

Abstract
Perineural α2-adrenoceptor activation relieves hypersensitivity induced by peripheral nerve injury or sciatic inflammatory neuritis. This effect is associated with a reduction in pro-inflammatory cytokines, as well as a reduction in local leukocyte number and their capacity to produce pro-inflammatory cytokines. Curiously, clonidine’s antinociceptive effect appears with a 2–3-day delay after injection. Previous observations have shown that α-adrenoceptor activation induces apoptosis in leukocytes, which would reduce leukocyte number. Additionally, macrophage scavenging of apoptotic cells results in a shift to an anti-inflammatory phenotype, with expression of transforming growth factor (TGF)-β1. We therefore examined the effects of perineural clonidine 24 hr and 3 days after its injection on apoptosis, TGF-β1 expression and lymphocyte and macrophage phenotype in acute sciatic inflammatory neuritis. Perineural clonidine reduced ipsilateral neuritis-induced hypersensitivity in a delayed manner (3 days after treatment), along with a reduction at this time in lymphocyte number and an increase in caspase- 3 and TGF-β1 expressing cells and macrophages co-expressing TGF-β1 in the sciatic nerve. One day after injection clonidine treatment was associated with a reduction in lymphocytes and pro-inflammatory Th-1 cells as well as increased numbers of caspase-3 and TGF-β1 expressing cells and macrophages co-expressing TGF-β1 in sciatic nerve. Clonidine’s effects were prevented by co-administration of an α2-adrenoceptor antagonist. These data suggest that α2-adrenoceptor activation in sciatic inflammatory neuritis increases local apoptosis and anti-inflammatory products early after treatment. This early effect likely underlies the delayed anti-inflammatory and anti-hypersensitivity effects of perineural clonidine in this setting.
Keywords: Immune response, α2-adrenoceptor, leukocytes, neuropathic pain, caspase-3, TGF-β1, IL-1β, TNFα, hypersensitivity, sciatic nerve
Introduction
Peripheral nerve injury induces chronic pain in part due to immune activation orchestrated by stimulation and migration of leukocytes and by their expression of pro-inflammatory products. Immune activation near healthy peripheral nerves is sufficient to produce pain states (Watkins et al., 1995) and peripheral immune modulation reduces hypersensitivity to somatic stimuli in several neuropathic pain models (Twining et al., 2004; Romero-Sandoval and Eisenach 2006).
Sympathetic nervous system activity or exogenously applied adrenoceptor (AR) agonists can modulate activity and responses of lymphocytes and macrophages (Moynihan et al., 2004), due to actions on multiple AR subtypes, including α2-ARs (Titnchi and Clark, 1984; Spengler et al., 1990; Josefsson et al., 1996). Sympathetic and α-AR activation have been suggested to contribute to peripheral nerve injury-induced pain (Sato and Perl 1991; Koltzenburg and McMahon. 1991; McLachlan et al., 1993). However, we have also shown that perineural α2-AR activation relieves neuropathic pain by modulating immune responses (Romero-Sandoval et al., 2005; Romero-Sandoval and Eisenach 2006).
Because of the potential relevance of understanding how immune response modulation in neuropathic pain conditions alters pain processing, we sought to further probe the mechanisms by which α2-ARs affect hypersensitivity and immune responses in the sciatic inflammatory neuropathy (SIN) model. This model, consisting of local application of zymosan on the sciatic nerve through an implanted catheter (Gazda et al., 2001; Chacur et al., 2001; Milligan et al., 1999), results in intense and bilateral hypersensitivity to mechanical stimuli, dependent upon interleukin (IL) -1, IL-6 and tumor necrosis factor (TNF) α, reactive oxygen species and complement expression and generation (Twining et al., 2004).
Perineural administration of the α2-AR agonist, clonidine, reverses hypersensitivity in the SIN model (Romero-Sandoval et al., 2005). Antinociception from clonidine is associated with a reduction in pro-inflammatory cytokines, macrophage and lymphocyte number in the inflammatory exudates, and capacity of the leukocytes present to produce pro-inflammatory cytokines in response to further challenge. Since clonidine’s antinociceptive effects occur in a delayed manner several days after transient drug exposure, we hypothesize that clonidine acutely alters immune responses, leading over time to reduced migration and leukocyte function.
We based our study on five previous observations: (1) when macrophages recognize and engulf apoptotic cells, they alter their phenotype to a primarily anti-inflammatory one, expressing TGFβ-1 (Fadok et al., 2000; Huynh et al., 2002), (2) α-AR agonists change the proportion and increase the number of apoptotic leukocytes (Stevenson et al., 2001), (3) clonidine produces apoptosis in the developing brainstem (Dygalo 2004) and in skin melanocytes (Uchida-Oka and Sugimoto, 2001), (4) clonidine increases TGF-β1 expression in sciatic nerve tissue in a mechanical nerve injury model of neuropathic pain (Lavand’homme and Eisenach, 2003) and (5) perineural clonidine treatment reduces pro-inflammatory but increases anti-inflammatory responses in immune cells in the inflammatory exudates in the SIN model (Romero-Sandoval et al., 2005). These observations suggest that α2-AR mediated acute apoptosis could underlie the delayed beneficial effects of perineural clonidine in neuritis. Here we tested whether perineural clonidine induced apoptosis and resulted in altered immune cell phenotype in the inflamed nerve of the SIN model, and examined its time course and pharmacologic mechanism of action.
Methods
Animals and surgery
After approval by the Animal Care and Use Committee (Wake Forest University School of Medicine, Winston- Salem, North Carolina) male Wistar rats (200–250 g at the day of surgery), underwent implantation of a gelfoam-attached silastic catheter near the sciatic nerve as previously described (Gazda et al., 2001; Chacur et al., 2001; Romero-Sandoval et al., 2005). Briefly, the left sciatic nerve was exposed and wrapped with gelfoam attached to the end of the catheter. The catheter was anchored to the muscles using 3-0 silk suture and passed subcutaneously, exiting at the base of the tail. The external end was held in a plastic structure protected by an aluminum frame and sealed with rubber and silicone glue.
At the end of the experiments, the animals were anesthetized with halothane and were sacrificed by decapitation. All catheters were dissected, and only animals with correctly placed catheters were included in the analysis.
Drugs and in vivo treatments
Drugs were administered in awake and freely moving animals through the catheter 4–5 days after surgery. The drugs used were: incomplete Freund’s adjuvant (IFA) as vehicle, zymosan (in vehicle, both from Sigma, St Louis, MO), clonidine (in saline, from Roxane, Columbus, OH) and BRL44408, an α2-AR antagonist (Tocris, Ellisville, MO).
The animals received a single dose of zymosan (40μg, 50 μl) to induce acute inflammatory neuritis and bilateral hypersensitivity as previously described (Milligan et al., 2003; Romero-Sandoval et al., 2005). Clonidine, which has a terminal elimination half life of 8–25 hr (Anavekar et al., 1982; Conway et al., 1992) was administered 3 hr after zymosan (when hypersensitivity was already evident) in a single dose (30 μg, 60μl, a dose previously shown to be effective to relieve hypersensitivity in this model; Romero-Sandoval et al., 2005) or together with BRL44408 (45 μg, 10 μl in saline, a dose previously shown to block clonidine’s anti-hypersensitive effects; Lavand’homme et al., 2002). Control groups received vehicle instead of zymosan, and saline instead clonidine. The groups studied were: vehicle plus saline (v+s, n=4), zymosan plus saline (z+s, n=4), zymosan plus clonidine (z+c, n=5) and zymosan plus BRL44408 plus clonidine (z+BR+c, n=5).
