

Abstract
More than 20 Synechococcus and Cyanobium isolates were obtained from central European subalpine lakes and sequenced for their 16S rRNA gene and part of the phycocyanin operon (cpc), specifically the intergenic spacer (IGS) between cpcB and cpcA, and corresponding flanking regions (cpcBA-IGS). Maximum-likelihood analyses revealed the existence of at least six to seven clusters of nonmarine picocyanobacteria within the picophytoplankton clade and support the conjecture of global dispersal for some closely related picocyanobacterial genotypes.
Due to their significance for global biogeochemical cycling, recent research has focused on deciphering the genotypic and phenotypic diversity of marine picocyanobacteria (17, 30, 38). Comparatively sparse information is available on the dispersal and diversity of freshwater picocyanobacteria, although their genotypic differences and ecological significance have been studied in great detail for some lakes (4, 35). Recently, Ernst et al. (10) suggested that strain clusters belonging to the nonmarine branch of the picophytoplankton clade sensu Urbach et al. (38) have undergone ecosystem-dependent adaptive radiations within several brackish, freshwater and saline (Antarctic) lake environments. This conclusion is an apparent contradiction of the common conjecture that many free-living microbial species have a global distribution because their small size and great abundance result in dispersal which is rarely (if ever) restricted by geographical boundaries (13, 14). This argument has been known for a long time and is commonly referred to as the “everything is everywhere” hypothesis (2).
It is at present difficult to test this hypothesis for picocyanobacteria, because the genetics and ecophysiology of most picocyanobacteria remain poorly understood, and, furthermore, there is often no simple correlation between genotypic and phenotypic diversity (10). The phylogenetic analysis of Ernst and colleagues (10) and their inferences on dispersal and evolution of freshwater picocyanobacteria were derived from a limited number of strains.
Based upon a refined analysis originating from a larger database (with >20 new rRNA gene sequences), we show that some closely related forms are widely dispersed and that taxon undersampling may have resulted in premature phylogenetic inferences in the analysis of Ernst et al. (10). The implications of our findings are discussed in relation to the description of (microbial) ecotypes adapted to environments where the potential for widespread dispersal is high.
Source, isolation, and maintenance of cultures.
To avoid culture bias resulting from the use of standard plating techniques—these may favor the isolation of, e.g., phycocyanin-rich strains (9), which comprise a small fraction of picocyanobacteria inhabiting subalpine lakes (42)—we used single-cell sorting (MW isolates [7]) and the dilution culture technique (MH isolates) to obtain isolates from the surface 20-m water column of the oligo-mesotrophic lakes Mondsee and Hallstättersee, both deep subalpine lakes located in Upper Austria. The nonaxenic isolates were maintained in BG11 (34) at 15°C and under low light (white light, 10 μmol quanta m−2 s−1, continuously supplied by Osram “cool-white” fluorescent tubes) (henceforth referred to as standard culture conditions).
Assignment to pigment groups.
A Zeiss Axioplan microscope was used to differentiate phycoerythrin (PE)- and phycocyanin (PC)-rich isolates based upon their epifluorescent characteristics under blue (Zeiss filter set 05) and green excitation (Zeiss filter set 14) (21, 43) in comparison to the reference strains BO 8801, BO 8807, and BO 8809 (11), grown under standard culture conditions. PE-rich isolates were characterized by their orange-red fluorescence under green excitation and their yellow-orange fluorescence under blue excitation. PC-rich isolates appeared purple-red or red at both green and blue excitation.
DNA isolation, PCR amplification, and sequencing.
Near-full-length 16S rRNA gene sequences were determined for 21 Synechococcus and Cyanobium isolates sensu Castenholtz (5). The less conserved intergenic spacer (IGS) between cpcB and cpcA, and corresponding flanking regions (henceforth referred to as cpcBA-IGS), were sequenced for 25 Synechococcus and Cyanobium isolates. These gene sequences have been targeted in previous phylogenetic studies of picocyanobacteria (29).
