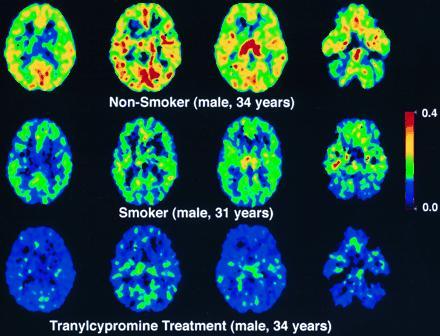
Pixel by pixel images of the model term which is a function of MAO A activity for a nonsmoker (Top row), a smoker (Middle row), and the same nonsmoker after treatment with tranylcypromine (Bottom row). The same four planes of the brain are shown for each subject and correspond to brain sections at 5.8 cm (level of the occipital cortex and the lateral ventricles), 5.1 cm (level of the occipital cortex and the lateral ventricles), 4.5 cm (level of the thalamus), and 1 cm (level of the lower temporal poles and the cerebellum) above the canthomeatal line (proceeding from left to right). The color scale represents values of λk3 [scales from 0.4 (red) to 0 (black)]. The values of λk3 for the thalamus are 0.319, 0.212, and 0.128 ccbrain (mlplasma)−1 min−1 for the nonsmoker, the smoker and the nonsmoker treated with tranylcypromine, respectively. The corresponding K1 values are 0.434, 0.421, 0.404 ml/cc/min.
In contrast to λk3, the plasma to brain transfer constant, K1, did not differ between nonsmokers and smokers for any brain region (Table 2). However, there was a significant main effect of gender on K1 for all brain regions with an average of 21% higher values for women (means 0.341 ± 0.066 versus 0.257 ± 0.016 for the global value for females and males respectively; ANOVA: F = 26, P < 0.0001; data for other brain regions not shown). There was not a smoking versus gender interaction effect on K1 (F = 0.2; P = 0.6).
To assess the reproducibility of repeated measures of [11C]clorgyline binding, three of the nonsmokers received a second PET scan 2 h after the first. The average change in K1 and λk3 for the nine brain regions examined for the three subjects was 9.5 ± 4.0% and 10.9 ± 5.0%, respectively (data not shown).
To assess the sensitivity of [11C]clorgyline binding to MAO inhibition, four of the nonsmokers were scanned at baseline and then treated with the nonselective MAO inhibitor drug, tranylcypromine (10 mg/day) for 3 days prior to a second scan. Tranylcypromine decreased λk3 by an average of 58% in all brain regions (Table 3; P < 0.0001, paired t test, two tail; see Fig. 1 for individual data on the thalamus and Fig. 2 for brain images) but did not change K1 (data not shown). Differences in λk3 were similar for all brain regions. The arterial plasma integral for labeled clorgyline after tranylcypromine was an average of 30 ± 3% greater than that at baseline (357 ± 64 and 465 ± 84 at baseline and after treatment; P < 0.0001, paired t test, two tail).
Table 3.
Effect of treatment with tranylcypromine on λk3 (n = 4)
| Brain region | Baseline | Tranylcypromine | % change*† |
|---|---|---|---|
| Global | 0.17 ± 0.05 | 0.08 ± 0.02 | −53 |
| Cingulate gyrus | 0.22 ± 0.05 | 0.10 ± 0.02 | −55 |
| Occipital cortex | 0.28 ± 0.05 | 0.11 ± 0.02 | −61 |
| Basal ganglia | 0.24 ± 0.07 | 0.11 ± 0.04 | −54 |
| Thalamus | 0.30 ± 0.08 | 0.12 ± 0.04 | −60 |
| Frontal cortex | 0.22 ± 0.06 | 0.08 ± 0.015 | −64 |
| Parietal cortex | 0.19 ± 0.03 | 0.08 ± 0.015 | −58 |
| Temporal cortex | 0.22 ± 0.04 | 0.08 ± 0.014 | −64 |
| Cerebellum | 0.13 ± 0.04 | 0.064 ± 0.022 | −51 |
% change refers to percent change of the model term λk3 (ccbrain (mlplasma)−1 min−1) from the baseline scan compared to the second scan.
P < 0.0001, paired t-test, two tail.
DISCUSSION
The main finding in this study is that smokers have reduced brain MAO A relative to nonsmokers as measured with [11C]clorgyline and PET. This result is consistent with the results of studies in animals exposed to tobacco smoke (14) and on peripheral measures of MAO A in heavy smokers (5). The reduction in brain MAO A was not accounted for by reduced brain blood flow since the plasma to brain transfer constant K1 did not differ between smokers and nonsmokers [though we did find a lower blood to brain transfer constant K1 in men relative to women which is likely to reflect the known lower cerebral blood flow in males than in females (28)].