Behavioral testing
Withdrawal threshold to von Frey filament (Stoelting, Wood Dale, IL) probing was determined using an up-down statistical method (Chaplan et al., 1994). Thresholds were measured ipsi- and contralaterally to neuritis before catheter implantation, 4–5 days after catheter implantation (immediately before zymosan or vehicle injection), 3 hr after zymosan injection and 1 or 3 days thereafter. Withdrawal thresholds were measured twice at each site, and the average used for data analysis.
Tissue preparation and Immunostaining
Animals (n=4 for all groups) on post-treatment day 1 or 3 were deeply anesthetized with pentobarbital and perfused transcardially with buffer (0.01M phosphate buffered saline + 1% sodium nitrite, 100 ml) followed by 4% paraformaldehyde (400 ml) at room temperature. The sciatic nerve (~1 cm) ipsilateral to neuritis was removed and postfixed in the same fixative for 2–3 hr followed by cryoprotection in 30% sucrose for 48–72 h at 4°C. Tissue was cut at a 16 μm thickness using a cryostat and sections immunolabeled on slides. After four washes with 0.01M phosphate buffered saline+0.15% Triton 100X (PBS + T), the slides were incubated in 50% alcohol for 45 min, washed four times with PBS + T and blocked with 1.5% normal goat serum. Slides were then incubated overnight at 4°C in primary antibodies, washed two times with PBS+T, then incubated for 1 h with fluorescein (1:200, West Grove, PA) or biotin (1:200, Vector, Burlingame, CA) plus Alexa 568 (1:400, Molecular Probes, Eugene, OR) secondary antibodies.
Apoptotic cells were identified using an antibody to cleaved caspase-3 (rabbit polyclonal antibody, 1:1000, Wako Pure Chemical Industries, Richmond, VA). Lymphocytes and monocytes/macrophages were identified using CD2 and ED1 antibodies, respectively (both mouse monoclonal antibodies from Serotec, Raleigh, NC; CD2 at 1:500 and ED1 at 1:200). We also co-localized caspase-3 with CD2 or ED1. We identified TGFβ-1 expressing cells using a rabbit polyclonal antibody (1:1000, Biovision, Mountain View, CA). We chose TGFβ-1, since others have shown that anti-inflammatory macrophages over-produce this factor (Fadok et al., 1998; Huynh et al., 2002), and considered cells which co-localized ED1 and TGFβ-1 as anti-inflammatory macrophages. We identified the anti-inflammatory cytokine, IL-10, also used to mark Th-2 lymphocytes, with a rabbit polyclonal antibody (1:500, Biosource, Camarillo, CA) and co-localized it with CD2 to identify anti-inflammatory Th-2 type cells. Finally, to identify pro-inflammatory Th-1 lymphocytes, we co-localized CD2 with a rabbit polyclonal antibody to signal transducer and activator of transcription 4 (STAT4, 1:400, Santa Cruz Biotechnology, Santa Cruz, CA).
Images of tissue sections were captured using a digital camera attached to a Leica Axioplan2 light microscope, and a semi-quantitative method was used for image analysis. The number of positively stained objects in a 570 × 330 μm area was determined using image analysis software and a fixed intensity threshold. The average of 3–4 sections per animal was used for analysis. Number or proportions are expressed per 570 × 340 μm area of the sciatic nerve.
Leukocyte collection and in vitro treatment
Gelfoam surrounding the sciatic nerve was retrieved 3 hr after zymosan treatment (n=6). As previously described (Gazda et al., 2001), the gelfoam was dissected and mechanically dissociated in Hanks’ Balanced Salt Solution (HBSS, 20 mL) by means of micro forceps in a plastic Petri dish. The suspension was filtered using a 70-micron nylon mesh using plastic syringe filter holders. Cells were counted using a hemocytometer and their viability was evaluated using trypan blue extrusion. Only preparations with >90% viable cells were used. Cells were re-suspended at 1×107 cells/mL in HBSS. Immediately after collection, cells were diluted to 1×106 cells/ml in Roswell Park Memorial Institute medium (RPMI 1640 + 10% fetal bovine serum + 1% penicillin and streptomycin + 1% L-glutamine) in polypropylene RNAse, DNAse and pyrogen free tubes. Cells were incubated with saline (control group) or clonidine (1 and 100 μM) overnight (14–16 hr) at 37°C and 5% CO2. Supernatants were frozen at −80°C until cytokine measurement assay.
Cytokine measurement assay
Samples of supernatants were slowly melted and 50μL were immediately used for cytokine detection. Following the instructions given by the manufacturer, a 3-plex bead-based immunoassay cytokine kit for rat (Bio Rad, Hercules, CA) was used for simultaneous detection of IL-10, IL-1β, and TNFα concentrations. This technique has been validated previously (Romero-Sandoval et al., 2005). Multi-wavelength fluorescence and cytokine concentrations were determined with a luminometer (Luminex 100TM system, Luminex, Austin, TX), and BioRad software (BioRad, Hercules, CA). Assays were performed in duplicate, with the average used for analysis
Statistical analysis
Data are presented as mean ± s.e.m. The effects of catheter implantation or drug treatments on withdrawal threshold were determined using a two way ANOVA followed by Dunnett’s test. Comparisons of cytokine concentrations and immunolabeling were performed using t tests or when normality failed, Mann Whitney U tests. Proportions were evaluated using Fisher’s exact test. A P value <0.05 was considered significant.
Results
Behavioral experiments
Unilateral IFA administration produced a slight but significant reduction in withdrawal threshold ipsilateral to injection without any change contralaterally. Subsequent saline administration did not further alter withdrawal threshold in either hind paw. Unilateral zymosan injection induced a bilateral reduction in withdrawal threshold within 3 hr after its administration which remained present 1 and 3 days thereafter. Perineural saline treatment did not modify this zymosan-induced hypersensitivity. Perineural clonidine significantly diminished zymosan-induced hypersensitivity 3 days after treatment, but not earlier, and this effect was prevented by co-administration of BRL44408 (Fig. 1).
Figure 1.
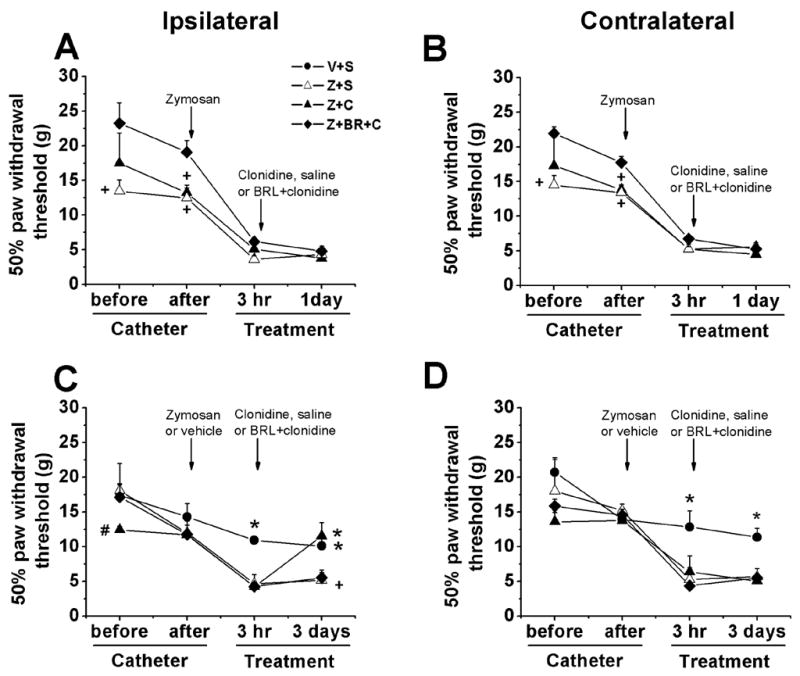
Mechanical withdrawal threshold ipsilateral and contralateral to neuritis. Withdrawal threshold to von Frey stimulation ipsilateral (A) and contralateral (B) to neuritis before and after catheter implantation, 24 hr after zymosan (40 μg) plus saline (Z+S), zymosan plus clonidine (30 μg; Z+C) and zymosan plus BRL44408 (45 μg) plus clonidine (30 μg; Z+BR+C). Withdrawal threshold to von Frey stimulation ipsilateral (C) and contralateral (D) to neuritis before and after catheter implantation, and 3 days after zymosan (40 μg) plus saline, vehicle plus saline (V+S), zymosan plus clonidine (30 μg) and zymosan plus BRL44408 (45 μg) plus clonidine (30 μg). Withdrawal thresholds were significant reduced (P<0.05) 3 hr after zymosan injections compared to the value observed before catheter implantation. *P<0.05 compared to zymosan plus saline group; #P<0.05 compared to vehicle or zymosan plus saline group. *P<0.05 compared to zymosan plus saline group. +P<0.05 compared to zymosan plus BRL44408 plus clonidine group.