DNA was extracted from 10 ml of culture material using the FastDNA kit and FastPrep instrument (Bio 101). 16S rRNA gene and cpcBA-IGS sequences were amplified using cyanobacterium-specific primer pairs, 16S5′F (AGAGTTTGATCCTGGCTCAG) and B23S5′R (CTTCGCCTCTGTGTGCCTAGGT) (19, 32) and cpcBF(UFP) (TAGTGTAAAACGACGGCCAGTTGYYTKCGCGACATGGA) and cpcAR(URP) (TAGCAGGAAACAGCTATGACGTGGTGTARGGGAAYTT) (29), respectively. PCRs were done in 25-μl volumes, with final concentrations of reactants as follows: for the 16S rRNA gene PCR, 0.2 mM (each) deoxynucleoside triphosphate, 0.4 μM (each) primer, ca. 100 ng of template DNA, 1 mg of bovine serum albumin ml−1, 1.5 mM MgSO4, and 0.4 to 0.8 U of high-fidelity SuperTaq (Ambion) polymerase; for the cpcBA-IGS PCR, 0.3 mM (each) deoxynucleoside triphosphate, 0.5 μM (each) primer, 10 to 100 ng of template DNA, 2.5 mM MgCl2, and 0.5 U of Taq (Qiagen) polymerase. Cycling parameters are given in the work of Scheldeman et al. (32) (for 16S rRNA gene PCR) and Robertson et al. (29) (for cpcBA-IGS PCR). PCRs were performed in multiples, and products were pooled, purified using the QIAquick PCR purificaton kit (Qiagen), and then sequenced (VBC Genomics) bidirectionally.
Sequence alignment and phylogenetic analyses.
CLUSTALX (37) and the ARB (http://www.arb-home.de) automatic alignment tool (Fast Aligner, version 1.03) were used to produce working alignments of the cpcBA-IGS and 16S rRNA gene sequences, respectively. The final alignments were obtained by manual refinement, taking into account structural constraints (16S rRNA gene sequences: secondary structure models available from the Comparative RNA Web Site (http://www.rna.icmb.utexas.edu); cpcBA-IGS sequences: amino acid alignment given as Table 23 in the supplement to Bickel et al. (3). After the removal of hypervariable (50% conservation filter implemented in ARB using the filter by base frequency function; see Ludwig and Klenk [20] for a discussion on the use of filters in phylogenetic analysis) and other potentially misleading sites, the alignments used in phylogenetic analyses consisted of 1,383 (16S rRNA gene sequences) and 362 (cpcBA-IGS sequences) nucleotide positions. Pairwise nucleotide sequence identities were calculated from the full-length alignments using uncorrected distances and excluding sequences from putative coisolates (i.e., isolates originating from the same water sample and identical in both 16S rRNA gene and cpcBA-IGS sequence sets), poorly aligned columns (i.e., most of the IGS between cpcB and cpcA), and sites with ambiguous (i.e., codes M, R, W, Y, etc.) bases. Maximum-likelihood (12) trees were inferred with TREEFINDER (http://www.treefinder.de), assuming the GTR + Γ model of nucleotide substitution. For both the 16S rRNA gene and cpcBA-IGS data sets, GTR + Γ was the best-fit model of nucleotide substitution according to analyses performed with ModelTest (27).
Synechococcus PCC 6301 (formerly Anacystis nidulans) was chosen as the outgroup since it forms, together with PCC 7943 and PCC 7942, a paraphyletic group closely related to but well separated from the picophytoplankton clade sensu Urbach et al. (38) (10; N. D. Crosbie, unpublished analyses).
The cpcBA-IGS sequence set was examined for recombination events using a substitution method sensu Posada (26) as implemented in Geneconv 1.81 (S. A. Sawyer [http://www.math.wustl.edu/∼sawyer]). The global permutation P values based on BLAST-like global scores (10,000 replicates) smaller than 0.05 were considered as evidence of recombination. A multiple-comparison correction (Bonferroni) is already built into the P values. The default value of the parameter gscale (gscale = 0) was used.
Results of phylogenetic analyses.
