

Abstract
By transfecting fibroblast cells with an HIV-1-MN molecular clone with a deletion of the major packaging sequence, we have developed a stable HIV-1 packaging cell line, Ψ422. Ψ422 cells form syncytia with CD4-positive cells, correctly express HIV-1 structural proteins, and produce a large amount of mature particles with normal reverse transcriptase activity. Yet these particles, in which RNA was not detected by reverse transcriptase–PCR, are not infectious. When stably transfected with an HIV-1-based retroviral vector, the Ψ422 cell line produces virions capable of transducing CD4-positive cells with high efficiency (up to 105 cells per ml). The availability of this stable noninfectious HIV-1 packaging cell line capable of generating high-titer HIV-1 vectors represents a new step toward the use of an HIV-1 delivery system in gene therapy.
Keywords: gene therapy, AIDS, gene delivery system, retroviral vector, intracellular immunization
Gene therapeutics is the latest addition to the armamentarium for AIDS treatment (1). It can target viral RNAs (ribozymes, antisense RNAs), viral proteins (RNA decoys, trans-dominant viral proteins, intracellular single-chain antibodies, soluble CD4), infectible cells (suicide genes), or the immune system (in vivo immunization). Yet these various possibilities are hampered by the limitations of the delivery systems currently used in gene therapy. For instance, the extensively used murine retroviral vectors transduce human peripheral blood lymphocytes poorly, and they fail to transduce nondividing cells such as monocytes/macrophages, which are known to be reservoirs for HIV. An appealing alternative would be to utilize HIV-based delivery systems, which would ensure optimal cell targeting and intracellular colocalization of target and effector molecules. In addition, HIV-derived vectors would be packaged by the wild-type virions of the patient in vivo and thereby be rescued and disseminated to a larger pool of potentially HIV-infectible cells. Finally, some of the regulatory elements in the vector [e.g., trans-activation response element (TAR), Rev-responsive element (RRE), and packaging signal sequences] would themselves be antagonistic to HIV replication.
Most of the efforts to develop HIV-based delivery systems have so far resulted in extremely low transduction efficiencies (2, 3, 4, 5). We now report a new packaging cell line, which when transfected with an HIV-based vector, enables the transduction of CD4-positive (CD4+) cells with high efficiency.
METHODS
Packaging Plasmid Construction.
We inserted the HIV-1-MN-ST.1 genome (6) into the commercial PCR II vector (Invitrogen). To delete the major packaging site (Fig. 1), the first 743 nucleotides of the HIV-1-MN long terminal repeat (LTR) [nucleotide numbering according to Myers et al. (7)] were amplified by PCR, introducing an AscI restriction site at the 3′ end of the PCR product by using the oligomers 5′-TGGATGGGTTAATTTACTCCC-3′ and 5′-GGCGCGCCTCACCAGTCGCCGCCCC-3′. This PCR product was cloned in the PCR II vector, which contains a unique ApaI restriction site (plasmid pLTR, Fig. 2). Then a gag-pol fragment (from nucleotide 1 in gag to nucleotide 360 in pol) was amplified, introducing an AscI restriction site at the 5′ end of the PCR fragment by using the oligomers 5′-GGGGCGCGCCGAGAGAGATGGGGTGCGAGAGCGTCGG-3′ and 5′-CTGATCATACTGTCTTACTTTG-3′. The resulting PCR fragment, containing a unique ApaI restriction site, was inserted into the PCR II vector (plasmid pGAG). Plasmids pLTR and pGAG were digested by AscI and ApaI, and the AscI–ApaI fragment of pGAG, corresponding to the first 1230 bases of gag was inserted into the pLTR clone (plasmid pLTR/GAG). Thus 37 nucleotides, starting from 6 nucleotides downstream the 5′ major splice donor to 7 nucleotides upstream of the beginning of gag (Fig. 1) have been deleted in the pLTR/GAG construct. Finally, an ApaI–ApaI fragment of the parental pMN clone corresponding to the viral genome downstream from the ApaI site in gag was inserted into pLTR/GAG to generate the plasmid ΔΨ.
Figure 1.
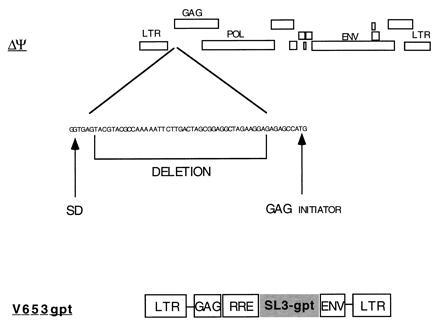
Structures of the ΔΨ packaging plasmid and the V653gpt vector. The positions of the major splice donor site (SD) and of the RRE are indicated.
