

Abstract
Human CD34 cells express low levels of the DNA repair protein O6-alkylguanine-DNA alkyltransferase (AGT) and are sensitive to 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU). Gene transfer of the AGT gene, methylguanine DNA methyltransferase (MGMT), results in only modest BCNU resistance. Recently, an AGT inhibitor, O6-benzylguanine (BG), entered clinical trials. In preclinical studies, BG potentiated the cytotoxic effect of BCNU in tumors but increased toxicity to normal CD34 cells. We transferred a mutant MGMT containing a glycine-to-alanine mutation at position 156, resulting in marked resistance to BG, into Chinese hamster cells; the K562 cell line and human CD34 cells used the retroviral backbone MFG. In each instance, cells expressed increased AGT and were much more resistant to the combination of BG and BCNU than the parental cells or cells transduced with wild-type MGMT. Furthermore, the transduction efficiency in human CD34 cells was in excess of 70%, and the proportion of CD34 transduced cells resistant to the combination was >30%. Thus, retroviral-mediated transduction of a mutant MGMT into CD34 cells appears to be an effective way to induce selective resistance to a drug combination designed to overcome a significant resistance mechanism to nitrosoureas in tumors.
Transfer of a drug resistance gene such as dihydrofolate reductase (1, 2), multiple drug resistance-1 (3, 4, 5), and aldehyde dehydrogenase (6) into hematopoietic progenitors is a proposed method to protect the bone marrow from the toxic effects of chemotherapeutic agents, improving tolerance to anticancer agents, and perhaps providing a means for dominant selection of a second therapeutic gene. We (7, 8) and others (9, 10) have recently advocated the use of methylguanine DNA methyltransferase gene (MGMT), the gene that encodes the DNA repair protein O6-alkylguanine-DNA alkyltransferase (AGT), in this setting for a number of reasons. First, AGT confers resistance to multiple alkylating agents including nitrosoureas, dacarbazine, temozolomide, and procarbazine. Second, hematopoietic cells and in particular CD34 hematopoietic progenitors contain low levels of this protein (11, 12), explaining the common finding of dose-limiting, cumulative myelosuppression. Third, there are clear animal and clinical examples of delayed hematopoietic toxicity with the use of these agents including marrow hypoplasia and secondary leukemias (10, 13), suggesting that the sensitivity to nitrosoureas and related compounds extends to early hematopoietic progenitors.
The mechanism of action of the alkyltransferase is unique among DNA repair enzymes. The protein serves as the acceptor of DNA-alkylations at the O6 position of guanine, the site of one of the most cytotoxic lesions formed by both chloroethylating and methylating agents (14, 15). Repair proceeds by covalent transfer of the adduct to the active site of the protein, a “suicide” process. Cytotoxicity of unrepaired O6-chloroethylguanine lesions is due to intramolecular rearrangement to O6-N1-ethanoguanine followed by interstrand crosslink formation (16), whereas that of the methylating agents is due to recognition of the O6-methylguanine·cytosine or O6-methylguanine·thymine (formed after replication) base mispair by the mismatch repair complex and induction of aberrant repair processes leading to multiple DNA strand breaks (17).
We have recently shown that overexpression of MGMT protects hematopoietic cells from the toxic effects of nitrosoureas both in vitro and in vivo. MGMT overexpression in thymic T cells of the mouse prevents methylnitrosourea-induced “secondary” T-cell lymphoma/leukemias (18) and results in rapid loss of the offending O6-methylguanine DNA adduct from cortical T-lymphocytes (19, 20), the target for the induction of the lymphoma/leukemias. In retroviral gene transfer studies, murine bone marrow cells transduced with human MGMT showed 40-fold overexpression of AGT and an increase in 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) resistance in hematopoietic progenitors at least 37 weeks after transplantation into isogeneic mice (7). BCNU treatment of transplanted mice increased the proportion of hematopoietic progenitors carrying the provirus and the mean level of expression of human MGMT (21). Likewise, Maze et al. (10) showed a survival advantage to mice transplanted with MGMT-transduced marrow cells and repeatedly exposed to BCNU. Finally, MGMT gene transfer into human CD34 cells resulted in a slight increase in clonogenic survival in vitro at high concentrations of BCNU (8).
Although MGMT overexpression increases BCNU resistance in normal murine and human hematopoietic cells, the effect has been quite modest, raising questions about its therapeutic utility. Two recent advances have shifted our approach toward therapeutic transfer of MGMT into human CD34 cells. First, O6-benzylguanine (BG) was described as a potent inactivator of the AGT capable of enhancing the antitumor efficacy of BCNU in cell lines and in human tumor xenografts (14, 22, 23, 24, 25). Since many tumors express high levels of AGT and are resistant to BCNU and related compounds, BG represents a significant advance in the approach to biochemical modulation of tumor drug resistance (25). Not surprisingly, in preclinical toxicology, myelosuppression was dose limiting with BG and BCNU (26). In early phase 1 trials with BG alone, we have observed significant inhibition of the alkyltransferase in human lymphocyte and tumor samples without toxicity (27); however, it appears likely that as the dose of BCNU is increased, myelosuppression will be dose limiting.