The ability of PET and labeled clorgyline to measure brain MAO A is supported by prior studies in mice (17), by the reduction in the binding of labeled clorgyline by treatment for 3 days with the nonselective MAO inhibitor tranylcypromine (10 mg per day; Table 3), and by the general similarity in the relative regional distribution of carbon-11 as determined by PET with the regional distribution of MAO A in human brain postmortem. MAO A has been reported to be relatively high in thalamus (29, 30) and in occipital cortex (31), with intermediate values in basal ganglia, frontal, and temporal cortices (29, 32) and a low concentration in cerebellum (31), though this comparison is limited by the lack of a single postmortem study measuring the same regions sampled by PET.
MAO A inhibition would be predicted to spare norepinephrine and serotonin, both of which are MAO A-specific substrates, in addition to dopamine, which is a substrate for both MAO subtypes. The monoamines norepinephrine and serotonin are linked to mood as is evidenced by the effectiveness of the tricyclic antidepressants and selective serotonin reuptake inhibitors in the treatment of depression (12). Since MAO A inhibitors are also effective antidepressants, the inhibition of human brain MAO A by smoke may contribute to the difficulty of smoking cessation in depression (2). More specifically, withdrawal from cigarettes would not only represent withdrawal from nicotine but also withdrawal from MAO A inhibitory substances in smoke.
A question that remains is the extent to which the level of MAO A inhibition achieved by cigarette smoke is clinically significant. Estimates of the degree to which MAO A needs to be inhibited for clinical improvement vary from 20% to 80% (as assessed from peripheral measures of monoamine metabolites) (5, 33, 34). The level of MAO A inhibition which we measured in smokers (28%) is in the low range of these values (assuming that peripheral measures of monoamine metabolites reflect brain MAO A levels). Since the time interval between the last cigarette and the PET scan averaged 2.7 h, which is longer than the typical recycle time of 1 h or less for the smoker, it is possible that the degree of inhibition was underestimated in our study. However, future studies are required to determine if the level of MAO A inhibition achieved with chronic exposure to cigarette smoke is associated with antidepressant effects.
We recently reported that MAO B was inhibited by about 40% in smokers relative to nonsmokers and former smokers (13). This finding, coupled with the results of the present study, raises the question of whether chronic, partial reduction of both MAO A and B by smoke enhances neurotransmitter activity. Studies in animals have reported that the simultaneous inhibition of both MAO subtypes produces large increases in serotonin outflow (35). Since both MAO A and B break down dopamine, their simultaneous inhibition by smoke may combine to enhance brain dopamine which has been implicated in reward and reinforcement (36). This may enhance the behavioral and addictive properties of nicotine and other substances of abuse by preventing the breakdown of dopamine and other neurotransmitters which are released by abused substances (37). This may be relevant to the comorbidity between smoking and alcoholism and the addiction to other substances (38).
Though MAO B inhibition by smoke has been discussed as a mechanism which may account for the decreased risk of Parkinson disease in smokers (39), MAO A may also play a role in the link between Parkinson disease and smoking because it is compartmentalized within dopaminergic neurons. This may place dopaminergic neurons at risk for damage from reactive oxygen species resulting from MAO A generated hydrogen peroxide (40).
Though this study only measured the effects of cigarette smoke on brain MAO A, it is possible that smoke may also inhibit MAO A in peripheral organs. MAO A inhibition in the liver is of particular importance because of its role in breaking down vasoactive amines associated with hypertension. Future studies are needed to determine whether liver MAO A is inhibited by cigarette smoke and its potential contribution to toxicity associated with cigarette smoke.
In conclusion, we report here the first direct observation in the human brain that tobacco smoke exposure is associated with a reduction in MAO A. Since MAO A breaks down monoamines linked to mood, this study suggests that MAO A inhibition needs to be considered as a link between tobacco smoke exposure and depression. It also provides support for a recent trial using MAO inhibitor therapy for smoking cessation (41) Even though this study suggests a mechanism by which smoking may provide relief in depressed individuals, the adverse effects of smoking are overwhelming (42). Thus the challenge remains to understand the neurochemical effects of smoke that contribute to smoking behavior and epidemiology and to use this knowledge to develop better therapies for smoking cessation, particular in that subgroup of individuals who consistently relapse.
Acknowledgments
We are grateful to Robert Carciello, Richard Ferrieri, Donald Warner, Payton King, Noelwah Netusil, and Carol Redvanly for their advice and assistance and to the subjects who volunteered for this study. This research was carried out at Brookhaven National Laboratory under Contract DE-AC02-76CH00016 with the U.S. Department of Energy and supported by its Office of Health and Environmental Research and also by the National Institutes of Health (National Institute of Neurological Diseases and Stroke, Grant NS 15380, and National Institute on Drug Abuse, Grant DA 06891).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: MAO A and B, monoamine oxidase A and B; PET, positron-emission tomography.