Figure 2.
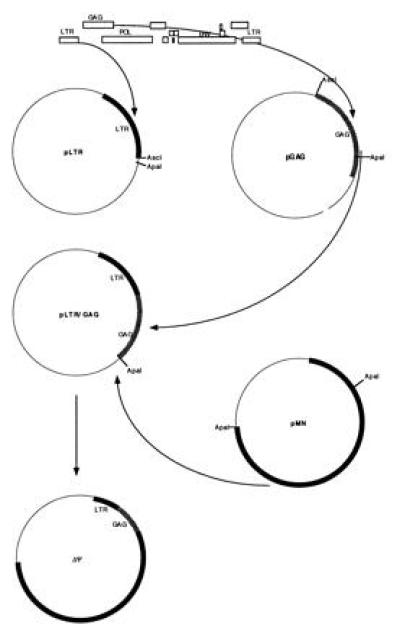
Diagram of the construction of the ΔΨ packaging plasmid. To delete the major packaging signal of HIV, we have subcloned the LTR (plasmid pLTR) and the gag gene (plasmid pGAG), leaving a 37-base gap between these two fragments. These two fragments have been ligated together (plasmid pLTR/GAG) and with the rest of the HIV genome (plasmid ΔΨ).
Vector Construction.
The vector V653gpt (Fig. 1) was obtained by replacement of the neomycin phosphotransferase (neor) gene on the previously described V653RSN plasmid (8) with the xanthine–guanine phosphoribosyltransferase (gpt) gene. The gpt gene was amplified by PCR, introducing an XhoI site at the 5′ end and a SalI site at the 3′ end of the PCR product by using the oligomers 5′-CCGCTCGAGATGAGCGAAAAATACATCGTC-3′ and 5′-CGGTCGACTTAGCGGCCGCGACCGGAGATTGGCGGG-3′. V653RSN was digested with SalI, which recognizes two sites in the sequences flanking the neor gene, and ligated with the PCR product digested with XhoI and SalI.
Transfection.
To generate the HIV-1 packaging cell line, HeLa cells were transfected by the calcium phosphate method as follows: A subconfluent HeLa cell culture in a six-well plate (Costar) was transfected with linearized and calcium phosphate-precipitated plasmid ΔΨ (10 μg) and plasmid SV-NEO, which contains the neor gene driven by the simian virus 40 promotor (0.6 μg), in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum (FCS), antibiotics, and glutamine (DMEM/10% FCS).
After 18 hr, wells were washed with Dulbecco’s phosphate-buffered saline (PBS), pH 7.8, incubated for 2 min at 20°C with 15% (vol/vol) glycerol solution in Hepes-buffered saline (50 mM Hepes, pH 7.1/280 mM NaCl/1.5 mM Na2HPO4), washed twice with PBS, and cultured in DMEM/10% FCS. At day 3, G418 (GIBCO/BRL) was added to the cell culture at 500 μg/ml. Ψ422 cells were transfected with 10 μg of V653gpt by the same method. Mycophenolic acid-resistant cells (g cells) were selected in culture for stable production of vector.
Protein Labeling and Immunoprecipitation.
Ten million cells were washed with PBS, incubated 1 hr in cysteine-free DMEM/10% FCS, and then labeled for 5 hr by the addition of 10 μl of l-[35S]cysteine [11 mCi (1 mCi = 37 MBq), 0.0102 mM]. The cells were washed again with PBS and lysed in 2 ml of 5 mM Tris·HCl, pH 8.0/150 mM NaCl/10 mM EDTA/0.1% SDS/1 mM phenylmethanesulfonyl fluoride/1% Triton X-100/1% deoxycholic acid. After a 1-hr incubation at 4°C, the lysate was centrifuged for 30 min at 14,000 rpm in an Eppendorf 5415 C centrifuge. One milliliter of supernatant was added to 6 μg of staphylococcal protein A-Sepharose (CL-4B, Pharmacia LKB) that had been preincubated with 10 μl of a pool of human HIV+ plasma. After 18 hr of incubation at 4°C, the Sepharose was washed, boiled for 10 min, and centrifuged. Twenty-five microliters of the supernatant was then subjected to electrophoresis on an SDS/polyacrylamide gel. After electrophoresis, the gel was dried and autoradiographed.
Reverse Transcriptase (RT) Activity Assay.