Second, a number of AGT point mutations that have been characterized retain DNA repair activity but are remarkably resistant to inactivation by BG. One of these, the glycine-to-alanine mutation at position 156 (G156A) in the human protein results in 240-fold resistance to BG (28). For this reason, we engineered the MFG retroviral vector to contain the G156A mutant MGMT (ΔMGMT) and studied its ability to confer resistance to the combination of BG and BCNU in human CD34 cells. Transduced cells are remarkably resistant to this combination compared with untransduced cells or cells transduced with wild-type MGMT (wtMGMT). Thus, our results support the potential therapeutic use of the ΔMGMT in vivo to protect hematopoietic cells from the combination of BG and BCNU.
METHODS
Retroviral Vectors.
Retroviral vectors pMFG-wtMGMT and pMFG-ΔMGMT were constructed by inserting the wild-type and G156A mutant human MGMT cDNA coding sequences, respectively, into the unique NcoI and BamHI restriction sites of pMFG (Paul Robbins, University of Pittsburgh; 29). The NcoI containing the ATG start codon and BamHI sites at the 5′ and 3′ termini of human MGMT cDNA sequence, respectively, were generated by PCR amplification with primers 5′-p, 5′-CTTGGAACCATGGACAAGGATTGTGAAA-3′; and 3′-p, 5′-CTTAGGATCCCATCCGATGCAGTGTTACACG-3′. ΔMGMT cDNA was generated by site-directed PCR mutagenesis in two steps using oligonucleotides containing GCC in place of GGC at codon 156. Sequences were confirmed by the dideoxynucleotide chain termination method (fmol DNA sequencing system, Promega).
Transfection of the Vector Constructs into CHO Cells.
A total of 6 μg of each plasmid (pMFG-wtMGMT or pMFG-ΔMGMT) was cotransfected into 1.8 × 106 CHO cells with 0.6 μg of pSV2neo plasmid DNA with LipofectAMINE (GIBCO/BRL), selected in G418 (1 g/liter), and screened for AGT activity.
Virus-Producing Cells.
pMFG-wtMGMT or pMFG-ΔMGMT and pSV2neo DNA were cotransfected into the packaging line GP + E86, followed by viral supernatant infection of the amphotropic cell line GP + envAm12 (30) (Arthur Bank, Columbia University). To increase titer, a supernatant “ping-pong” method was used (31). The amphotropic ΨCRIP MFG-lacZ cell line was from Paul Robbins. Titer was estimated from supernatants collected after six daily media changes (7) by infecting 1 × 105 K562 cells (as described below) with limiting dilutions of viral supernatant.
K562 Transduction.
The human chronic myelogenous leukemia cell line K562 was retrovirally transduced as described (7). Briefly, wtMGMT and ΔMGMT producers were treated with 10 μg/ml mitomycin C and replated. Twenty-four hours later, K562 cells were added in the presence of human interleukin 3 (IL-3) (100 units/ml), human granulocyte–macrophage colony-stimulating factor (GM-CSF) (200 units/ml), and polybrene (8 μg/ml) and were collected after a 48-hr coculture.
CD34 Transduction.
Peripheral blood mononuclear cells were obtained by apheresis from adult patients treated with cyclophosphamide and/or granulocyte-CSF under an Institutional Review Board-approved protocol (32). CD34 progenitors were isolated using the Ceprate LC Stem Cell Concentrator (Shelly Helmfeld, CellPro, Bothell, WA), followed by an incubation with biotinylated anti-CD34 antibody, passage over an avidin column, and elution by gentle agitation. Recovered cells had an average purity of 57%. For coculture infection, fresh CD34 cells (1 × 105 cells/ml) were cultured for 96 hr in Iscove’s modified Dulbecco’s medium (GIBCO/BRL) containing 20% heat-inactivated fetal calf serum and supplemented with human stem cell factor (100 ng/ml, Amgen Biologicals), IL-3 (100 units/ml) and IL-6 (100 units/ml) (both from Sandoz Pharmaceutical), and protamine sulfate (4 μg/ml, Sigma) and then were cultured with MFG-ΔMGMT, MFG-wtMGMT, or MFG-lacZ producers. At 48 hr, half of the medium was replaced. For retroviral supernatant infection, CD34 cells were prepared as above and cultured on human bone marrow stroma initiated as follows. Bone marrow mononuclear cells were cultured in myeloid long-term culture medium (Stem Cell Technologies, Vancouver, B.C., Canada) at 2 × 106 cells/ml for 3 days at 37°C/5% CO2 and transferred to 33°C/5% CO2, passaged once, and irradiated with 15 Gy 24 hr prior to use. ΔMGMT or lacZ retroviral supernatant collected from confluent producer cells as described (7) was changed at 24-hr intervals and cultured with CD34 cells as described above. After 72 hr, cells were removed from the stromal layer with cell dissociation buffer (GIBCO/BRL).