Phylogenetic analyses of the 16S rRNA gene sequences revealed a high pairwise similarity for all freshwater picocyanobacteria (Fig. 1). Overall, the near-full-length 16S rRNA gene sequences obtained from 67 inland water picocyanobacterial isolates differed by a maximum of 5% in pairwise comparisons (average pairwise sequence identity, 98%; calculations excluded isolate PS-845 and isolates from Antarctic saline lakes). Our cluster designations (and their nomenclature) are based on the groups proposed originally by Robertson et al. (29), with modifications according to Ernst et al. (10).
FIG. 1.
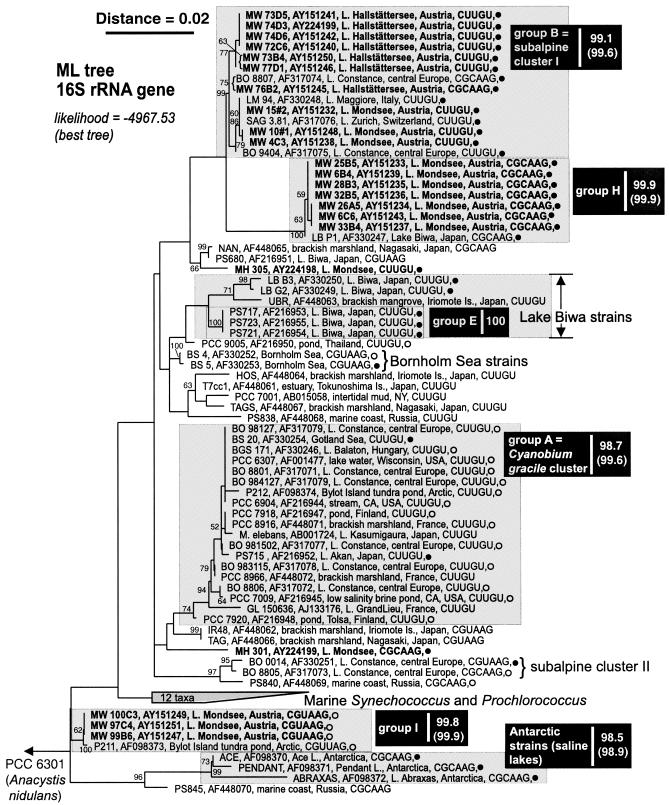
Maximum-likelihood tree of the picophytoplankton clade sensu Urbach et al. (38), inferred from 16S rRNA gene sequences (1,383 nt positions). Terminal branches display isolate and GenBank accession numbers (given in bold font for sequences determined in this study), isolation details (location, habitat), helix 49 tetraloop motif, and pigment group (•, PE rich; ○, PC rich). Numbers at nodes indicate the percent bootstrap frequency (1,000 replicates) obtained from neighbor-joining (31) trees calculated by TREECON for Windows (39) assuming a Jukes-Cantor model of nucleotide substitution. Bootstrap values of <50% are not shown. Minimum and mean (parentheses) pairwise percent similarities are shown on the right-hand side of each group label. Sources: results herein, references 10, 28, 29, and 40; Warwick Vincent, personal communication (isolation details for P211 and P212); and Watanabe, personal communication (isolation details for NAN, UBR, HOS, T7cc1, TAG, TAGS, IR48 PS-838, PS-840, and PS-845). The Ernst et al. (10) cluster designations subalpine cluster I, Lake Biwa strains, Bornholm Sea strains, Cyanobium gracile cluster, and subalpine cluster II are also provided. The outgroup was Synechococcus PCC 6301 (formerly Anacystis nidulans).
Many of our new PE-rich isolates from Lakes Mondsee and Hallstättersee clustered with isolates obtained from other subalpine lakes, falling into two groups, B (= subalpine cluster I) and H. The new group, H, consists of seven closely related (average pairwise sequence similarity for the 16S rRNA gene, 99.9%) isolates from L. Mondsee and one isolate from Japanese Lake Biwa. Additionally we propose the new group I, which contains three PC-rich isolates from L. Mondsee and one PC-rich isolate from an Arctic tundra pond. Isolates forming group H and those obtained from L. Hallstättersee exhibited a greater tendency to form cell aggregations under standard culture conditions. All isolates obtained in this study were coccoid with the exception of MH 305, MH 307, and MW 76B2, which formed rods. The latter isolate appears to be identical to BO 8807, which also exhibits a rod morphology (10).