Leukocyte presence in nerve 3 days after zymosan with or without clonidine
We first focused on leukocyte presence 3 days after clonidine, since this was the time of maximum behavioral effect. In the absence of clonidine, zymosan resulted 3 days after injection in a large increase in the number of ED1 expressing macrophages and CD2 expressing T lymphocytes (Fig. 2A, Fig. 2B, Fig. 3A). Zymosan treatment also caused an increase in the number of apoptotic cells labeled with cleaved capase-3 (Fig. 3A). The identity of these cells is uncertain, since fewer than 5% of them co-localized with ED1 and fewer than 25% co-localized with CD2 (Fig. 3C). Zymosan increased the number of CD2 positive cells which co-expressed cleaved caspase-3, although this was only a small proportion (10% of CD 2 positive cells, Fig. 3D), and zymosan did not increase the proportion or number of ED1 cells which co-expressed caspase-3 (Fig. 3E). Zymosan treatment increased the number of cells labeled with anti-inflammatory cytokines, IL-10 and TGF-β1 (Fig. 3B, Fig. 3F). We hypothesized that macrophages were likely sources of TGF-β1. Less than 5% of TGF-β1 labeled cells co-expressed ED1 (Fig. 3G), but this proportion was increased by zymosan to nearly 25% (Fig. 3B). Less than 5% of ED1 labeled cells co-expressed TGF-β1 and zymosan did not change this proportion (Fig. 3H). Zymosan did not modify the number or proportion of CD2 positive cells which co-expressed either IL-10 (Th-2 lymphocytes, data not shown) or STAT4 (Th-1 lymphocytes, data not shown). In summary, perineural zymosan increased macrophage and T lymphocyte number in sciatic nerve 3 days after injection. It also increased the number of apoptotic cells and those expressing anti-inflammatory factors, but the large majority of each were neither macrophages nor T lymphocytes.
Figure 2.
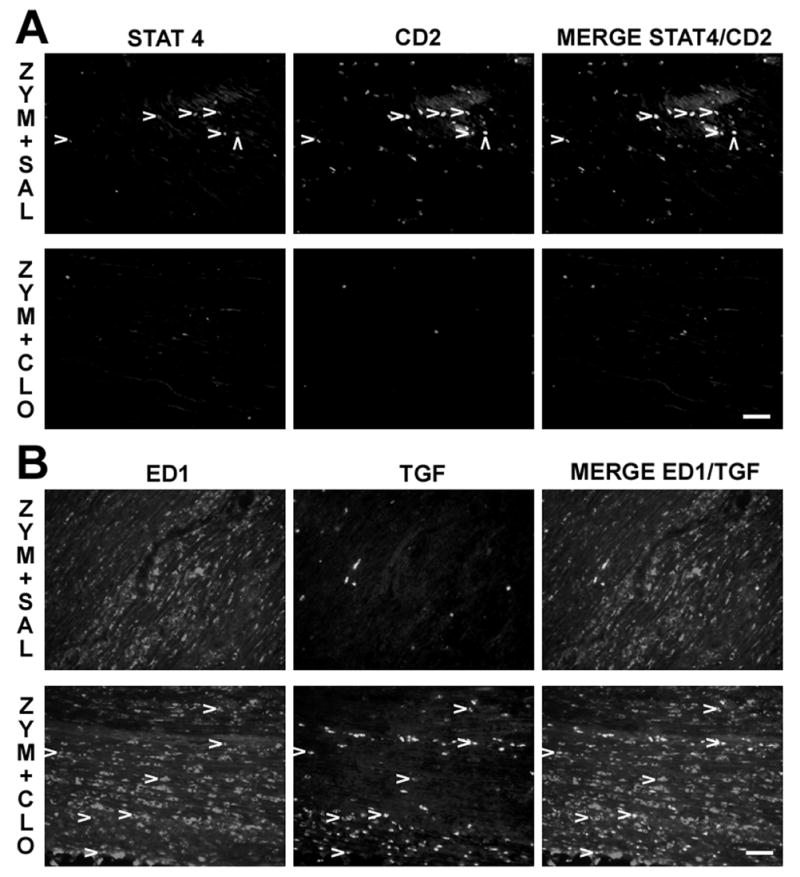
Clonidine reversal of zymosan’s effects in apoptotic cells, lymphocytes, TGF-β1 (TGF) and anti-inflammatory macrophages in sciatic nerve 3 days after treatments. A. Immunostaining of STAT4, CD2 and co-localization of both (arrow heads) in the sciatic nerve in zymosan plus saline (upper panel, ZYM+SAL) and zymosan plus clonidine group (lower panel, ZYM+CLO). B. Immunostaining of ED1, TGF-β1 and co-localization of both (arrow heads) in the sciatic nerve in zymosan plus saline (upper panel) and zymosan plus clonidine group (lower panel). Scale bar = 50 μm.
Figure 3.