In Vitro BG/BCNU Treatment.
Cells were resuspended in serum-free medium containing 100 units/ml GM-CSF with or without 10 μM BG and incubated at 37°C for 1 hr (12). BCNU was added to the cells for 2 hr (7), after which cells were incubated for 7–10 days in methylcellulose (Stem Cell Technologies) containing either stem cell factor, IL-3, hemin, erythropoietin, and GM-CSF (8) (for CD34 cells) or GM-CSF and IL-3 (for K562 cells) in triplicate, with or without 5 μM BG.
Immunoassay.
Cytospin preparations were stained for AGT using the monoclonal antibody mT3.1 (D. Bigner, Duke University) and biotin/avidin horseradish peroxidase system (Vector Laboratories). Western blots were done as described (33), and the bound antibody was detected by the chemiluminescence ECL kit (Amersham). AGT activity was correlated to densitometric band intensity using MGMT-transduced K562 cells (7).
AGT Assay.
AGT activity was measured as [3H]methyl groups removed from [3H]O6-methylguanine present in [3H]methylnitrosourea-treated alkylated calf thymus DNA. The alkylated [3H-methyl]O6-methylguanine and N7-methylguanine bases were separated by HPLC and quantified by liquid scintillation. AGT activity was expressed as fmol O6-methylguanine removed per μg of DNA (34).
PCR Provirus Analysis.
DNA was isolated from single colonies as described (7). A 152-bp human MGMT fragment that amplifies only the provirus and not endogenous sequences and a 295-bp human dystrophin fragment were amplified and separated by 2% agarose gel.
Reverse Transcription–PCR Analysis.
RNA was prepared from 5 to 10 methylcellulose colonies as described (7). In the presence of reverse transcriptase, a 497-bp fragment was amplified using the sense primer (5′-TGGTACCTCACCCTTACCGAGTC-3′) containing sequences of the MFG proviral backbone and the antisense primer (5′-ACACCTGTCTGGTGAACGACTCT-3′) specific to human MGMT.
Helper Virus Assay.
After retroviral supernatant infection of K562 and NIH 3T3 cells, supernatants were collected and cultured with fresh NIH 3T3 cells or NIH lac cells. The former were analyzed for the presence of proviral sequences by PCR. The latter were analyzed for transmission of lac plus virus into naive NIH 3T3 cells. Replication-competent virus was not detected in supernatants tested by either assay.
RESULTS
Expression of AGT and BG Resistance in CHO Cells.
ΔAGT protein levels and activity was compared with wtAGT in transfected CHO cells by enzyme assay and Western blot analysis. AGT activity was 3.5 fmol/μg DNA in ΔMGMT-transfected CHO cells compared with 84 fmol/μg DNA in wtMGMT transfectants, providing independent confirmation that the G156A mutant AGT protein is functional in mammalian cells (35). In CHO cells the EC50 for ΔAGT inhibition was ≈30 μM BG compared with <0.1 μM BG for wtAGT (Fig. 1A).
Figure 1.
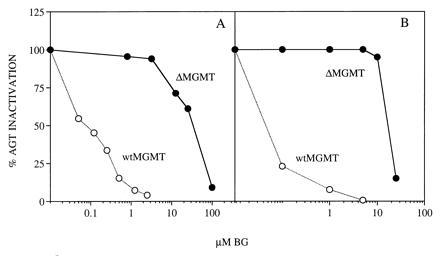
BG inactivation of wtAGT and ΔAGT. wtMGMT or ΔMGMT transfected CHO (A) or transduced K562 (B) cells were exposed to BG for 1 hr. Cells were harvested immediately, and AGT activity in protein extracts was determined by biochemical assay.
Titer.
MFG-ΔMGMT titer from serial dilutions of Am-12 supernatant was estimated by immunohistochemical detection of infected K562 cells (Fig. 2). A clone with a titer of 5 × 105 AGT-positive infectious particles per ml was used for further experiments. Of note, ΔAGT staining was nuclear, indicating that the mutant protein retained its nuclear localization.
Figure 2.
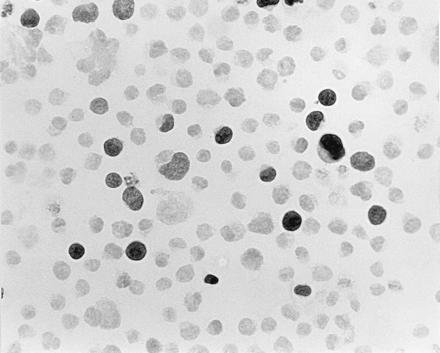
Immunohistochemical detection of AGT in ΔMGMT-transduced K562 cells. K562 cells (1 × 105) were infected with dilutions of amphotropic supernatant collected from Am12ΔMGMT clone 20. At a 1:50 dilution of supernatant, ≈10% of the cells expressed nuclear human AGT when reacted with monoclonal antibody mT3.1.