A maximum-likelihood tree derived from the cpcBA-IGS gene sequences of 59 isolates, including 25 new Synechococcus and Cyanobium isolates from subalpine lakes, was constructed based upon an unambiguous alignment of 362 nucleotides (nt) (Fig. 2). The overall pattern was consistent with the 16S rRNA gene-based phylogeny (Fig. 1)—for example, the new groups H and I were evident in both trees—but demonstrates, however, that subalpine cluster I (10) is not exclusive for subalpine central European lakes (Fig. 2).
FIG. 2.

Maximum-likelihood tree of the picophytoplankton clade sensu Urbach et al. (38), inferred from cpcBA-IGS gene sequences (362 nt positions). Terminal branches display isolate and GenBank accession numbers (given in bold font for sequences determined in this study), isolation details (location, habitat), IGS length (number of nucleotides), and pigment group (•, PE rich; ○, PC rich). Numbers at nodes indicate the percent bootstrap frequency (1,000 replicates) obtained from neighbor-joining (31) trees calculated by TREECON for Windows (39) assuming a Jukes-Cantor model of nucleotide substitution. Bootstrap values of <50% are not shown. Minimum and mean (parentheses) pairwise percent similarities are shown on the right-hand side of each group label. Sources: results herein and reference 29. The Ernst et al. (10) cluster designations subalpine cluster I and Cyanobium gracile cluster are also provided. The outgroup was Synechococcus PCC 6301 (formerly Anacystis nidulans).
Isolates obtained from widely separated locations shared up to 99.87% identity in 16S rRNA gene sequences (e.g., the PC-rich isolates P211 and MW 100C3 differed in two nt positions; Fig. 1, bottom) and up to 99.72% in cpcBA-IGS sequences (e.g., the PE-rich isolates PS-714 and BO 8807 differed in one nucleotide position; Fig. 2, top).
As originally observed by Robertson et al. (29), the length of the IGS separating cpcB and cpcA exhibited a strong relationship with phylogenetic groups (Fig. 2). Additionally, two isolates (PS-727 and PS-729) having an IGS length inconsistent with their initial (i.e., in the work of Robertson et al. [29]) classification in group B, were clearly separated in our phylogenetic analyses of the larger cpcBA-IGS data set (cf. Robertson et al. [29]).
With respect to terminal branching patterns, the results suggest a high degree of congruence between 16S rRNA gene- and cpcBA-IGS-based phylogenies of the nonmarine members of the picophytoplankton clade, though with less well delineated subgroups in the more conserved 16S rRNA gene phylogeny (29; this study). Contrary to the observations of Ernst et al. (10) and in agreement with Robertson et al. (29), we found a moderate and strong correlation between phylogenetic clusters and pigment group assignment in the 16S rRNA gene- and cpcBA-IGS-based phylogenetic analyses, respectively. We confirm, however, that both pigment types (i.e., PE- or PC-rich) can be found in some closely related isolates (e.g., isolates forming group A).
In the 16S rRNA gene sequence set, a pattern suggesting motif exchange (e.g., compare the helix 49 motif pattern of isolates BO 8807 and MW 76B2 [group B] with the helix 49 motif pattern of group H isolates—see Fig. 1), involving the terminal loop of helix 49 and its closing base pair (reference 10 and results herein), could have resulted from horizontal transfer (the “simplified complexity hypothesis”; see reference 41) and/or from endogenous processes (15). From X-ray structures, helix 49 appears not to be in contact with any other molecules within the ribosome and belongs to the group of rRNA hairpins exhibiting variable length and variable loop sequences (16).