To measure viral RT activity, culture supernatant (0.50 ml) was mixed with 0.24 ml of 30% (wt/wt) polyethylene glycol and 20 μl of 4 M NaCl, and centrifuged at 14,000 rpm for 30 min. The pellet was resuspended in 10 μl of 10 mM Tris·HCl, pH 7.8/100 mM NaCl/1 mM EDTA/0.1% Triton X-100, and 40 μl of a mixture containing 62.5 mM Tris·HCl at pH 7.8, 25 mM MgCl2, 25 mM KCl, 2.5 mM dithiothreitol, 31.25 μg/ml poly(rA)·oligo(dT)12–18 (Pharmacia), and 6.25 μM [3H]dTTP (specific activity 10 Ci/mmol). After a 1-hr incubation at 37°C the mixture was spotted on DE81 paper (Whatman), air-dried, washed three times with 5% sodium pyrophosphate and twice more with water, and air-dried again. Radioactivity was measured by a scintillation spectrometer (Beckman).
RT-PCR Assay.
Before HIV RNA detection, cell culture supernatant was treated with DNase I (Boehringer Mannheim) for 1 hr (500 μl containing 10 units of enzyme and 50 μl of 200 mM Tris·HCl, pH 8.3/500 mM KCl/25 mM MgCl2/1 mg of bovine serum albumin per ml). Then 500 μl of 6 M guanidine thiocyanate/50 mM Tris·HCl, pH 8.3, and 5 μl of glass milk (GeneClean II, Bio 101) was added, and the mixture was rotated for 90 min at 20°C. After three washings, the RNA in the pellet was air-dried and eluted with 5 μl of water for 3 min at 50°C. To this 5 μl of RNA, 2 units of Tth DNA polymerase (Boehringer Mannheim) in 1 μl of 10× RT buffer (Boehringer Mannheim), 0.8 μg of HIV gag oligonucleotide SK39, and 1 μl containing each dNTP at 2 mM were added. The RNA in this mixture was then reverse transcribed (4.5 μl of the mixture and 0.5 μl of 9 mM MnCl2 incubated 20 min at 65°C) or not (4.5 μl of the mixture and 0.5 μl of water on ice for 20 min). Finally, for the PCR, 2 μl of 10× PCR buffer (Boehringer Mannheim), 2.5 μl of 7.5 mM EGTA, 12.5 μl of H2O, 0.8 μg of HIV gag oligonucleotide SK38, and 2 μl containing each dNTP at 2 mM were added. After 5 min at 94°C, 30 cycles were performed: denaturation for 1 min at 94°C, hybridization for 1 min at 52°C, and polymerization for 30 sec at 72°C. The amplified products were electrophoresed in a 2% agarose gel, blotted onto a GeneScreenPlus nylon membrane (NEN) by salt transfer, and hybridized with an α-32P-labeled (Random Primed DNA-labeling kit; Boehringer Mannheim) HIV-1 gag fragment. Labeled RT-PCR fragments were visualized by autoradiography.
p24 Assay.
HIV-1 p24 concentration was determined in the culture supernatants by using a commercial ELISA kit (Coulter).
Infectivity Assay.
To determine the infectivity of HIV-1 virions, serial dilutions of culture supernatants were incubated in quadruplicate with 4 × 104 Molt4/8 cells in 200 μl of RPMI medium 1640/10% FCS, cultured for 24 days, washed, and then cultured for 4 additional days. The percentage of cultures positive for HIV-1 p24 expression was then determined for each viral input. At day 28, PCR was also carried out to detect HIV-1 gag sequences. Molt4/8 cells (2 × 104) were resuspended in 25 μl of 100 mM KCl/10 mM Tris·HCl, pH 8.3/2.5 mM MgCl2, lysed in 25 μl of 10 mM Tris·HCl, pH 8.3/2.5 mM MgCl2/1% Tween 20/1% Nonidet P-40, and treated with proteinase K. To the cell lysate 0.4 μg of the gag oligomers SK38 (gag position 1543–1570) and SK39 (gag position 1630–1657), 2.5 μl containing each dNTP at 2 mM, 2.5 μl of 25 mM MgCl2, 0.5 unit of Taq DNA polymerase, 2.5 μl of 10× PCR buffer (Promega), and water for a final volume of 25 μl were added. After 5 min at 94°C, 35 cycles were performed: denaturation for 1 min at 94°C, hybridization for 1 min at 52°C, and polymerization for 30 sec at 72°C. As a positive control, PCR was performed on the same cellular extracts, using oligomers specific for β-actin: 5′-TACATGGCTGGGGTGTTGAA-3′ and 5′-TCCCACTCCTACGAGAGAA-3′. The PCR fragments were then analyzed as described for the RT-PCR assay.