Expression and Drug Resistance in K562 Cells.
K562 cells were retrovirally infected by coculture with wtMGMT or ΔMGMT Am-12 producer cells. AGT activity and resistance to BG were compared in the two cell cultures without prior selection. Mean AGT levels were 25.7 fmol/μg DNA in wtMGMT-transduced cells, 2.7 fmol/μg DNA in ΔMGMT-transduced cells, and undetectable in uninfected cells. The EC50 for BG inactivation of AGT was <0.1 μM in wtMGMT compared with ≈18 μM in ΔMGMT-transduced cells. At 5 μM BG, ΔAGT-containing cells retained >90% activity whereas wtAGT was undetectable (Fig. 1B). To determine whether ΔAGT expression could increase tolerance to BG and BCNU, transduced K562 cells were exposed to 10 μM BG and/or various concentrations of BCNU and plated in methylcellulose. Clonogenic ΔMGMT and wtMGMT cells were resistant to BCNU alone. In contrast, ΔMGMT-transduced cells were significantly more resistant to BG and BCNU than wtMGMT-transduced or untransduced cells (Fig. 3). After BG treatment, the BCNU IC50 was 11.3 vs. 4 vs. 1.3 μM and the IC90 was >30 vs. 16 vs. 5 μM, for ΔMGMT, wtMGMT, and untransduced cells, respectively. Furthermore, ΔMGMT-transduced K562 cells maintained 20% clonogenic survival at 30 μM BCNU and BG compared with <1% of cells transduced with wtMGMT. Proviral integration was assessed by PCR amplification of a 152-bp MGMT fragment and identified in 22 of 33 colonies (67%) (data not shown). AGT immunoreactive protein levels detected in pooled colonies from wtMGMT-infected K562 cells were 10-fold higher than in ΔMGMT-transduced cells (Fig. 4).
Figure 3.
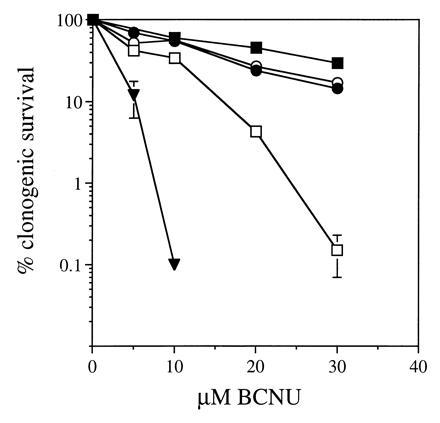
Clonogenic survival of transduced K562 cells after treatment with BG and/or BCNU. K562 cells transduced with wtMGMT (▪), ΔMGMT (•), and untransduced cells (▾) were treated with 0–40 μM BCNU only; wtMGMT (□) and ΔMGMT (○) were treated with 10 μM BG followed by 0–30 μM BCNU. Cells were plated in methylcellulose in triplicate, and colonies were enumerated in 7 days. Error bars represent mean ± SD.
Figure 4.
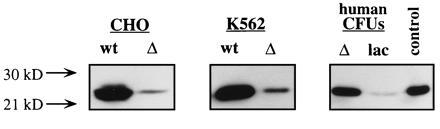
Western blot analysis of human AGT expression. Protein extracts from CHO cells (lanes 1 and 2) transfected with wtMGMT and ΔMGMT and K562 cells (lanes 3 and 4) and pooled human hematopoietic progenitors (lanes 5 and 6) transduced with wtMGMT, ΔMGMT, or lacZ were separated on a 10% SDS/PAGE gel. Blots were immunoreacted with monoclonal antibody mT3.1, and the 22-kDa human AGT was visualized by chemiluminescent substrate. AGT activity was estimated by correlation of band intensity to a protein extract expressing known levels of AGT (lane 7).
Expression and Drug Resistance in Human CD34 Cells.
The nonadherent cell count in cocultures of human CD34 cells and MFG-ΔMGMT, wtMGMT, or MFG-lacZ producers increased 5-fold over 96 hr due to growth factor stimulation. After infection, cells were treated with either BCNU alone or with BCNU and 10 μM BG and then plated in methylcellulose. wtMGMT and ΔMGMT-transduced CD34 cells had only a small increase in resistance to BCNU alone compared with lacZ-transduced cells as we have reported for wtMGMT (8). However, after pretreatment with 10 μM BG to deplete wtAGT, a striking resistance to BCNU was observed in ΔMGMT-transduced CD34 cells (Fig. 5A). The divergence between the lacZ-, wtMGMT-, and ΔMGMT-transduced clonogenic progenitor cell survival increased as the dose of BCNU increased. Thus, relative to the clonogenic survival of cells transduced with lacZ, the survival of ΔMGMT was 73.7 ± 10.6 at the IC50 and 49.0 ± 24.2 at the IC90. Approximately 25% of ΔMGMT-transduced clones were resistant to 10 μM BG and 10 μM BCNU, a dose that killed >99% of lacZ-transduced hematopoietic progenitor cells. Additionally, ΔMGMT-transduced progenitors maintained 20% survival compared with 1% of wtMGMT-transduced progenitors at 20 μM BCNU.