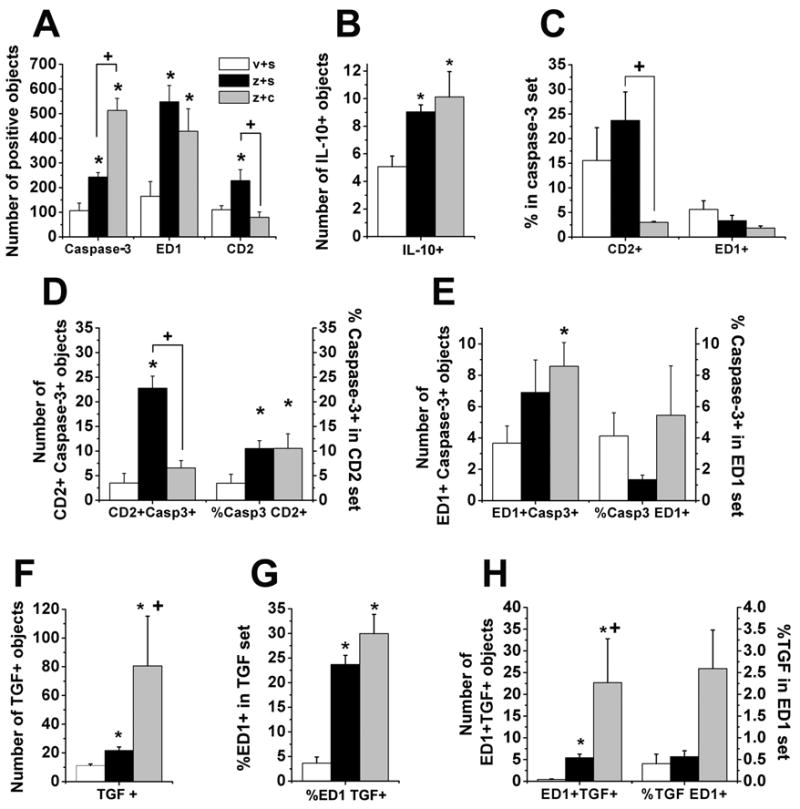
Zymosan’s effects and clonidine reversal of zymosan’s effects in apoptotic cells, lymphocytes, TGF-β1 (TGF) and anti-inflammatory macrophages in sciatic nerve 3 days after IFA plus saline (v+s), zymosan plus saline (z+s) or clonidine (z+c). A. Quantification of caspase- 3 (Casp3), ED1 and CD2 immunostaining. *P<0.05 comparison vs. IFA plus saline group; +P<0.05 comparison vs. zymosan plus saline group. B. Quantification of IL-10 expression. *P<0.05 comparison vs. IFA plus saline group. C. Quantification of apoptotic cells analyzed as proportion of co-expression of CD2 or ED1 in the caspase-3 set. +P<0.05 comparison vs. zymosan plus saline group. D. Quantification of apoptotic lymphocytes analyzed as number of CD2 and caspase-3 co-localization immunostaining and proportion of caspase-3 co-expression in the CD2 set. *P<0.05 comparison vs. IFA plus saline group; +P<0.05 comparison vs. zymosan plus saline group. E. Quantification of apoptotic macrophages analyzed as number of ED1 and caspase-3 co-localization immunostaining and proportion of caspase-3 co-expression in the ED1 set. *P<0.05 comparison vs. IFA plus saline group; +P<0.05 comparison vs. zymosan plus saline group. F. Quantification of TGF-β1 expression. *P<0.05 comparison vs. IFA plus saline group ; +P<0.05 comparison vs. zymosan plus saline group. G. Quantification of apoptotic macrophages analyzed as number of ED1 and caspase-3 co-localization immunostaining and proportion of caspase-3 co-expression in the ED1 set. *P<0.05 comparison vs. IFA plus saline group. H. Quantification of anti-inflammatory macrophages analyzed as number of ED1 and TGF-β1 co-localization immunostaining and proportion of co-expression of TGF-β1 in the ED1 set. *P<0.05 comparison vs. IFA plus saline group; +P<0.05 comparison vs. zymosan plus saline group.
Perineural clonidine, administered 3 hr after zymosan, significantly reduced the number of CD2 expressing cells in sciatic nerve 3 days later, returning it to control levels (Fig. 2A, Fig. 3A), and significantly increased the number of apoptotic cells (Fig. 2C, Fig. 3A). As in the zymosan alone group, the large majority of apoptotic cells in the animals receiving zymosan followed by clonidine failed to co-express ED1 (Fig. 3C). Apoptotic cells in the animals receiving zymosan followed by clonidine co-localized with CD2 in a smaller proportion and number than the zymosan alone group (Fig. 3C, Fig. 3D). However, clonidine did not modify the proportion of T lymphocytes which co-expressed caspase-3 induced by zymosan (10% of CD 2 positive cells, Fig. 3D), or the proportion of ED1 co-expressing cleaved caspase-3 (<6% of ED1 positive cells, Fig. 3E). Clonidine increased the number, but not the proportion of TGF-β1 labeled cells co-expressing ED1 (Fig. 3G, Fig. 3H). As with zymosan alone, over 95% of ED1 expressing cells were not marked by TGF-β (Fig. 3B). Clonidine did not induce a significant change in the number or proportion of CD2 positive cells which co-expressed either IL-10 (Th-2 lymphocytes, data not shown) or STAT4 (Th-1 lymphocytes, Fig. 2A).
Leukocyte presence in nerve 1 day after zymosan with or without clonidine
The reduction in CD2 expressing cells in sciatic nerve 3 days after perineural clonidine suggested that clonidine might be reducing lymphocyte number by inducing apoptosis of these cells shortly after its injection, with their disappearance 3 days later. We therefore examined the effect of perineural zymosan plus saline or plus clonidine on leukocytes in sciatic nerve 24 hr after zymosan injection. Consistent with this hypothesis, at this earlier time clonidine significantly increased the number of cells expressing cleaved caspase-3 and increased the proportion of CD2 positive cells co-expressing cleaved caspase-3 to nearly 25% of CD2 positive cells (Fig. 4A, Fig. 4B, Fig. 4E). Clonidine treatment also reduced the number as well as proportion of pro-inflammatory Th-1 lymphocytes, as measured by co-expression of CD2 and STAT4 (Fig. 4F, Fig. 5A). Clonidine treatment did not modify the number of IL-10 expressing cells (data not shown) or the number as well as proportion of anti-inflammatory Th-2 lymphocytes, as measured by co-expression of CD2 and IL-10 (data not shown). Finally, <10% TFG-β1 cells co-expressed ED1 in zymosan alone group, and clonidine treatment increased this proportion to nearly 35% (Fig. 4C, Fig. 5B). Clonidine also increased the number of TGF-β1 profiles, however over 90% of ED1 expressing cells did not co-express TGF-β1 (Fig. 4D). In summary, examination of sciatic nerve 1 day after zymosan plus clonidine compared to 3 days revealed a larger increase of lymphocytes undergoing apoptosis and less pro-inflammatory lymphocytes and more anti-inflammatory macrophages.
Figure 4.
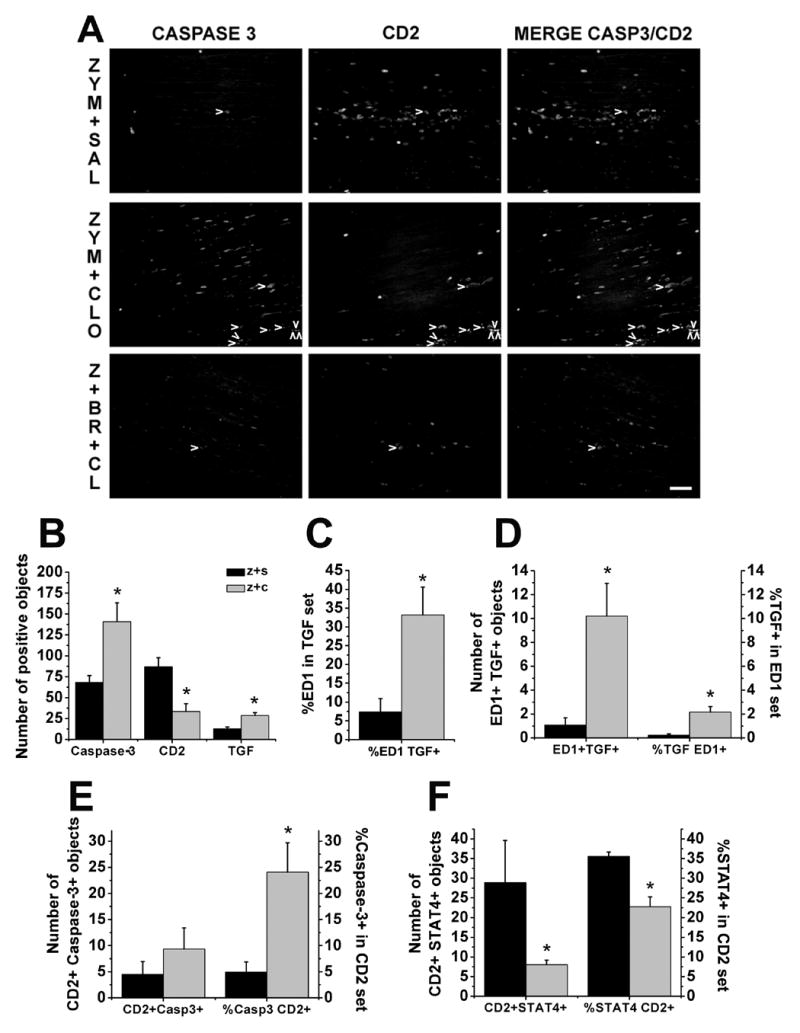
Clonidine’s effects and BRL44408 reversal of clonidine’s effects in apoptotic cells, lymphocytes, TGF-β1 (TGF), anti-inflammatory macrophages and Th-1 type lymphocytes in sciatic nerve 24 hr after treatments. A. Immunostaining of caspase-3 (Casp3), CD2 and co-localization of both (arrow heads) in the sciatic nerve 24 hr after zymosan plus saline (upper panel, ZYM+SAL), zymosan plus clonidine (middle panel, ZYM+CLO) and zymosan plus BRL44408 plus clonidine group (lower panel, Z+BR+CL). Scale bar = 50 μm. B. Quantification of caspase-3, CD2 and and TGF-β1 immunostaining in zymosan plus saline (z+s) or clonidine (z+c) groups. *P<0.05 comparison vs. zymosan plus saline group. C. Quantification of anti-inflammatory macrophages analyzed as proportion of ED1 co-expression in the TGF-β1 set. *P<0.05 comparison vs. zymosan plus saline group. D. Quantification of anti-inflammatory macrophages analyzed as number of ED1 and TGF-β1 co-localization immunostaining and proportion of TGF-β1 co-expression in the ED1 set. *P<0.05 comparison vs. zymosan plus saline group. E. Quantification of apoptotic lymphocytes analyzed as number of CD2 and caspase-3 co-localization immunostaining and proportion of caspase-3 co-expression in the CD2 set. *P<0.05 comparison vs. zymosan plus saline group. F. Quantification of Th-1 type lymphocytes analyzed as number of CD2 and caspase-3 co-localization immunostaining and proportion of STAT 4 co-expression in the CD2 set. *P<0.05 comparison vs. zymosan plus saline group.