References
- 1.Wald N J, Hackshaw A K. Br Med Bull. 1996;52:3–11. doi: 10.1093/oxfordjournals.bmb.a011530. [DOI] [PubMed] [Google Scholar]
- 2.Glassman A H, Helzer J E, Covey L S, Cottler L B, Stetner F, Tipp J E, Johnson J. J Am Med Assoc. 1990;264:1546–1549. [PubMed] [Google Scholar]
- 3.Breslau N, Kilbey M, Andreski P. Arch Gen Psychiatry. 1993;50:31–35. doi: 10.1001/archpsyc.1993.01820130033006. [DOI] [PubMed] [Google Scholar]
- 4.Boulton A A, Yu P H, Tipton K. Lancet. 1988;i:114–115. doi: 10.1016/s0140-6736(88)90308-x. [DOI] [PubMed] [Google Scholar]
- 5.Berlin I, Said S, Spreus-Varoquaux O, Olivares R, Launay J-M, Puech A J. Biol Psychiatry. 1995;38:756–761. doi: 10.1016/0006-3223(95)00084-4. [DOI] [PubMed] [Google Scholar]
- 6.Selikoff I J, Robitzek E H, Ornstein G G. J Am Med Assoc. 1952;150:973–980. doi: 10.1001/jama.1952.03680100015006. [DOI] [PubMed] [Google Scholar]
- 7.Zeller E A, Barsky J, Berman E R. J Biol Chem. 1955;214:267–274. [PubMed] [Google Scholar]
- 8.Berry M D, Juorio A V, Paterson I A. Prog Neurobiol. 1994;42:375–391. doi: 10.1016/0301-0082(94)90081-7. [DOI] [PubMed] [Google Scholar]
- 9.Bach A W J, Lin N C, Johnson D L, Abell C W, Bembenek M E, Kwan S-W, Seeburg P H, Shih J C. Proc Natl Acad Sci USA. 1988;85:4934–4938. doi: 10.1073/pnas.85.13.4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riederer P, Konradi C, Schay V, Kienzl E, Birkmayer G, Danielczyk W, Sofic E, Youdim M B H. Adv Neurol. 1986;45:111–118. [PubMed] [Google Scholar]
- 11.Glover V, Elsworth J D, Sandler M. J Neural Transm Suppl. 1980;16:163–172. doi: 10.1007/978-3-7091-8582-7_18. [DOI] [PubMed] [Google Scholar]
- 12.Caldecott-Hazard S, Schneider L S. Synapse. 1992;10:141–168. doi: 10.1002/syn.890100209. [DOI] [PubMed] [Google Scholar]
- 13.Fowler J S, Wang G-J, Volkow N D, Pappas N, Logan J, MacGregor R R, Alexoff D, Wolf A P, Warner D, Cilento R, Zezulkova I. Nature (London) 1996;379:733–736. doi: 10.1038/379733a0. [DOI] [PubMed] [Google Scholar]
- 14.Pavlin R, Sket D. Farm Vestn. 1993;44:185–192. [Google Scholar]
- 15.Yu P H, Boulton A A. Life Sci. 1987;41:675–682. doi: 10.1016/0024-3205(87)90446-2. [DOI] [PubMed] [Google Scholar]
- 16.Carr L A, Basham J K. Life Sci. 1991;48:1173–1177. doi: 10.1016/0024-3205(91)90455-k. [DOI] [PubMed] [Google Scholar]
- 17.MacGregor R R, Halldin C, Fowler J S, Wolf A P, Arnett C D, Langström B, Alexoff D. Biochem Pharmacol. 1985;34:3207–3210. doi: 10.1016/0006-2952(85)90173-x. [DOI] [PubMed] [Google Scholar]
- 18.Fowler J S, MacGregor R R, Wolf A P, Arnett C D, Dewey S L, Schlyer D, Christman D, Logan J, Smith M, Sachs H, Aquilonius S M, Bjurling P, Halldin C, Hartwig P, Leenders K L, Lundquist H, Oreland L, Stalnacke C-G, Langström B. Science. 1987;235:481–485. doi: 10.1126/science.3099392. [DOI] [PubMed] [Google Scholar]
- 19.Fowler J S, Wang G-J, Logan J, Xie S, Volkow N D, MacGregor R R, Schlyer D J, Pappas N, Alexoff D L, Patlak C, Wolf A P. J Nucl Med. 1995;36:1255–1262. [PubMed] [Google Scholar]
- 20.MacGregor R R, Fowler J S, Wolf A P, Langström B, Halldin C. J Labelled Compd Radiopharm. 1988;25:1–9. [Google Scholar]
- 21.Matsui T, Hirano A. An Atlas of the Human Brain for Computerized Tomography. Stuttgart, Germany: Gustav Fischer; 1978. [Google Scholar]