Figure 5.
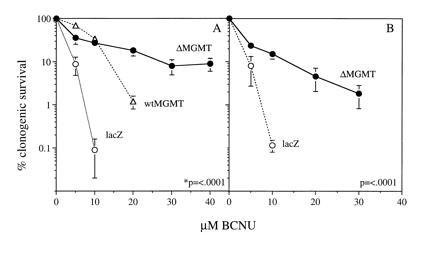
Clonogenic survival of transduced CD34 cells following treatment with BG plus BCNU. CD34 cells were transduced by coculture method (A) or retroviral supernatant in the presence of allogenic stroma (B). Transduced cells were treated with 10 μM BG and 0–40 μM BCNU and plated in methylcellulose in triplicate, and colonies were enumerated in 7–10 days. Data points represent the mean ± SEM of five experiments (A) and three experiments (B) from separate donors. P < 0.001 for the comparison ΔMGMT vs. lacZ.
We also transduced CD34 cells with retroviral supernatant in the presence of human bone marrow stroma, an approach currently used in clinical protocols. Similar to results obtained by coculture transduction, ΔMGMT-transduced cells were significantly more resistant to the drug combination than those transduced with lacZ (Fig. 5B). The BCNU IC50 was 3 ± 0.5 vs. 1.5 ± 0.72, and the IC90 was 13.3 ± 1.5 vs. 3.6 ± 1.5 for ΔMGMT and lacZ, respectively. Following ΔMGMT infection, PCR amplification of proviral MGMT was observed in 19 of 27 (70%) individual erythroid burst-forming unit (BFU-E) and granulocyte/macrophage colony-forming unit (CFU-GM) colonies. ΔMGMT mRNA expression was detected in pooled colony samples by reverse transcription–PCR (Fig. 6). AGT levels detected by Western blot were 6-fold higher in ΔMGMT-transduced progenitor colonies than lacZ-transduced progenitors (Fig. 4).
Figure 6.
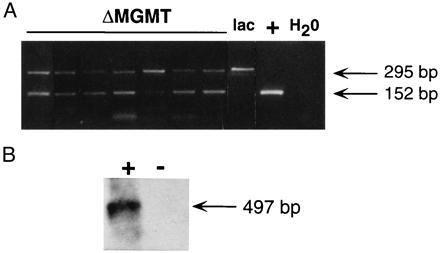
ΔMGMT integration and expression in human hematopoietic progenitor colonies. (A) Representative PCR amplification of a 152-bp MGMT fragment and a 295-bp human dystrophin fragment as an internal control. Analysis of seven MGMT and one lacZ transduced individual progenitor colonies obtained from methylcellulose on day 10 are shown. An MFG-ΔMGMT plasmid was used as a positive control. (B) Reverse transcription–PCR amplification from pooled methylcellulose colonies as described above. After DNase treatment, a 497-bp retroviral-specific fragment was amplified in the presence of reverse transcriptase (+) but not in the absence of reverse transcriptase (−), indicating complete digestion of DNA. PCR product was detected by hybridization with a 32P random primed MGMT cDNA.
DISCUSSION
A drug resistance gene selectively expressed in hematopoietic progenitors may provide a distinct therapeutic advantage during antineoplastic chemotherapy. In these studies, expression of the mutant drug resistance protein, G156AΔAGT, in human CD34 cells following retroviral transduction increased resistance to the combination of BG and BCNU above that seen in CD34 cells which express either endogenous or transduced wtAGT.
Enthusiasm for wtMGMT as a drug resistance gene emerged because cell lines lacking AGT and sensitive to BCNU became resistant after wtMGMT transduction (36). However, transduction of cells expressing endogenous AGT results in less enhancement of BCNU resistance than transduction of nonexpressing cells (37). In murine systems, Allay et al. (7) and Moritz et al. (38) found only about a 2-fold increase in BCNU resistance after retroviral-mediated gene transfer of wtMGMT into hematopoietic cells. Since transduced genes often are expressed at lower levels in human CD34 cells than in cell lines (7), it is not surprising that transduction of CD34 cells with wtMGMT had little effect on BCNU resistance except at high drug concentrations (8).