Figure 5.
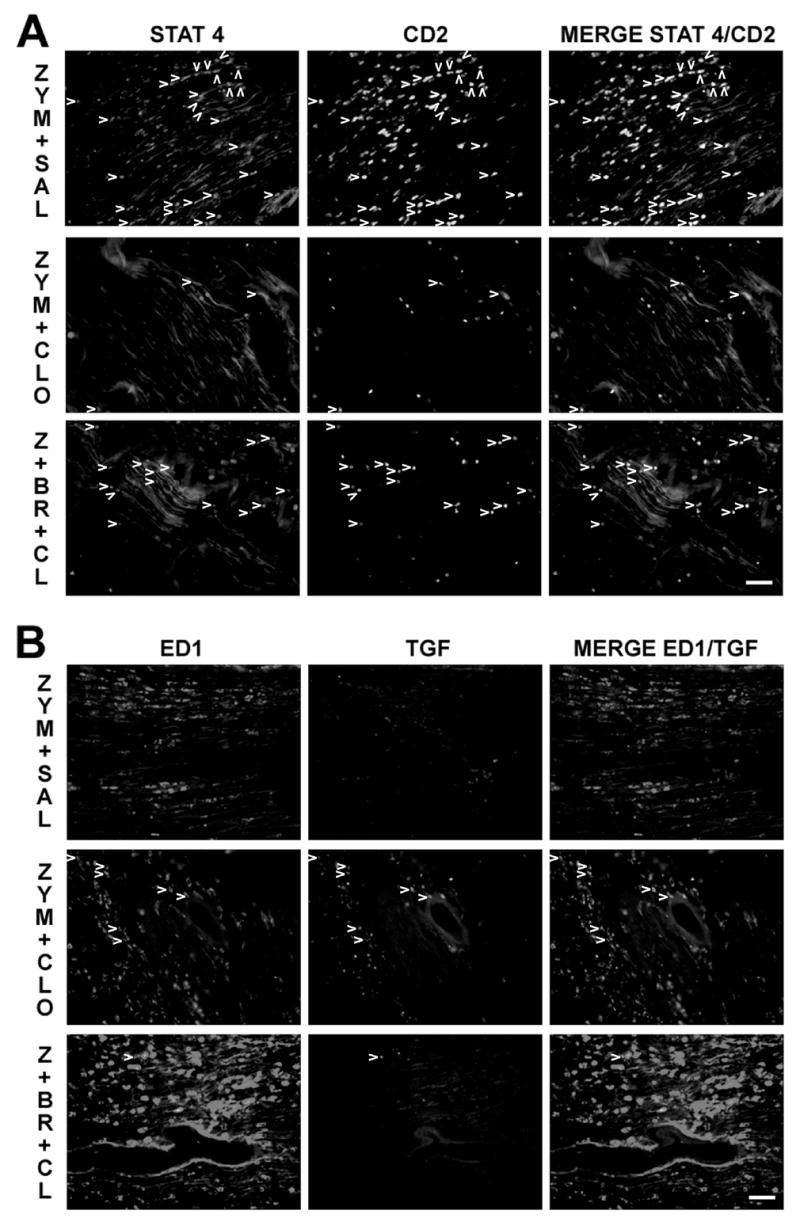
Clonidine’s effects and BRL44408 reversal of clonidine’s effects in lymphocytes, TGF-β1 (TGF), anti-inflammatory macrophages and Th-1 type lymphocytes in sciatic nerve 24 hr after treatments. A. Immunostaining of STAT4, CD2 and co-localization of both (arrow heads) in the sciatic nerve 24 hr after zymosan plus saline (upper panel, ZYM+SAL), zymosan plus clonidine (middle panel, ZYM+CLO) and zymosan plus BRL44408 plus clonidine group (lower panel, Z+BR+CL). B. Immunostaining of ED1, TGF-β1 and co-localization of both (arrow heads) in the sciatic nerve 24 hr after zymosan plus saline (upper panel) zymosan plus clonidine (middle panel) and zymosan plus BRL44408 plus clonidine group (lower panel). Scale bar = 50 μm.
Involvement of α2-adrenoceptors in clonidine’s effects
The α2-adrenoceptor antagonist, BRL44408, did not modify the reduction in CD2 immunostaining by clonidine 24 hr after its injection (data not shown). BRL44408 blocked clonidine-induced increase in the number of cleaved caspase-3 and TGF-β1 expressing cells 24 hr after its injection (Fig. 6A). BRL44408 also blocked the clonidine-induced increase in proportion of lymphocytes co-labeled with cleaved caspase-3 (Fig. 6B) and the clonidine-induced increase in macrophages co-expressing TGF-β1 (Fig. 6C). Finally, BRL44408 blocked the reduction of pro-inflammatory Th-1 type lymphocytes by clonidine (Fig. 6D).
Figure 6.