- 22.Koeppe R A, Holthoff A, Frey K A, Kilbourn M R, Kuhl D A. J Cerebr Blood Flow Metab. 1991;111:735–744. doi: 10.1038/jcbfm.1991.130. [DOI] [PubMed] [Google Scholar]
- 23.Logan J, Dewey S L, Wolf A P, Fowler J S, Brodie J D, Angrist B, Volkow N D, Gatley S J. Synapse. 1991;9:195–207. doi: 10.1002/syn.890090306. [DOI] [PubMed] [Google Scholar]
- 24.Fowler J S, Volkow N D, Logan J, Schlyer D J, MacGregor R R, Wang G-J, Wolf A P, Pappas N, Alexoff D, Shea C, Gatley S J, Dorflinger E, Yoo K, Morawsky L, Fazzini E. Neurology. 1993;43:1984–1992. doi: 10.1212/wnl.43.10.1984. [DOI] [PubMed] [Google Scholar]
- 25.Patlak C, Fenstermacher J D, Blasberg R G. J Cerebr Blood Flow Metab. 1983;3:1–7. doi: 10.1038/jcbfm.1983.1. [DOI] [PubMed] [Google Scholar]
- 26.Blomqvist G. J Cerebr Blood Flow Metab. 1984;4:629–632. doi: 10.1038/jcbfm.1984.89. [DOI] [PubMed] [Google Scholar]
- 27.Walpole R E, Myers R H. Probability and Statistics for Engineers and Scientists. 2nd Ed. New York: Macmillan; 1978. pp. 256–259. [Google Scholar]
- 28.Esposito G, Van Horn J D, Weinberger D R, Berman K F. J Nucl Med. 1996;37:559–564. [PubMed] [Google Scholar]
- 29.Oreland L, Arai Y, Stenstrom A, Fowler C J. Mod Probl Pharmacopsychiatry. 1983;19:246–254. doi: 10.1159/000407521. [DOI] [PubMed] [Google Scholar]
- 30.Rao V L R, Giguere J-F, Layrargues G P, Butterworth R F. Brain Res. 1993;621:349–352. doi: 10.1016/0006-8993(93)90126-8. [DOI] [PubMed] [Google Scholar]
- 31.Glover V, Elsworth J D, Sandler M. J Neural Transm Suppl. 1980;16:163–172. doi: 10.1007/978-3-7091-8582-7_18. [DOI] [PubMed] [Google Scholar]
- 32.Saura J, Bleuel Z, Ulrich J, Mendelowitsch A, Chen K, Shih J C, Malherbe P, Da Prada M, Richards J G. Neuroscience. 1996;70:755–774. doi: 10.1016/s0306-4522(96)83013-2. [DOI] [PubMed] [Google Scholar]
- 33.McDaniel K. Clin Neuropharmacol. 1986;9:207–234. doi: 10.1097/00002826-198606000-00001. [DOI] [PubMed] [Google Scholar]
- 34.Berlin I, Zimmer R, Thiede H-M, Payan C, Hergueta T, Robin L, Puech A J. Br J Clin Pharmacol. 1990;30:805–816. doi: 10.1111/j.1365-2125.1990.tb05445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Celada P, Bel N, Artigas F. J Neural Transm Suppl. 1994;41:357–363. doi: 10.1007/978-3-7091-9324-2_47. [DOI] [PubMed] [Google Scholar]
- 36.Di Chiara G, Imperato A. Proc Natl Acad Sci USA. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pontieri F E, Tanda G, Orzi F, Di Chiara G. Nature (London) 1996;382:255–257. doi: 10.1038/382255a0. [DOI] [PubMed] [Google Scholar]
- 38.Henningfield J E, Clayton R, Pollen W. Br J Addict. 1990;85:279–292. doi: 10.1111/j.1360-0443.1990.tb03084.x. [DOI] [PubMed] [Google Scholar]
- 39.Morens D M, Grandinetti A, Reed M D, White L R, Ross G W. Neurology. 1995;45:1041–1051. doi: 10.1212/wnl.45.6.1041. [DOI] [PubMed] [Google Scholar]
- 40.Olanow C W. Trends Neurosci. 1993;16:439–444. doi: 10.1016/0166-2236(93)90070-3. [DOI] [PubMed] [Google Scholar]
- 41.Berlin I, Said S, Spreus-Varquaux A. Clin Pharmacol Ther. 1995;58:444–452. doi: 10.1016/0009-9236(95)90058-6. [DOI] [PubMed] [Google Scholar]
- 42.Henningfield J E, Schuh L M, Jarvik M E. In: Psychopharmacology, The Fourth Generation of Progress. Bloom F E, Kupfer D J, editors. New York: Raven; 1995. pp. 1715–1729. [Google Scholar]