ΔMGMT gene transfer into CD34-derived colonies was efficient (70% by PCR of proviral sequences) and similar to the transduction rates reported in CD34 cells for dihydrofolate reductase using producer cell coculture and cytokine stimulation (2). Transduction of ΔMGMT into CD34 cells resulted in enhancement of clonogenic survival after BG and BCNU treatment. Remarkably, >30% of colony-forming progenitors appeared very resistant to BG and BCNU, compared with none of the lacZ-transduced cells or the nontransduced progenitors as reported (12). This degree of drug resistance compares favorably with that reported after multidrug resistance gene transfer (4, 5).
Recent evidence has shown that BG is an effective modulator of tumor–BCNU resistance in preclinical models and is well tolerated in early clinical studies (23, 27). However, it seems likely that the dose-limiting toxicity of BG and BCNU will be myelosuppression (26). Following reintroduction of ΔMGMT-transduced CD34 cells in vivo, a significant survival advantage is expected with less rather than more myelosuppression after repeated doses of BG and BCNU. Furthermore, since chloroethylnitrosoureas, procarbazine, and dacarbazine have been implicated in secondary leukemias (15, 39), it is possible that BG depletion of AGT will increase this risk, limiting the utility of the combination. Thus, another rationale for the use of ΔMGMT gene transfer is to protect hematopoietic cells sufficiently to prevent late oncogenic events.
These results suggest that ΔMGMT cDNA is a better drug-resistance gene candidate than wtMGMT because the relative protection of ΔMGMT-transduced CD34 cells against BG and BCNU is greater than observed with wtMGMT and BCNU alone (8, 10, 38). It is of interest that overexpression of wtMGMT slightly improves resistance to BG and BCNU, perhaps because of the increased rate of synthesis of AGT from the transgene. Since AGT has a much higher rate of reaction with O6-alkylguanine DNA adducts compared with BG (40), AGT molecules synthesized after BCNU-induced DNA damage may preferentially repair the adduct rather than be inactivated by BG, which slightly increases resistance to BCNU. Nonetheless, our results indicate remarkable protection from BG and BCNU in hematopoietic cells expressing ΔMGMT, and it would not appear appropriate to pursue the use of wtMGMT in this context. It is noteworthy that we observed higher levels of wild-type immunoreactive and functional AGT compared with mutant AGT levels in transfected CHO cells as well as transduced hematopoietic cells. This is in contrast to Loktionova and Pegg (35), who demonstrate comparable mutant and wild-type AGT levels in CHO cells. One explanation may be that the previous study compared clones selected in 80 μM BCNU, whereas our transfectants were selected based on their resistance to G418 only and not to BCNU.
Transfer of the bacterial ada gene, which encodes one of two bacterial alkyltransferases, into murine hematopoietic progenitors is another approach to BG-resistant alklytransferase gene transfer into hematopoietic cells. The ada-encoded alkyltransferase is resistant to BG when expressed in cell-free extracts and mammalian cells (41). Moore et al. (42) described the crystal structure of the Ada protein in which the active site cysteine is buried and must be exposed to react with the adduct at O6 of guanine. This conformational change may be hindered by the presence of threonine at the site comparable to Gly-156 in mammalian AGTs and may explain the marked resistance to reaction with BG in both the Ada protein and ΔAGT.
Using retroviral gene transfer of the ada gene, Harris et al. (9) noted increased resistance of murine hematopoietic cells to BG and BCNU in vitro and a survival advantage for transplanted mice treated with the combination. Differences in survival between transduced and nontransduced cells were not as high in the Harris study as those noted here. First, there is evidence that the bacterial protein is not well-nuclear-localized and may not be an efficient DNA repair protein in eukaryotic cells even if overexpressed (37). Second, mouse AGT has a higher EC50 for BG than human AGT (43), so the degree of protection noted in mouse cells cannot be directly converted to that expected in human CD34 cells. Finally, it is possible that an immune response would develop against the bacterial alkyltransferase in vivo that would not be expected with human ΔAGT.
As a result of low AGT levels in early hematopoietic progenitors (12), these cells are susceptible to cumulative cytotoxicity from nitrosoureas and related alkylating agents. Furthermore, homozygous disruption of MGMT in mice results in severe myelosuppression and subsequent death within 17 days of treatment with methylnitrosourea (44). Conversely, overexpression of MGMT in transgenic animals provides protection against T-cell leukemias (18). Our previous studies have shown that AGT levels are not increased in CD34 cells after drug or cytokine exposure (12, 45, 46), suggesting that overexpression of ΔMGMT mediated by gene therapy may be the only approach to increase nitrosourea resistance in progenitor cells.
In summary, overexpression of ΔMGMT in CD34 cells results in resistance to the chemotherapeutic combination of BG and BCNU, suggesting that gene therapy with ΔMGMT rather than wtMGMT will provide significant, selective protection of hematopoietic cells compared with tumor cells in the clinical setting.