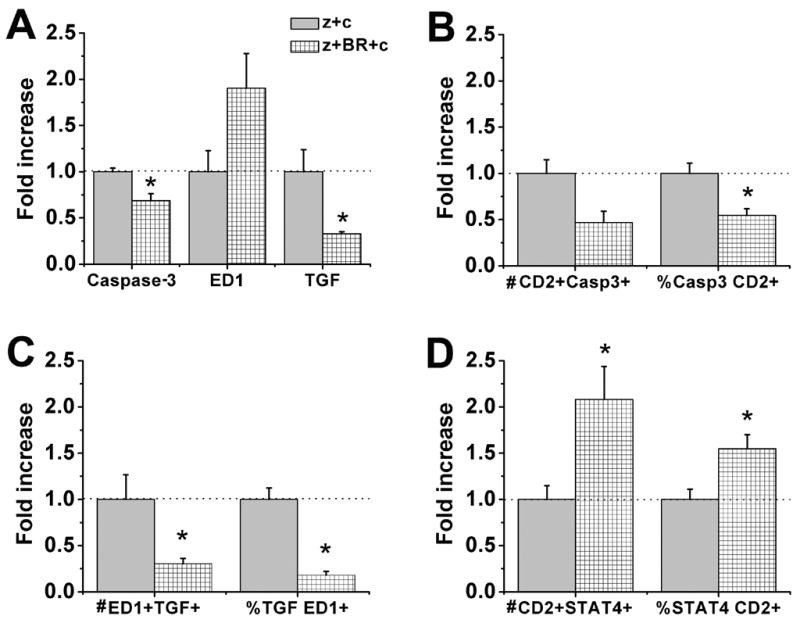
BRL44408 reversal of clonidine’s effect in apoptotic cells, lymphocytes, TGF-β1 (TGF), anti-inflammatory macrophages and Th-1 type lymphocytes in sciatic nerve 24 hr after treatments. Normalized data of zymosan plus clonidine (z+c) and zymosan plus BRL44408 and clonidine (z+BR+c). A. BRL44408 reversal of clonidine’s effect in caspase-3, ED1 and TGF-β1 immunostaining. B. BRL44408 reversal of clonidine’s effect in apoptotic lymphocytes analyzed as number of CD2 and caspase-3 co-localization immunostaining and proportion of caspase-3 co-expression in the CD2 set. C. BRL44408 reversal of clonidine’s effect in anti-inflammatory macrophages analyzed as number of ED1 and TGF-β1 co-localization immunostaining and proportion of TGF-β1 co-expression in the ED1 set. D. BRL44408 reversal of clonidine’s effect in pro-inflammatory Th-1 type lymphocytes analyzed as number of CD2 and caspase-3 co-localization immunostaining and proportion of STAT 4co-expression in the CD2 set. *P<0.05 comparison vs. zymosan plus clonidine group.
Lack of clonidine’s effect in cytokine production
Clonidine (1–100 μg, ex vivo) did not modify the production of IL-1β, IL-10 and TNFα in leukocytes previously activated in vivo with zymosan (Fig. 6).
Discussion
This study confirms the delayed antinociceptive effects of clonidine in acute sciatic neuritis, paralleled by a reduction in leukocyte number. Whereas previous studies examined the leukocytes in the inflammatory exudate captured in the gelfoam placed adjacent to the nerve (Romero-Sandoval et al., 2005), the current study examined cells in the sciatic nerve itself, which arguably are more important to pain and hypersensitivity. The current data suggest that clonidine, by an action on α2-adrenoceptors, acutely increases lymphocyte apoptosis and reduces pro-inflammatory and increases anti-inflammatory phenotypes of immune and other cells.
Zymosan and clonidine’s effects in hypersensitivity, leukocytosis and cell phenotype
Zymosan activates nuclear factor-kB by binding to Toll-like receptor 2 and -6 (Underhill et al., 1999; Young et al., 2001) which likely contributes to the production of pro-inflammatory cytokines (IL-1, IL-6, TNFα) and, in turn, to the leukocytosis observed in this and our previous studies (Romero-Sandoval et al., 2005). Both zymosan-induced leukocytosis and over-expression of pro-inflammatory cytokines play key roles in zymosan-induced hypersensitivity in the days after generation of the SIN model (Twining et al., 2004; Romero-Sandoval et al., 2005). Supporting this, the current study shows that clonidine reduced zymosan-induced hypersensitivity ipsilateral to neuritis, reduced the number of lymphocytes and induces an increase in presumed anti-inflammatory macrophages invading the sciatic nerve.
On the other hand, we did not observe a significant reduction in the number of macrophages after clonidine treatment, which contrasts with our previous study. The sensitivity of the different techniques used in both studies (immunohistochemistry vs. flow cytometry) may be the reason of this discrepancy, which suggests that stronger changes may be necessary in order to be detected by immunohistochemistry. Alternatively, there could be a differential effect of clonidine on inflammatory exudates surrounding tissue and in cells within the neural tissue itself.
Antinociception induced by clonidine as observed in the current study contrasts with expectations from previous studies that suggest that α-AR activation may contribute to peripheral nerve injury-induced pain (Sato and Perl 1991; Koltzenburg and McMahon. 1991; McLachlan et al., 1993). However, there is extensive evidence to support antinociception induced by activation of peri-neural α2-ARs (Lavand’homme et al., 2002; Lavand’homme and Eisenach, 2003). Our current findings confirm that peri-neural clonidine reduced behavioral hypersensitivity in neuritis and peripheral nerve injury pain models (Romero-Sandoval et al., 2005; Romero-Sandoval and Eisenach 2006).
Clonidine did not modify zymosan-induced mirror-image pain, a phenomenon driven by spinal glial activation and pro-inflammatory cytokines (Milligan et al., 2003). Our findings suggest that peripheral immune modulation produced by clonidine is not sufficient to change the immune response at the spinal cord, a hypothesis that is being tested in our laboratory.
Effects on anti-inflammatory factors
Zymosan increased IL-10 expression, an anti-inflammatory cytokine actively involved in the later resolution of inflammation in nerve injury, by reducing pro-inflammatory cytokines (Be’eri H et al., 1998). IL-10 increases in response to the increase in pro-inflammatory cytokine expression (de Waal Malefyt 2001). Additionally, zymosan increased TGF-β1 expression, an anti-inflammatory factor which is increased in response to apoptotic cell engulfment and leads to the resolution of inflammation (Fadok et al., 2000; Huynh et al., 2002). Therefore it is possible that IL-10 and TGF-β1 may be involved in the later resolution of the inflammatory response in the SIN model. Interestingly, clonidine did not alter zymosan-induced IL-10 and significantly increased TGF-β1 expression, further favoring the local anti-inflammatory environment and confirming our previous observations (Romero-Sandoval et al., 2005).
Clonidine modulates immune response by acting on α2-ARs
Clonidine, 24 hr after treatment, reduced the number of lymphocytes and, in a receptor-specific manner reduced the number of pro-inflammatory Th-1 cells and increased the expression of TGF-β1 and the number of presumed anti-inflammatory macrophages present in the sciatic nerve compared to the zymosan plus saline group. Despite these overall anti-inflammatory effects, clonidine did not modify zymosan-induced hypersensitivity at this earlier time. The reasons for this discrepancy are uncertain. Probably, the involvement of other cell types in this earlier stage may account for this discrepancy. Schwann cells, for example, are an important source of cytokines in the early phase of Wallerian degeneration (Reichert et al., 1994), and are likely important in neuritis-induced hypersensitivity. The lack of anti-hypersensitivity effects of clonidine 24 hr after its injection could be due to a lack of effect on these cells. Supporting this, we have observed that peripheral nerve injury induces the expression of α2-ARa in epineural connective tissue and in infiltrating macrophages and lymphocytes, but not in Schwann cells (Lavand’homme et al., 2002).
Zymosan-induced hypersensitivity at early phases depends primarily on cytokine production rather than on cell migration, since 3 and 24 hr after zymosan treatment leukocyte number does not increase in the area of injection, while resident leukocytes increase IL-1 and TNFα production (Gazda et al., 2001). Interestingly, and using the same conditions as previously described (Gazda et al. 2001), ex vivo clonidine did not acutely modify the capacity of in vivo zymosan-activated leukocytes to produce pro-inflammatory (TNFα and IL-1β) or anti-inflammatory (IL-10) cytokines. It is possible that peri-neural clonidine is not directly altering cytokine production by leukocytes in the early stage of neuritis, however we recognize that isolated cells may behave differently in ex vivo experiments.