Transduction.
Supernatants of mycophenolic acid-resistant (gpt-expressing) cells (called g cells) were filtered (pore size 0.22 μm), diluted 1:10, and added for 24 hr to 30% confluent HeLa-T4 cells cultivated in 1 ml of DMEM/10% FCS (six-well plate, Costar). Two days later, the HeLa-T4 cells were selected in DMEM supplemented with 10% dialyzed FCS, 250 μg/ml xanthine, 15 μg/ml hypoxanthine, 10 μg/ml thymidine, 2 μg/ml aminopterin, 25 μg/ml mycophenolic acid, and 150 μg/ml l-glutamine. After 4 weeks, resistant cells were counted under an inverted optical microscope.
Southern Blotting.
Genomic DNA (10 μg) extracted by using the QIAamp blood kit (Qiagen, Chatsworth, CA), was digested overnight with 25 units of the restriction enzyme DraIII, electrophoresed on a 0.7% agarose gel, blotted onto a GeneScreenPlus nylon membrane by salt transfer, and hybridized with an α-32P-labeled gpt fragment.
PCR Detection of the Transduced Vector.
PCR was performed on 200 ng of DNA from transduced HeLa-T4 cells or Ψ422 cells (negative control), or on 2 pg of V653gpt plasmid mixed with 200 ng of DNA from HeLa-T4 cells (positive control) as described for the infectivity assay. The LTR-RRE fragment of the vector was amplified by using the oligomers 5′-TTTTCGCGAGCGGCCGCCGGAAGGGCTAATTCACTCC-3′ and 5′-GGTATCTTTCCACAGCTAGG-3′; the RRE-gpt fragment, by using 5′-GGAGCTTTGTTCCTTGGG-3′ and 5′-ATCAACCAGCGGACGACCAG-3′; and the gpt-LTR fragment, by using 5′-AGCTACGATCACGACAACCA-3′ and 5′-TTTTCGCGAGCGGCCGCTGCTAGAGATTTCCACACTG-3′. Hybridization was performed for 1 min at 60°C, and polymerization was for 2 min and 30 sec at 72°C. The PCR fragments were then analyzed as described for the RT-PCR assay, the LTR-RRE and the gpt-LTR fragments being hybridized with an HIV-LTR probe, and the RRE-gpt fragment with a gpt probe.
RESULTS
Design of an HIV Packaging Cell Line.
We have chosen the HIV-1-MN-ST.1 molecular clone to make a packaging cell line because of its efficiency of infection in both monocytes and T cells (6). The packaging cell expression vector ΔΨ was constructed by deleting 37 nucleotides between the major splice donor site and the beginning of the gag gene (Fig. 1), a sequence within the previously defined major packaging site for HIV-1 (9, 10, 11, 12, 13). For this deletion, PCR fragments derived from the LTR and gag were ligated first to each other and subsequently with the rest of the viral genome to generate the ΔΨ clone (see Methods and Fig. 2). HeLa cells were cotransfected with ΔΨ and a plasmid containing the neor gene and were cultured in the presence of neomycin (G418). A G418-resistant clone, Ψ422, which produced 20 ng/ml of p24Gag antigen was isolated and further analyzed. This clone was able to induce the formation of syncytia after an overnight coculture with uninfected CD4+ human T cells Molt4/8 (data not shown). To determine if HIV-1 structural proteins were correctly expressed in the Ψ422 cells, we metabolically labeled the cells and immunoprecipitated the viral proteins in the cell lysate. As shown in Fig. 3, Ψ422 cells synthesize the major HIV-1 structural proteins: the envelope precursor gp160 and the external envelope glycoprotein gp120, and the p55 Gag immature protein and its p25 subproduct. Electron microscopy (Fig. 4) showed the production of a large amount of virions by the Ψ422 cell line, including many mature particles. These particles have RT specific activity (4947 cpm/ml per 50 ng of p24) comparable to that of wild-type MN virions (5364 cpm/ml per 50 ng of p24). However, these particles are not infectious, as shown in Fig. 5. CD4+ T cells Molt4/8 exposed to the equivalent of 1 ng of p24 of Ψ422 virions neither produced any Gag protein (Fig. 5A) nor expressed any gag sequence (Fig. 5B) 28 days later. In the same experiment, HIV production was detected in cell cultures exposed to 0.1 pg of wild-type HIV-1-MN virions. The lack of infectivity of the Ψ422 viral particles correlated with a drastic reduction in the RNA content in these particles (Fig. 6). We failed to detect viral RNA by RT-PCR in 140 pg of p24 equivalent of Ψ422 virions, whereas HIV RNA was detected in 35 pg of p24 equivalent of wild-type MN virions. The Ψ422 cell line has been cultured for several months, stably producing noninfectious virions.