Deep-branching patterns were sensitive to hypervariable sites (i.e., those removed by a 50% conservation filter) and the inclusion or exclusion of the helix 49 tetraloop motif (6 nt positions in the alignment) in the 16S rRNA gene sequence matrix subjected to phylogenetic analyses. Notably, PE-rich Antarctic isolates obtained from marine-derived saline lakes (28) and PS-845 (isolation details suggest a marine origin; M. M. Watanabe, personal communication) clustered with PC-rich freshwater isolates obtained from the Arctic (Bylot Island ponds) and central Europe (Lakes Mondsee and Constance) when the motif was included (results not shown). When excluded (either separately or together with hypervariable sites), the Antarctic isolates clustered together with PS-845 at the base of the picophytoplankton clade (Fig. 1). Although we could find no evidence of recombination in the set of picocyanobacterial cpcBA-IGS sequences (P > 0.05), the potential for spurious phylogenies (18, 22) was minimized by restricting analyses to an alignment made almost entirely from the flanking regions. A similar approach was taken by Robertson et al. (29).
Global dispersal versus ecosystem-dependent radiation.
While certain groups of freshwater picocyanobacteria may have arisen in a specific ecosystem type (e.g., deep subalpine lakes), the frequency and widespread nature of microbial dispersal (13) would all but ensure that newly adapted strains and their progenitors are dispersed to widely separated locations. Given conditions suitable for growth, the population of “old” and “new” arrivals would increase, but bias originating from selective sampling of picocyanobacterial strains (for example, by fluorescence in situ hybridization, quantitative PCR, culture, or clone libraries) must be taken into account before safe conclusions regarding the geographical restriction of ecotypes sensu Cohan (6) can be made. Although our analyses confirm that most gene clusters do contain isolates originating with nearby locations (10), there is also evidence for widespread dispersal of some closely related taxa. This is illustrated by cluster B, the so-called subalpine cluster I (10), which contains isolates obtained from Irish Lough Neagh (isolate PS-714) and several Japanese lakes of very different type from the oligo-mesotrophic, deep subalpine lakes (e.g., Fig. 2). Lake Kasumigaura (from which PS-725 was isolated), for instance, is the second-largest hypertrophic lake in Japan, with a surface area of 220 km2 and a mean water depth of 4 m (36).
Prokaryote speciation is thought to have produced ecotypes recognizable as tightly clustered groups in sequence-based phylogenies (6, 23). Because phylogenetic analyses are sensitive, however, to the character and number of sequences included in the analysis and on the particular algorithm used to perform the clustering (25), robust ecotypes might be defined from a relatively small number of 16S rRNA gene sequences originating from a relatively small number of locations and environments. At the other extreme, the collection of a large number of more variable sequences [e.g., cpcBA-IGS (29; this study) or ITS sequences (10)] from a great many locations and environments will be needed. The present analysis suggests that there are at least seven clusters within the nonmarine picocyanobacteria forming the picophytoplankton clade sensu Urbach et al. (38). Whether these gene clusters should be given species rank remains debatable until more ecophysiological evidence on representative strains has been accumulated.
Although sequence-based clustering methods guide the formulation of testable hypotheses, the practical challenge for microbial ecologists will be to define ecotypes using methods which accurately predict ecological equivalence, which in turn requires careful consideration of the number and nature (genotypic and phenotypic) of arbiters (e.g., growth rate, grazing susceptibility, subtractive hybridization [1, 33], or gene expression) chosen to test ecotype boundaries—i.e., which strains should be included or excluded. This is especially important given that apparently minor differences in picocyanobacterial genotypes can obscure ecologically significant differences (8, 10). A quantitative appraisal of the “everything (microbial) is everywhere” hypothesis will require that putative ecotypes are empirically defined. Limited phylogenetic information in the 16S rRNA gene (24), taxon undersampling, and the potential “erosion” of phylogenies due to horizontal transfer of genetic information mean that a mature understanding of picocyanobacterial ecotypes, their effective and potential biogeography (or lack thereof), will require that more importance is given to quantifying the limitations of (i) sequence acquisition, depending on the extent and nature of sampling, (ii) the number and choice of gene or gene-fragment(s), and (iii) methods of phylogenetic analysis.