BRL44408 reversal of clonidine’s effects on pro-inflammatory Th-1 cells, TGF-β1 expression and presumed anti-inflammatory macrophages acutely after its injection suggests that clonidine is acting on α2-ARs in this setting at this time point. Similarly, we have previously shown that transient BRL44408 exposure in the sciatic nerve reverses clonidine-induced antinociception 3 days after the treatment (Romero-Sandoval et al., 2005). Furthermore, BRL44408 also reverses the reduction by clonidine of the cytokine content in leukocytes in the inflammatory exudates induced as well as clonidine’s behavioral effects 3 days after treatment (Romero-Sandoval et al., 2005). Therefore, clonidine’s effects at the later period are likely a consequence of its acute effects described in this study.
When BRL4408 was co-administered with clonidine in the presence of zymosan, we observed an unexpected numerical increase in ED1 expressing cells. This finding could be considered to suggest an involvement of α2-ARs in cell migration at the earlier stage of neuritis, however this effect was not statistically significant. Additionally, clonidine did not modify the number of ED1 expressing macrophages in the presence of zymosan, therefore, we speculate that this observation could be due to a nonspecific effect of BRL44408.
Clonidine’s effect in apoptosis
To further understand how clonidine is inducing such changes in the immune response to acute neuritis we investigated its effects on apoptosis. This was based on previous observations that ingestion by macrophages of apoptotic cells actively suppresses their production of pro-inflammatory growth factors, cytokines, chemokines and eicosanoids (Fadok et al., 1998; McDonald et al., 1999; Huynh et al., 2002; Fadok et al., 2000), that α-AR agonists (including clonidine) induce apoptosis in several cell types including leukocytes (Stevenson et al., 2001; Uchida-Oka and Sugimoto, 2001; Dygalo et al., 2004), and that clonidine induces an increase in TGF-β1 expression in neuropathic pain conditions (Lavand’homme and Eisenach 2003), which could reflect a phenotypic switch by macrophages when ingesting apoptotic cells.
Zymosan induced an increase in apoptotic cells 3 days after its injection. However, clonidine further increased the number of apoptotic cells, both at this time and at 24 hr after injection. This effect was prevented by the co-administered α2-AR antagonist, in accordance with clonidine-induced apoptosis in developing brainstem (Dygalo et al., 2004) and in skin melanocytes by an action on α2-ARs to decrease cAMP-PKA signaling (Uchida-Oka and Sugimoto, 2001).
The majority of caspase-3 expressing cells in zymosan-inflamed sciatic nerve were neither macrophages nor lymphocytes. Zymosan increases cytokine production, and TNFα and IL-1β induce neutrophil apoptosis by means of caspase-3, -8 and -9 mechanisms (Salamone et al., 2004). It is conceivable that some of these cells were neutrophils.
Clonidine induced a ~4-fold higher proportion of apoptotic lymphocytes compared to zymosan plus saline group by an action on α2-ARs. This would provide an alternative explanation for the reduction in the number of lymphocytes induced by clonidine. BRL44408 blocked clonidine’s effect in apoptotic lymphocytes, but not clonidine’s effect in the number of lymphocytes. This agrees with previous data showing that α-AR agonists induce apoptosis in lymphocytes (Stevenson et al., 2001), but these data suggest that other mechanisms than induction of apoptosis must be operative in the reduction in lymphocyte number following clonidine treatment.
Clonidine-induced apoptosis, a possible key effect
Engulfment of apoptotic cells shifts macrophage phenotype to produce less pro-inflammatory cytokines and more anti-inflammatory factors, which in turn leads to resolution of inflammation (Fadok et al., 2000; Huynh et al., 2002). Additionally, TGF-β1 also modulates Th-1 type lymphocytes (Gorelik et al., 2002). In accordance with this, we also observed that clonidine, by acting on α2A-ARs, induced a reduction of Th-1 type cells and an increase of macrophages expressing TGF-β1, suggesting they are anti-inflammatory macrophages. Therefore, we speculate that the pro-apoptotic effects of zymosan may contribute to the resolution of inflammation, and that the pro-apoptotic effects of clonidine may be the key effect which drives the more rapid resolution of the neuritis-induced hypersensitivity following perineural clonidine treatment.
3. Conclusions
Our data are in accordance with previous observations that clonidine is effective to relieve hypersensitivity in neuritis as well as in peripheral nerve injury (Romero-Sandoval et al., 2005; Romero-Sandoval and Eisenach 2006), suggesting that clonidine may be effective to treat patients in chronic pain conditions due to nerve injury. In conclusion, this study complements our previous observations and demonstrates that perineural clonidine induces an anti-inflammatory immune response. We speculate that this is due in part to an acute induction of apoptosis in lymphocytes and other cells which reduces lymphocyte number and increases TGFβ-1 expression by macrophages and other cells to resolve the inflammatory process and hypersensitivity. These effects of clonidine are blocked by co-administration of a specific antagonist, consistent with an action on α2-ARs. This study enriches our understanding regarding the mechanism of action by which α2-AR activation may induce antinociceptive effects in persistent pain, produces new testable hypothesis regarding the role of apoptosis and anti-inflammatory factors in neuropathic pain, and provides the rationale for novel therapeutic strategies for such conditions.
Figure 7.
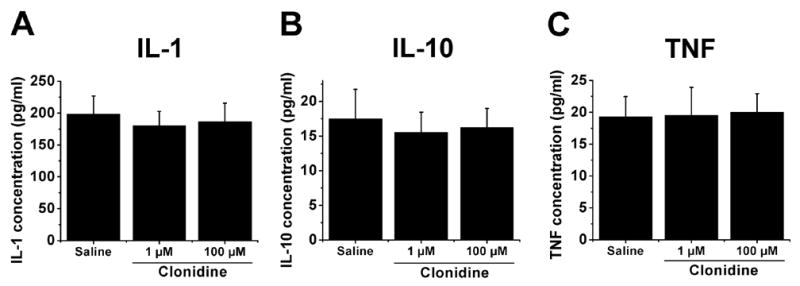
Lack of effect of clonidine in leukocytes cytokines production. IL-1β (IL-1, A), IL-10 (B) and TNFα (TNF, C) production by leukocytes activated in vivo with zymosan and retrieved 3 hr later from the gelfoam and incubated overnight in the absence (saline) or presence of clonidine in 1–100 μM concentrations.