Figure 3.

HIV proteins in Ψ422 cells. 35S-labeled viral proteins from HeLa cells (negative control, lane 1), HIV-1-MN-infected HeLa-T4 cells (positive control, lane 2), or Ψ422 cells (lane 3) were immunoprecipitated with a pool of AIDS sera and resolved by electrophoresis on an SDS/polyacrylamide gel. Positions of molecular mass markers are indicated on the right.
Figure 4.
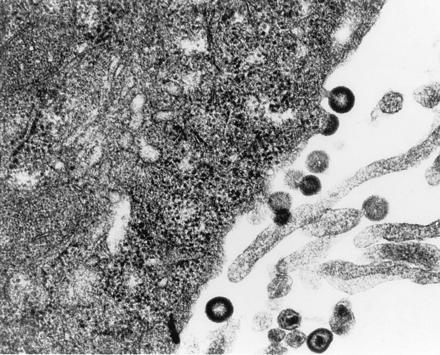
Electron micrograph of Ψ422 cells showing viral particles with normal lentivirus morphology. (×29,600.)
Figure 5.
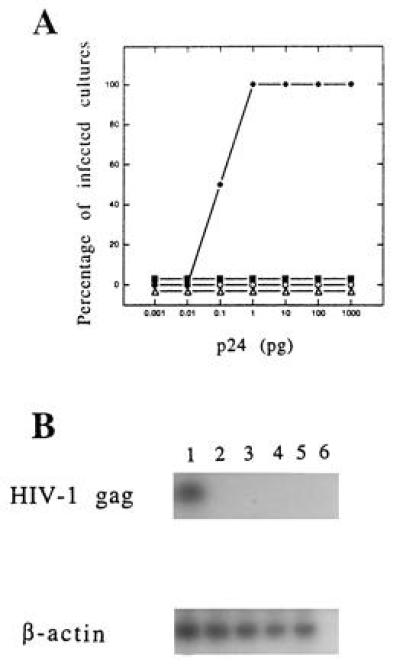
Lack of infectivity of the Ψ422 and g cell line supernatants. (A) Quadruplicate cultures of Molt4/8 cells were exposed to various amounts (0.001 to 1000 pg of p24 antigen) of wild-type HIV-1-MN (♦) or mutant (Ψ422, ▪; gV, ○; gZ, ▵) virions. At day 28 cells were tested for HIV-1 infection by p24 capture assay. The percentage of cultures which were HIV p24-positive is represented for each viral input. (B) In parallel, cells exposed to 0.1 pg of HIV-1-MN (lane 1) or to 1000 pg of Ψ422 (lane 2), gV (lane 3), or gZ (lane 4) virions for 28 days were lysed and submitted to PCR using gag-specific oligomers. A 114-nucleotide HIV-1 gag sequence was amplified only in cells infected with wild-type HIV-1-MN (Upper). As a positive control the β-actin gene was detected in the same cell lysates by PCR using specific oligomers (Lower). PCR was performed on noninfected Molt4/8 cells (lane 5) and on buffer (lane 6) as negative controls.
Figure 6.

Nucleic acid content of viral particles. RNA was extracted from various amounts (140, 35, or 8 pg of p24 antigen) of wild-type HIV-1-MN or Ψ422 virions, amplified by PCR with or without prior reverse transcription, electrophoresed on an agarose gel, blotted, and hybridized with an HIV-1 specific-probe.
Use of Ψ422 Cell Line for Gene Transfer.