Nucleotide sequence accession numbers.
GenBank accession codes for sequences determined in this study are AY151211 to AY151251 and AY224198 to AY224207.
Acknowledgments
We thank P. Stadler for technical assistance; M. W. Hahn for providing isolates MH 301, MH 305, and MH 307; A. Wilmotte and B. R. Robertson for providing details of unpublished primers; and W. F. Vincent and M. M. Watanabe for information on several isolates included in the phylogenetic analyses. Comments from M. W. Hahn and R. Kurmayer helped to improve an earlier draft of the manuscript.
This work was supported by the Austrian Science Fund (FWF P14238-Bio).
REFERENCES
- 1.Agron, P. G., M. Macht, L. Radnedge, E. W. Skowronski, W. Miller, and G. L. Andersen. 2002. Use of subtractive hybridization for comprehensive surveys of prokaryotic genome differences. FEMS Microbiol. Lett. 211:175-182. [DOI] [PubMed] [Google Scholar]
- 2.Amann, R., and R. Roselló-Mora. 2001. Mikrobiologische Aspekte der Biodiversität, p. 161-180. In P. Janich, M. Gutmann, and K Prieβ (ed.), Biodiversität. Springer, Berlin, Germany.
- 3.Bickel, P. J., K. J. Kechris, P. C. Spector, G. J. Wedemayer, and A. N. Glazer. 2002. Finding important sites in protein sequences. Proc. Natl. Acad. Sci. USA 99:14764-14771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Callieri, C., and J. G. Stockner. 2002. Freshwater autotrophic picoplankton: a review. J. Limnol. 61:1-14. [Google Scholar]
- 5.Castenholz, R. W. 2001. Phylum BX. Cyanobacteria, oxygenic photosynthetic bacteria, p. 473-599. In D. R. Boone and R. W. Castenholz (ed.), Bergey's manual of systematic bacteriology, vol. 1. Springer, New York, N.Y.
- 6.Cohan, F. M. 2001. Bacterial species and speciation. Syst. Biol. 50:513-524. [DOI] [PubMed] [Google Scholar]
- 7.Crosbie, N. D., M. Pöckl, and T. Weisse. Rapid establishment of clonal isolates of freshwater autotrophic picoplankton by single-cell and single-colony sorting. J. Microbiol. Methods, in press. [DOI] [PubMed]
- 8.Crosbie, N. D., K. Teubner, and T. Weisse. Flow-cytometric mapping provides novel insights into the seasonal and vertical distributions of freshwater autotrophic picoplankton. Aquat. Microb. Ecol., in press.
- 9.Ernst, A. 1991. Cyanobacterial picoplankton from Lake Constance. I. Isolation by fluorescence characteristics. J. Plankton Res. 13:1307-1312. [Google Scholar]
- 10.Ernst, A., S. Becker, U. I. A. Wollenzien, and C. Postius. 2003. Ecosystem-dependent adaptive radiations of picocyanobacteria inferred from 16S rRNA and ITS-1 sequence analysis. Microbiology 149:217-228. [DOI] [PubMed] [Google Scholar]
- 11.Ernst, A., G. Sandmann, C. Postius, S. Brass, U. Kenter, and P. Böger. 1992. Cyanobacterial picoplankton from Lake Constance. 2. Classification of isolates by morphology and pigment composition. Bot. Acta 105:161-167. [Google Scholar]
- 12.Felsenstein, J. 1981. Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17:368-376. [DOI] [PubMed] [Google Scholar]
- 13.Finlay, B. J. 2002. Global dispersal of free-living microbial eukaryote species. Science 296:1061-1063. [DOI] [PubMed] [Google Scholar]
- 14.Finlay, B. J., S. C. Maberly, and J. I. Cooper. 1997. Microbial diversity and ecosystem function. Oikos 80:209-213. [Google Scholar]