Acknowledgments
We thank Dr. Hillard Lazarus for providing the CD34 cells, Dr. Darell Bigner for the mT3.1 monoclonal antibody for detection of AGT, and Dr. Anthony Pegg for insightful comments during the course of these studies. This work was supported in part by Public Health Service Grants R01ES06288, UO1CA75525, R01CA63193, and P30CA43703.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: BG, O6-benzylguanine; AGT, O6-alkylguanine-DNA alkyltransferase protein; MGMT, methylguanine DNA methyltransferase gene; wtMGMT, wild-type MGMT; ΔMGMT, G156A mutant MGMT; BCNU, 1,3-bis(2-chloroethyl)-1-nitrosourea; GM-CSF, granulocyte–macrophage colony-stimulating factor; IL, interleukin.
References
- 1.Corey C, DeSilva A, Holland C, Williams D. Blood. 1990;75:337–343. [PubMed] [Google Scholar]
- 2.Flasshove M, Banerjee D, Mineishi S, Li M, Bertino J, Moore M. Blood. 1995;85:566–574. [PubMed] [Google Scholar]
- 3.Sorrentino B, Brandt S, Bodine D, Gottesman M, Pastan I, Cline A, Nienhuis A. Science. 1992;257:99–103. doi: 10.1126/science.1352414. [DOI] [PubMed] [Google Scholar]
- 4.Ward M, Ayello J, Reiss R, Ursi G, Richardson C, Hesdorffer C, Bank A. Clin Cancer Res. 1996;2:873–882. [PubMed] [Google Scholar]
- 5.Bertolini F, DeMonte L, Corsini C, Lazzari L, Lauri E, Soligo D, Ward M, Bank A, Malavasi F. Br J Haematol. 1994;88:318–324. doi: 10.1111/j.1365-2141.1994.tb05025.x. [DOI] [PubMed] [Google Scholar]
- 6.Magni M, Shammah S, Schiro R, Bregni M, Siena S, DiNicola M, Dalla-Favera R, Gianni A. Blood. 1996;87:1097–1102. [PubMed] [Google Scholar]
- 7.Allay J, Dumenco L, Koç O, Liu L, Gerson S. Blood. 1995;85:3342–3351. [PubMed] [Google Scholar]
- 8.Allay J, Koç O, Davis B, Gerson S. Clin Cancer Res. 1996;2:1353–1359. [PubMed] [Google Scholar]
- 9.Harris L, Marathi U, Edwards C, Houghton P, Srivastava D, Vanin E, Sorentino B, Brent T. Clin Cancer Res. 1995;1:1359–1365. [PubMed] [Google Scholar]
- 10.Maze R, Carney J, Kelley M, Glassner B, Williams D, Samson L. Proc Natl Acad Sci USA. 1996;93:206–210. doi: 10.1073/pnas.93.1.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerson S L, Miller K, Berger N A. J Clin Invest. 1985;76:2106–2114. doi: 10.1172/JCI112215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gerson S, Phillips W, Kastan M, Dumenco L, Donovan C. Blood. 1996;88:1649–1655. [PubMed] [Google Scholar]
- 13.Devereux S, Selassie T G, Hudson G V, Hudson B V, Linch D C. Br Med J. 1990;301:1077–1080. doi: 10.1136/bmj.301.6760.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerson S L, Willson J K. Hematol Oncol Clin North Am. 1995;9:431–450. [PubMed] [Google Scholar]
- 15.Pegg A E. Cancer Res. 1990;50:6119–6129. [PubMed] [Google Scholar]
- 16.Brent T P, Lestrud S O, Smith D G, Remack J S. Cancer Res. 1987;47:3384–3387. [PubMed] [Google Scholar]
- 17.Karran P, Macpherson P, Ceccotti S, Dogliotti E, Griffin S, Bignam M. J Biol Chem. 1993;268:15878–15886. [PubMed] [Google Scholar]
- 18.Dumenco L L, Allay E, Norton K, Gerson S L. Science. 1993;259:219–222. doi: 10.1126/science.8421782. [DOI] [PubMed] [Google Scholar]
- 19.Zaidi N, Allay E, Ayi T, Li B, Dumenco L, Sy M, Gerson S. Carcinogenesis. 1995;16:1047–1053. doi: 10.1093/carcin/16.5.1047. [DOI] [PubMed] [Google Scholar]
- 20.Liu L, Allay E, Dumenco L, Gerson S. Cancer Res. 1994;54:4648–4652. [PubMed] [Google Scholar]
- 21.Allay J, Davis B, Gerson S. Blood. 1995;86:113a. (abstr.). [Google Scholar]
- 22.Gerson S, Berger N, Arce C. Biochem Pharmacol. 1992;43:1101–1107. doi: 10.1016/0006-2952(92)90618-s. [DOI] [PubMed] [Google Scholar]