In addition to the main Ψ sequence encompassing the major splice donor site and the beginning of gag, other sequences within the HIV-1 genome could play a role in the packaging of viral RNA. The first hundred nucleotides of the gag gene may be directly or indirectly involved in the binding of the viral RNA to the nucleocapsid of the virion (2, 8, 14, 15, 16). A stretch within the env gene, including the RRE, has been reported to also contain a packaging signal (3, 8). Thus, Parolin et al. (8) have designed an optimal HIV-1 vector, V653RSN, containing the first 653 nucleotides of gag and an RRE-containing fragment of env. We have replaced the neor gene in V653RSN with the gpt gene and transfected this new plasmid, V653gpt, into Ψ422 cells. gpt can serve as a positive selectable marker in media containing mycophenolic acid, an inhibitor of de novo synthesis of guanine monophosphate (17). Moreover, gpt can also serve as a negative selectable marker, or a suicide gene, when 8-thioxanthine is used as a counter-selective agent (18). Several mycophenolic acid-resistant cell lines (g lines) were isolated from the transfected Ψ422 cells. Since these cells presumably produced the packaged gpt vector, their filtered supernatants were used to transduce HeLa-T4 cells. Transduction efficiency was evaluated 4 weeks later by counting the mycophenolic acid-resistant HeLa-T4 cells. After this selection all the HeLa-T4 cells transduced with plain medium, Ψ422 cell supernatant, or V653gpt-transfected HeLa cell supernatant were dead. In contrast, up to 105 resistant cells were obtained after transduction with 1 ml of supernatants from two independent vector-producing cell lines (Table 1). The capacity of these two g cell lines, gV and gZ, to transfer mycophenolic acid resistance was dependent on the presence of the CD4 receptor on target cells. HeLa CD4− cells incubated with g cell supernatant died within 3 weeks in the presence of mycophenolic acid (Table 1).
Table 1.
Transduction capacity of g cell lines
| Cell supernatant | Target cell | Exp. | Titer, TU/ml |
|---|---|---|---|
| gV | HeLa-T4 | 1 | 1.2 × 104 |
| 2 | 1.0 × 104 | ||
| 3 | 4.6 × 104 | ||
| HeLa | 0 | ||
| gZ | HeLa-T4 | 1 | 1.0 × 104 |
| 2 | 1.3 × 104 | ||
| 3 | 1.4 × 105 | ||
| HeLa | 0 |
TU, transducing unit(s).
We next examined if replication-competent retroviruses could have arisen through recombination between viral sequences in the packaging cell line and the vector. Therefore, the infectivity of gV and gZ supernatants was tested on Molt4/8 cells as described for Ψ422 cell supernatant. Again, no trace of viral protein (Fig. 5A) or DNA (Fig. 5B) was detected after 28 days of culture. Even after addition of fresh Molt4/8 cells, the mutant virion cultures remained HIV-p24-negative 2 months later.
To determine if the transfer of the gpt gene was accomplished without rearrangement, the integrity of the V653gpt vector in the transduced cells was checked. First, genomic DNA extracted from transduced cells was digested with DraIII (a restriction enzyme able to cut only once in each LTR of the vector), blotted, and hybridized with a gpt-specific probe. Fig. 7A shows that within the digested cellular DNA a unique fragment contained the gpt gene and that this fragment has the expected size for the vector sequences flanked by the two DraIII restriction sites. To further assess the absence of any recombination, we used PCR as a more sensitive assay. Three overlapping fragments of the vector were amplified from DNA of transduced cells, blotted, and hybridized with specific probes. PCR was also carried out on Ψ422 cells as a negative control and on V653gpt plasmid as a positive control. The size and the specificity of the PCR products shown in Fig. 7 demonstrate that the V653gpt vector was transferred without detectable modification.
Figure 7.
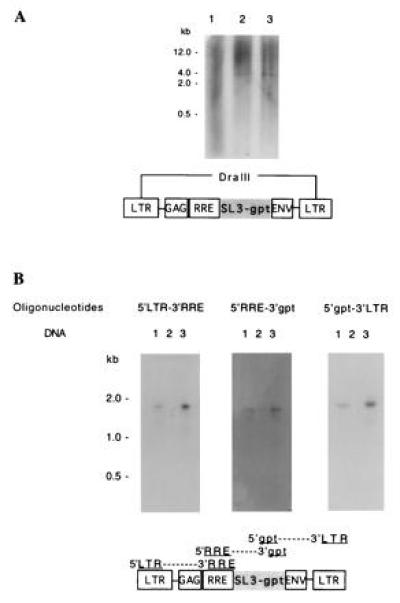
Analysis of the V653gpt vector in the transduced cells. (A) Total DNA from HeLa-T4 cells (negative control, lane 1), transduced HeLa-T4 cells (lane 2), and plasmid V653gpt mixed with HeLa-T4 cellular DNA (positive control, lane 3) were analyzed by Southern blot hybridization after DraIII digestion. DraIII cuts the vector twice, releasing a 4-kb fragment as shown on the diagram. (B) PCRs using the three pairs of oligonucleotides shown on the V653gpt vector diagram were carried out on genomic DNA extracted from transduced HeLa-T4 cells (lanes 1), Ψ422 cells (negative control, lanes 2), or V653gpt plasmid (positive control, lanes 3). The PCR fragments were separated by electrophoresis on a gel, blotted, and hybridized with an HIV-1 LTR probe (oligonucleotides 5′LTR-3′RRE and 5′gpt-3′LTR), or a gpt probe (oligonucleotides 5′RRE-3′gpt).