Acknowledgments
This work was supported in part by National Institutes of Health Grants GM35523 and NS41386. We thank Julie Wieseler Frank for help with experimental details.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anavekar SN, Jarrott B, Toscano M, Louis WJ. Pharmacokinetic and pharmacodynamic studies of oral clonidine in normotensive subjects. Eur J Clin Pharmacol. 1982;23:1–5. doi: 10.1007/BF01061368. [DOI] [PubMed] [Google Scholar]
- Be’eri H, Reichert F, Saada A, Rotshenker S. The cytokine network of wallerian degeneration: IL-10 and GM-CSF. Eur J Neurosci. 1998;10:2707–2713. [PubMed] [Google Scholar]
- Chacur M, Milligan ED, Gazda LS, Armstrong C, Wang H, Tracey KJ, Maier SF, Watkins LR. A new model of sciatic inflammatory neuritis (SIN): induction of unilateral and bilateral mechanical allodynia following acute unilateral peri-sciatic immune activation in rats. Pain. 2001;94:231–244. doi: 10.1016/S0304-3959(01)00354-2. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Conway EL, Anavekar SN, Howes LG, Louis WJ. Pharmacokinetic comparison of a slow-release clonidine with a conventional formulation after acute and chronic administration in hypertensives. J Clin Pharmacol. 1992;32:427–433. doi: 10.1002/j.1552-4604.1992.tb03858.x. [DOI] [PubMed] [Google Scholar]
- de Waal Malefyt R. In: IL-10, Cytokine Reference. Oppenhheim JJ, Feldman M, editors. New York: Academic; 2001. pp. 165–85. [Google Scholar]
- Dygalo NN, Bannova AV, Kalinina TS, Shishkina GT. Clonidine increases caspase-3 mRNA level and DNA fragmentation in the developing rat brainstem. Brain Res Dev Brain Res. 2004;152:225–231. doi: 10.1016/j.devbrainres.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Rose DM, Pearson A, Ezekewitz RA, Henson PM. A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature. 2000;405:85–90. doi: 10.1038/35011084. [DOI] [PubMed] [Google Scholar]
- Gazda LS, Milligan ED, Hansen MK, Twining CM, Poulos NM, Chacur M, O’Connor KA, Armstrong C, Maier SF, Watkins LR, Myers RR. Sciatic inflammatory neuritis (SIN): behavioral allodynia is paralleled by peri-sciatic proinflammatory cytokine and superoxide production. J Peripher Nerv Syst. 2001;6:111–129. doi: 10.1046/j.1529-8027.2001.006001111.x. [DOI] [PubMed] [Google Scholar]
- Gorelik L, Constant S, Flavell RA. Mechanism of transforming growth factor beta-induced inhibition of T helper type 1 differentiation. J Exp Med. 2002;195:1499–1505. doi: 10.1084/jem.20012076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest. 2002;109:41–50. doi: 10.1172/JCI11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefsson E, Bergquist J, Ekman R, Tarkowski A. Catecholamines are synthesized by mouse lymphocytes and regulate function of these cells by induction of apoptosis. Immunology. 1996;88:140–146. doi: 10.1046/j.1365-2567.1996.d01-653.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koltzenburg M, McMahon SB. The enigmatic role of the sympathetic nervous system in chronic pain. Trends Pharmacol Sci. 1991;12:399–402. doi: 10.1016/0165-6147(91)90615-y. [DOI] [PubMed] [Google Scholar]
- Lavand’homme PM, Eisenach JC. Perioperative administration of the alpha2-adrenoceptor agonist clonidine at the site of nerve injury reduces the development of mechanical hypersensitivity and modulates local cytokine expression. Pain. 2003;105:247–254. doi: 10.1016/s0304-3959(03)00221-5. [DOI] [PubMed] [Google Scholar]
- Lavand’homme PM, Ma W, De Kock M, Eisenach JC. Perineural alpha(2A)-adrenoceptor activation inhibits spinal cord neuroplasticity and tactile allodynia after nerve injury. Anesthesiology. 2002;97:972–80. doi: 10.1097/00000542-200210000-00033. [DOI] [PubMed] [Google Scholar]
- McDonald PP, Fadok VA, Bratton D, Henson PM. Transcriptional and translational regulation of inflammatory mediator production by endogenous TGF-beta in macrophages that have ingested apoptotic cells. J Immunol. 1999;163:6164–6172. [PubMed] [Google Scholar]
- McLachlan EM, Janig W, Devor M, Michaelis M. Peripheral nerve injury triggers noradrenergic sprouting within dorsal root ganglia. Nature. 1993;363:543–546. doi: 10.1038/363543a0. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Hinde JL, Mehmert KK, Maier SF, Watkins LR. A method for increasing the viability of the external portion of lumbar catheters placed in the spinal subarachnoid space of rats. J Neurosci Methods. 1999;90:81–86. doi: 10.1016/s0165-0270(99)00075-8. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Twining C, Chacur M, Biedenkapp J, O’Connor K, Poole S, Tracey K, Martin D, Maier SF, Watkins LR. Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. J Neurosci. 2003;23:1026–1040. doi: 10.1523/JNEUROSCI.23-03-01026.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynihan J, Kruszewska B, Madden K, Callahan T. Sympathetic nervous system regulation of immunity. J Neuroimmunol. 2004;147:87–90. doi: 10.1016/j.jneuroim.2003.10.018. [DOI] [PubMed] [Google Scholar]
- Reichert F, Saada A, Rotshenker S. Peripheral nerve injury induces Schwann cells to express two macrophage phenotypes: Phagocytosis and the galactosespecific lectin MAC-2. J Neurosci. 1994;14:3231–3245. doi: 10.1523/JNEUROSCI.14-05-03231.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Sandoval A, Eisenach JC. Perineural clonidine reduces mechanical hypersensitivity and cytokine production in established nerve injury. Anesthesiology. 2006;104:351–5. doi: 10.1097/00000542-200602000-00022. [DOI] [PubMed] [Google Scholar]
- Romero-Sandoval EA, McCall C, Eisenach JC. Alpha2-adrenoceptor stimulation transforms immune responses in neuritis and blocks neuritis-induced pain. J Neurosci. 2005;25:8988–8994. doi: 10.1523/JNEUROSCI.2995-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamone G, Trevani A, Martinez D, Vermeulen M, Gamberale R, Fernandez-Calotti P, Raiden S, Giordano M, Geffner J. Analysis of the mechanisms involved in the stimulation of neutrophil apoptosis by tumour necrosis factor-alpha. Immunology 2004. 2004;113:355–62. doi: 10.1111/j.1365-2567.2004.01973.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato J, Perl ER. Adrenergic excitation of cutaneous pain receptors induced by peripheral nerve injury. Science. 1991;251:1608–1610. doi: 10.1126/science.2011742. [DOI] [PubMed] [Google Scholar]
- Spengler RN, Allen RM, Remick DG, Strieter RM, Kunkel SL. Stimulation of alpha-adrenergic receptor augments the production of macrophage-derived tumor necrosis factor. J Immunol. 1990;145:1430–1434. [PubMed] [Google Scholar]
- Stevenson JR, Westermann J, Liebmann PM, Hortner M, Rinner I, Felsner P, Wolfler A, Schauenstein K. Prolonged alpha-adrenergic stimulation causes changes in leukocyte distribution and lymphocyte apoptosis in the rat. J Neuroimmunol. 2001;120:50–57. doi: 10.1016/s0165-5728(01)00417-9. [DOI] [PubMed] [Google Scholar]
- Titnchi S, Clark B. Alpha-2 adrenoceptors in human lymphocytes: direct characterisation by [3H]-yohimbine binding. Biochem Biophys Res Commun. 1984;121:1–7. doi: 10.1016/0006-291x(84)90679-x. [DOI] [PubMed] [Google Scholar]
- Twining CM, Sloane EM, Milligan ED, Chacur M, Martin D, Poole S, Marsh H, Maier SF, Watkins LR. Peri-sciatic proinflammatory cytokines, reactive oxygen species, and complement induce mirror-image neuropathic pain in rats. Pain. 2004;110:299–309. doi: 10.1016/j.pain.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Uchida-Oka N, Sugimoto M. Norepinephrine induces apoptosis in skin melanophores by attenuating cAMP-PKA signals via alpha2-adrenoceptors in the medaka, Oryzias latipes. Pigment Cell Res. 2001;14:356–361. doi: 10.1034/j.1600-0749.2001.140507.x. [DOI] [PubMed] [Google Scholar]
- Underhill DM, Ozinsky A, Hajjar AM, Stevens A, Wilson CB, Bassetti M, Aderem A. The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature. 1999;401:811–815. doi: 10.1038/44605. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Maier SF, Goehler LE. Immune activation: the role of proinflammatory cytokines in inflammation, illness responses and pathological pain states. Pain. 1995;63:289–302. doi: 10.1016/0304-3959(95)00186-7. [DOI] [PubMed] [Google Scholar]
- Young SH, Ye J, Frazer DG, Shi X, Castranova V. Molecular mechanism of tumor necrosis factor-alpha production in 133-beta-glucan (zymosan)-activated macrophages. J Biol Chem. 2001;276:20781–20787. doi: 10.1074/jbc.M101111200. [DOI] [PubMed] [Google Scholar]