- 15.Gutell, R. R., N. Larsen, and C. R. Woese. 1994. Lessons from an evolving rRNA: 16S and 23S rRNA structures from a comparative perspective. Microbiol. Rev. 58:10-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hedenstierna, K. O. F., J. L. Siefer, G. E. Fox, and E. J. Murgola. 2000. Co-conservation of rRNA tetraloop sequences and helix length suggests involvement of the tetraloops in higher-order interactions. Biochimie 82:221-227. [DOI] [PubMed] [Google Scholar]
- 17.Honda, D., A. Yokota, and J. Sugiyama. 1999. Detection of seven major evolutionary lineages in cyanobacteria based on 16S rRNA gene sequence analysis with new sequences of five marine Synechococcus strains. J. Mol. Evol. 48:723-739. [DOI] [PubMed] [Google Scholar]
- 18.Janson, S., and E. Granéli. 2002. Phylogenetic analyses of nitrogen-fixing cyanobacteria from the Baltic Sea reveal sequence anomalies in the phycocyanin operon. Int. J. Syst. Evol. Microbiol. 52:1397-1404. [DOI] [PubMed] [Google Scholar]
- 19.Lepere, C., A. Wilmotte, and B. Meyer. 2000. Molecular diversity of Microcystis strains (Cyanophyceae, Chroococcales) based on 16S rDNA sequences. Syst. Geogr. Plants 70:275-283. [Google Scholar]
- 20.Ludwig, W., and H. Klenk. 2001. Overview: a phylogenetic backbone and taxonomic framework for procaryotic systematics, p. 49-65. In D. R. Boone and R. W. Castenholz (ed.), Bergey's manual of systematic bacteriology, vol. 1. Springer, New York, N.Y.
- 21.MacIsaac, E. A., and J. G. Stockner. 1993. Enumeration of phototrophic picoplankton by autofluorescence microscopy, p. 187-197. In P. F. Kemp, B. F. Sherr, E. B. Sherr, and J. J. Cole (ed.), Handbook of methods in aquatic microbial ecology. Lewis Publishers, Boca Raton, Fla.
- 22.Manen, J., and J. Falquet. 2002. The cpcB-cpcA locus as a tool for the genetic characterization of the genus Arthrospira (Cyanobacteria): evidence for horizontal transfer. Int. J. Syst. Evol. Microbiol. 52:861-867. [DOI] [PubMed] [Google Scholar]
- 23.Palys, T., F. M. Cohan, and L. K. Nakamura. 1997. Discovery and classification of ecological diversity in the bacterial world: the role of DNA sequence data. Int. J. Syst. Bacteriol. 47:1145-1156. [DOI] [PubMed] [Google Scholar]
- 24.Palys, T., E. Berger, I. Mitrica, L. K. Nakamura, and F. M. Cohan. 2000. Protein-coding genes as molecular markers for ecologically distinct populations: the case of two Bacillus species. Int. J. Syst. Evol. Microbiol. 50:1021-1028. [DOI] [PubMed] [Google Scholar]
- 25.Pollock, D. D., D. J. Zwickl, J. A. McGuire, and D. M. Hillis. 2002. Increased taxon sampling is advantageous for phylogenetic inference. Syst. Biol. 51:664-671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Posada, D. 2002. Evaluation of methods for detecting recombination from DNA sequences: empirical data. Mol. Biol. Evol. 19:708-717. [DOI] [PubMed] [Google Scholar]
- 27.Posada, D., and K. A. Crandall. 1998. Modeltest: testing the model of DNA substitution. Bioinformatics 14:817-818. [DOI] [PubMed] [Google Scholar]
- 28.Rankin, L. M., P. D. Franzmann, T. A. McMeekin, and H. R. Burton. 1997. Seasonal distribution of picocyanobacteria in Ace Lake, a marine-derived Antarctic lake, p. 178-184. In B. Battaglia, J. Valencia, and D. W. H. Walton (ed.), Antarctic communities. Species, structure and survival. Cambridge University Press, Cambridge, United Kingdom.