DISCUSSION
We have established a stable, noninfectious HIV-1 packaging cell line capable of producing relatively high-titer vectors. The vector preparations were free of replication-competent virus. This point is a major consideration for a gene delivery system ultimately designed for human use. Two reasons may explain the lack of infectivity of our vector stocks. First, the design of the deletion in the major Ψ sequence of HIV-1 may be crucial to avoid packaging and thereby infectivity. In a recent study based on the mapping of chemical- and RNase-accessible sites, coupled with computerized sequence analysis, Clever et al. (19) proposed a structure for Ψ that comprises four independent stem loops. Our ΔΨ plasmid contains a 37-nucleotide deletion in the Ψ sequence which destroys two of these four loops and therefore is less likely to retain packageability. Aldovini et al. (10) have made two other deletions within the Ψ sequence, also resulting in the disruption of the same two loops, and obtained noninfectious viral particles. On the other hand, two independent groups have deleted sequences corresponding only to the third of the four loops and observed some infectivity in their packaging plasmid (9, 11). The second reason may be technical. Most of the studies have so far been carried out by cotransfecting the packaging and the vector plasmids, which may increase the chance of recombination and resulting infectivity (2, 4, 8, 12). To further improve the safety of our system, we have recently replaced the HIV-1 vector with a similar HIV-2 vector. This HIV-2 vector was packaged into Ψ422 and was able to transfer the gpt gene into HeLa-T4 cells. In this new hybrid system the absence of any sequence similarity between the HIV-1 packaging plasmid and the HIV-2 vector provides additional safety (unpublished results).
Most of the previously reported HIV-1 packaging systems had low titers, ranging from 102 to 103 TU/ml, and/or resulted from transient transfections (2, 4, 5, 8, 20). A group reported a transduction efficiency of 2 × 104 TU/ml, but in this study the packaging system was infectious wild-type HIV-1 (3). The only study describing a stable cell line doubly transfected with the packaging plasmid and the vector reported transduction efficiency as low as 102 TU/ml (5). Therefore, our titers of up to 105 TU/ml are significantly higher than previous values and may be the result of at least two factors. First, most of the packaging virions described so far have been derived from the HXBc2 plasmid and appeared abnormal, with unprocessed p55Gag protein (5), disproportionately low RT activity (5, 9), and/or immature morphology revealed by electron microscopy (10, 11). Some of these deficiencies may stem from the fact that HXBc2 lacks a full complement of the viral accessory genes— e.g., the vpr gene is truncated (20). vpr has been shown to be important for cellular permissiveness to HIV-1 replication in vitro (21) and for infection of rhesus monkeys with simian immunodeficiency virus in vivo (22). Therefore HXBc2 clones additionally deleted in the major Ψ sequence may yield partially disabled virions. Second, the vector we used here contains all three RNA sequences that have been suggested to be involved in optimal HIV-1 packaging, whereas others may have used vectors devoid of gag and/or env sequences.
The titers presently achievable with HIV vectors are still 1–2 logs lower than those of murine retroviral vectors so widely used for ex vivo gene therapy trials. However, two advantages of HIV vectors more than compensate for the lower titers for treatment of HIV infection. First, the HIV envelope allows specific targeting to CD4+ T cells and to noncycling T cells, macrophages, and glial cells in vivo. Second, the vector can be rescued in vivo by helper HIV-1, resulting in an effective amplification of the vector in vivo. These features combined with further development efforts may make in vivo gene delivery a feasible goal.
The establishment of stable cell lines producing a high titer of fully characterized transducing particles is a major step forward in the utilization of HIV-derived constructs for gene therapy. In addition to applications for therapy against HIV infection, the ability of HIV vectors to target CD4+ lymphocytes as well as nondividing cells such as cells of the monocyte/macrophage lineages, may render this vector/packaging system useful for targeting other hematopoietic disorders. We have recently transduced human blood-derived macrophages with HIV vectors delivered through our packaging cell line (unpublished results). Moreover, the unique capacity of HIV to infect quiescent cells may be exploited by forming pseudotyped hybrid virions with broad envelope tropism to target other noncycling cells, including the primitive hematopoietic stem cells (23).