- 23.Gerson S, Zborowska E, Norton K, Gordon N, Willson J. Biochem Pharmacol. 1993;45:483–491. doi: 10.1016/0006-2952(93)90086-c. [DOI] [PubMed] [Google Scholar]
- 24.Dolan M E, Mitchell R B, Mummert C, Moschel R C, Pegg A E. Cancer Res. 1991;51:3367–3372. [PubMed] [Google Scholar]
- 25.Dolan M E, Pegg A E, Biser N D, Moschel R C, English H F. Cancer Chemother Pharmacol. 1993;32:221–225. doi: 10.1007/BF00685839. [DOI] [PubMed] [Google Scholar]
- 26.Page J, Giles H D, Phillips W, Gerson S L, Smith A C, Tomaszewski J E. Proc Am Assoc Cancer Res. 1994;35:328. (abstr.). [Google Scholar]
- 27.Spiro T, Willson J, Haaga J, Hoppel C, Liu L, Majka S, Gerson S. Proc Am Soc Clin Ocol. 1996;15:177. (abstr.). [Google Scholar]
- 28.Crone T M, Goodtzova K, Edara S, Pegg A E. Cancer Res. 1994;54:6221–6267. [PubMed] [Google Scholar]
- 29.Ohashi T, Boggs S, Robbins P, Bahnson A, Patrene K, Wei F-S, Wei J-F, Li J, Lucht L, Fei Y, Clark S, Kimak M, He H, Mowery-Rushton P, Barranger J. Proc Natl Acad Sci USA. 1992;89:11332–11336. doi: 10.1073/pnas.89.23.11332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Markowitz D, Goff S, Bank A. J Virol. 1988;167:400–406. [PubMed] [Google Scholar]
- 31.Bodine D M, McDonagh K T, Brandt S J, Ney P A, Agricola B, Byrne E, Nienhuis A W. Proc Natl Acad Sci USA. 1990;87:3738–3742. doi: 10.1073/pnas.87.10.3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koç O, Gerson S, Fox R, Schupp J, Marko D, Steckley J, Lazarus H. Blood. 1995;86:985a. (abstr.). [Google Scholar]
- 33.Brent T P, von Wronski M, Pegram C N, Bigner D D. Cancer Res. 1990;50:58–61. [PubMed] [Google Scholar]
- 34.Gerson S, Trey J, Miller K, Berger N. Carcinogenesis. 1986;7:745–749. doi: 10.1093/carcin/7.5.745. [DOI] [PubMed] [Google Scholar]
- 35.Loktionova N, Pegg A. Cancer Res. 1996;56:1578–1583. [PubMed] [Google Scholar]
- 36.Gerson S, Markowitz S D, Willson J K V. Proc Am Assoc Cancer Res. 1993;34:271. (abstr.). [Google Scholar]
- 37.Dumenco L, Warman B, Hatzoglou M, Lim I K, Abboud SL, Gerson S L. Cancer Res. 1989;49:6044–6051. [PubMed] [Google Scholar]
- 38.Moritz T, Mackay W, Glassner B J, Williams D A, Samson L. Cancer Res. 1995;55:2608–2614. [PubMed] [Google Scholar]
- 39.Pedersen-Bjergaard J, Philip P, Pedersen N, Hou-Jensen K, Svejgaard A, Jensen G, Nissen N. Cancer. 1984;45:452–462. doi: 10.1002/1097-0142(19840801)54:3<452::aid-cncr2820540313>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 40.Pegg A, Boosalis M, Samson L. Biochemistry. 1993;32:11998–20006. doi: 10.1021/bi00096a009. [DOI] [PubMed] [Google Scholar]
- 41.Dolan M E, Pegg A E, Dumenco L L, Moschel R C, Gerson S L. Carcinogenesis. 1991;12:2305–2309. doi: 10.1093/carcin/12.12.2305. [DOI] [PubMed] [Google Scholar]
- 42.Moore M H, Gulbis J M, Dodson E J, Demple B, Moody P C E. EMBO J. 1994;13:1495–1501. doi: 10.1002/j.1460-2075.1994.tb06410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu L, Lee K, Wasan E, Gerson S. Cancer Res. 1996;56:1880–1885. [PubMed] [Google Scholar]
- 44.Tsuzuki T, Sakumi K, Shiraishi H K, Igarashi H, Iwakuma T, Tominaga Y, Zhang S, Shimizu S, Ishakawa T, Nakamura K, Nakao K, Katsuki M, Sekiguchi M. Carcinogenesis. 1996;17:1215–1220. doi: 10.1093/carcin/17.6.1215. [DOI] [PubMed] [Google Scholar]
- 45.Gerson S L, Trey J E, Miller K. Cancer Res. 1988;48:1521–1527. [PubMed] [Google Scholar]
- 46.Gerson S, Trey J, Miller K, Benjamin E. Cancer Res. 1987;47:89–95. [PubMed] [Google Scholar]