- 29.Robertson, B. R., N. Tezuka, and M. M. Watanabe. 2001. Phylogenetic analyses of Synechococcus strains (cyanobacteria) using sequences of the 16S rDNA and part of the phycocyanin operon reveal multiple evolutionary lines and reflect phycobilin content. Int. J. Syst. Evol. Microbiol. 51:861-871. [DOI] [PubMed] [Google Scholar]
- 30.Rocap, G., L. Distel, J. B. Waterbury, and S. W. Chisholm. 2002. Resolution of Prochlorococcus and Synechococcus ecotypes by using 16S-23S ribosomal DNA internal transcribed spacer sequences. Appl. Environ. Microbiol. 68:1180-1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saitou, N., and M. Nei. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4:406-425. [DOI] [PubMed] [Google Scholar]
- 32.Scheldman, P., D. Baurain, R. Bouhy, M. Scott, M. Mühling, B. A. Whitton, A. Belay, and A. Wilmotte. 1999. Arthrospira (‘Spirulina’) strains from four continents are resolved into only two clusters, based on amplified ribosomal DNA restriction analysis of the internally transcribed spacer. FEMS Microbiol. Lett. 172:213-222. [DOI] [PubMed] [Google Scholar]
- 33.Schmidt, K. D., T. Schmidtrose, U. Romling, and B. Tummler. 1998. Differential genome analysis of bacteria by genomic subtractive hybridization and pulsed-field gel electrophoresis. Electrophoresis 19:509-514. [DOI] [PubMed] [Google Scholar]
- 34.Stanier, R. Y., R. Kunisawa, M. Mandel, and G. Cohen-Bazier. 1971. Purification and properties of unicellular blue-green algae (order Chroococcales). Bacteriol. Rev. 35:171-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stockner, J., C. Callieri, and G. Cronberg. 2000. Picoplankton and other non-bloom forming cyanobacteria in lakes, p. 195-231. In B. A. Whitton and M. Potts (ed.), The ecology of cyanobacteria. Kluwer Academic Publishers, Dordrecht, The Netherlands.
- 36.Sugiara, N., B. Wei, and T. Maekawa. 2002. The discrimination of the response pattern of inter-phylum phytoplankton diversity to long-term eutrophication trends in lake Kasumigaura, Japan. Aquat. Ecosyst. Health Manag. 5:403-410. [Google Scholar]
- 37.Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin, and D. G. Higgins. 1997. The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 24:4876-4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Urbach, E., D. J. Scanlan, D. L. Distel, J. B. Waterbury, and S. W. Chisholm. 1998. Rapid diversification of marine picophytoplankton with dissimilar light-harvesting structures inferred from sequences of Prochlorococcus and Synechococcus (Cyanobacteria). J. Mol. Evol. 46:188-201. [DOI] [PubMed] [Google Scholar]
- 39.Van de Peer, Y., and R. De Watcher. 1994. TREECON: a software package for the construction and drawing of evolutionary trees. Comput. Appl. Biosci. 9:177-182. [DOI] [PubMed] [Google Scholar]
- 40.Vincent, W. F., J. P. Bowman, L. M. Rankin, and T. A. McMeekin. 2000. Phylogenetic diversity of picocyanobacteria in Arctic and Antarctic ecosystems, p. 317-322. In C. R. Bell, M. Brylinsky, and P. Johnson-Green (ed.), Microbial biosystems: new frontiers. Proceedings of the 8th International Symposium on Microbial Ecology. Atlantic Canada Society for Microbial Ecology, Halifax, Canada.
- 41.Wang, Y., and Z. Zhang. 2000. Comparative sequence analyses reveal frequent occurrence of short segments containing an abnormally high number of non-random base variations in bacterial rRNA genes. Microbiology 146:2845-2854. [DOI] [PubMed] [Google Scholar]
- 42.Weisse, T. 1988. Dynamics of autotrophic picoplankton in Lake Constance. J. Plankton Res. 10:1179-1188. [Google Scholar]
- 43.Weisse, T., and U. Kenter. 1991. Ecological characteristics of autotrophic picoplankton in a prealpine lake. Int. Rev. Hydrobiol. 76:493-504. [Google Scholar]