Acknowledgments
We are grateful to J. Sodroski for plasmid V653RSN and to D. Looney for electron microscopy. This work was supported in part by the National Institutes of Health Strategic Program of Innovative Research on AIDS Therapy awarded to F.W.-S. and the University of California at San Diego Center for AIDS Research. P.C. was supported by the Fogarty International Center, the French Ministère de la Recherche et de la Technologie and the Fondation pour la Recherche Médicale.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: RRE, Rev-responsive element; LTR, long terminal repeat; RT, reverse transcriptase; TU, transducing unit.
References
- 1.Yu M, Poeschla E, Wong-Staal F. Gene Ther. 1994;1:13–26. [PubMed] [Google Scholar]
- 2.Buchschacher G L, Panganiban A T. J Virol. 1992;66:2731–2739. doi: 10.1128/jvi.66.5.2731-2739.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Richardson J H, Child L A, Lever A M L. J Virol. 1993;67:3997–4005. doi: 10.1128/jvi.67.7.3997-4005.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rizvi T A, Panganiban A T. J Virol. 1993;67:2681–2688. doi: 10.1128/jvi.67.5.2681-2688.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carroll R, Lin J-T, Dacquel E J, Mosca J D, Burke D S, St. Louis D C. J Virol. 1994;68:6047–6051. doi: 10.1128/jvi.68.9.6047-6051.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lori F, Hall L, Lusso P, Popovic M, Marham P, Franchini G, Reitz M S., Jr J Virol. 1992;66:5553–5560. doi: 10.1128/jvi.66.9.5553-5560.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Myers G, Hahn B, Mellors J W, Henderson L E, Korber B, Jeang K-T, McCutchan F E, Pavlaki G N, editors. Human Retroviruses and AIDS: A Compilation and Analysis of Nucleic Acid and Amino Acid Sequences. Los Alamos, NM: Los Alamos National Laboratory; 1992. [Google Scholar]
- 8.Parolin C, Dorfman T, Palu G, Gottlinger H, Sodroski J. J Virol. 1994;68:3888–3895. doi: 10.1128/jvi.68.6.3888-3895.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lever A, Gottlinger H, Haseltine W, Sodroski J. J Virol. 1989;63:4085–4087. doi: 10.1128/jvi.63.9.4085-4087.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aldovini A, Young R A. J Virol. 1990;64:1920–1926. doi: 10.1128/jvi.64.5.1920-1926.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clavel F, Orenstein J. J Virol. 1990;64:5230–5234. doi: 10.1128/jvi.64.10.5230-5234.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poznansky M, Lever A, Bergeron L, Haseltine W, Sodroski J. J Virol. 1991;65:532–536. doi: 10.1128/jvi.65.1.532-536.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayashi T, Shioda T, Iwakura Y, Shibuta H. Virology. 1992;188:590–599. doi: 10.1016/0042-6822(92)90513-o. [DOI] [PubMed] [Google Scholar]
- 14.Luban J, Goff S P. J Virol. 1991;65:3203–3212. doi: 10.1128/jvi.65.6.3203-3212.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berkowitz R D, Luban J, Goff S P. J Virol. 1993;67:7190–7200. doi: 10.1128/jvi.67.12.7190-7200.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luban J, Goff S P. J Virol. 1994;68:3784–3793. doi: 10.1128/jvi.68.6.3784-3793.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mulligan R C, Berg P. Proc Natl Acad Sci USA. 1981;78:2072–2076. doi: 10.1073/pnas.78.4.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spring K J, Mattick J S, Don R H. Biochim Biophys Acta. 1994;1218:158–162. doi: 10.1016/0167-4781(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 19.Clever J, Sassetti C, Parslow T G. J Virol. 1995;69:2101–2109. doi: 10.1128/jvi.69.4.2101-2109.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ratner L, Fisher A, Jagodzinski L L, Mitsuya H, Liou R-S, Gallo R C, Wong-Staal F. AIDS Res Hum Retroviruses. 1987;3:57–69. doi: 10.1089/aid.1987.3.57. [DOI] [PubMed] [Google Scholar]
- 21.Levy D N, Refaeli Y, Weiner D B. J Virol. 1995;69:1243–1252. doi: 10.1128/jvi.69.2.1243-1252.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lang S M, Weeger M, Stahl-Hennig C, Coulibaly C, Hunsmann G, Muller J, Muller-Hermelink H, Fuchs D, Wachter H, Daniel M M, Desrosiers R C, Fleckenstein B. J Virol. 1993;67:902–912. doi: 10.1128/jvi.67.2.902-912.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage F H, Verma I M, Trono D. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]