

Abstract
The remarkable resistance of the urinary tract to infection has been attributed to its physical properties and the innate immune responses triggered by pattern recognition receptors lining the tract. We report a distinct TLR4 mediated mechanism in bladder epithelial cells (BECs) that abrogates bacterial invasion, a necessary step for successful infection. Compared to controls, uropathogenic type 1 fimbriated Escherichia coli and Klebsiella pneumoniae invaded BECs of TLR4 mutant mice in 10-fold or greater numbers. TLR4 mediated suppression of bacterial invasion was linked to increased intracellular cAMP levels which negatively impacted Rac-1 mediated mobilization of the cytoskeleton. Artificially increasing intracellular cAMP levels in BECs of TLR4 mutant mice restored resistance to type 1 fimbriated bacterial invasion. This finding reveals a novel function for TLR4 and another facet of bladder innate defense.
INTRODUCTION
In spite of its close proximity to the gastrointestinal tract which harbors a large reservoir of, primarily, Gram negative bacteria with the potential to cause infections, the urinary tract remains refractile to infection. Much of this resistance is attributable to the mechanical forces existing in the urinary tract and to the ability of the epithelial cells lining the tract to recognize incoming bacteria and to mount a rapid and effective immune response (Mulvey et al., 2000; Reid and Sobel, 1987).
The bladder, which comprises a major portion of the lower urinary tract, is a specialized structure employed for the storage of urine. Because of this role, the superficial BECs intrinsically are highly impermeable to pathogens (Truschel et al., 2002). On the apical surfaces of BECs are distinct proteins called uroplakins which along with a collection of lipids, cholesterol and sphingolipids, which constitute a distinct cellular entity called lipid rafts, provides a highly ordered lipid structure with very low membrane fluidity and permeability (Apodaca, 2004; Truschel et al., 2002). Another major impediment to bacterial colonization is the powerful flushing actions of urine which eliminates all bacteria that are not intimately associated with BECs (Reid and Sobel, 1987). In addition to these physical barriers, prospective pathogens have to survive various powerful antimicrobial actions initiated by BECs and other mucosal cells (Mulvey et al., 2000; Samuelsson et al., 2004). Present in the plasma membrane of the epithelial cells are pattern recognition receptors (PRRs) such as TLR4 that recognize lipopolysaccharide (LPS) on Gram negative bacteria and activate a sequence of intracellular signals resulting in activation of the transcriptional factor, NF-κB, and the production of several NF-κB dependent cytokines including chemoattractants that recruit phagocytic cells to clear the infection (Schilling et al., 2003; Svanborg et al., 2006). The importance of TLR4 in the innate immune response is evident from the findings that TLR4 mutant mice mount a poor cytokine and neutrophil response to urinary tract infections (UTIs). Consequently, in contrast to isogenic control mice, these mice fail to resolve their infections (Hagberg et al., 1984; Schilling et al., 2001; Svanborg et al., 2006).
The TLR4 signaling mechanisms in BECs leading to cytokine and neutrophil responses during UTIs now appear to be more complex and distinct from that seen in other cell types. Initial studies revealed a signaling cascade consistent to that described in other cells namely, engagement of TLR4 by LPS triggers a signaling pathway involving several intracytoplasmic and nuclear transcriptional factors (Fischer et al., 2006; Schilling et al., 2001). TLR4 activation first engages a set of adaptor family members, which initiates a sequence of signaling events, resulting eventually in the activation of the transcriptional factor NF-κB, and the expression of several immunomodulatory cytokines such as IL-6 and IL-8 (Kawai and Akira, 2006; Schilling et al., 2001). More recent studies now reveal that the vigorous and rapid TLR4 mediated IL-6 and IL-8 responses of BECs to UPEC is only partially attributable to the classical NF-κB pathway and that a second and more rapid signaling pathway is involved. This pathway possesses a number of prominent cellular secondary messengers, Ca2+ and cAMP, as well as a transcriptional factor, cAMP response element-binding protein (CREB) (Song et al., 2007). Although this distinct TLR4 mediated pathway appears to be present only in BECs (Song et al., 2007), its specific contribution to bladder defense is unknown.
Uropathogens that overcome the defenses of the urinary tract typically do so by seeking refuge within the bladder epithelium. In their intracellular location, these pathogens avoid the clearing actions of both urine flow and recruited phagocytes. The most common mode of bacterial invasion of BECs involves type 1 fimbriae which are filamentous appendages expressed by E. coli, K. pneumoniae and various other uropathogenic enterobacteria (Abraham et al., 1988; Hagberg et al., 1983; Hagberg et al., 1981). Binding of FimH, a mannose binding lectin at the distal tips of the fimbriae to uroplakin 1a on the luminal surface of BECs, triggers a distinct series of signaling reactions that culminates in the entry of the bacteria into the dynamic subapical pool of discoid vesicles called fusiform vesicles found in BECs (Bishop et al., 2007). These fusiform vesicles are membrane-rich nondegradative compartments that serve to increase bladder volume by fusing with luminal plasma membrane of BECs (Truschel et al., 2002). Since the plasma membrane of BECs as well as fusiform vesicles are highly enriched in cholesterol, sphingolipids, and glycolipids (Apodaca, 2004), there is growing realization that UPEC invasion of BECs is lipid raft dependent. This notion has been supported by the finding that specific disruptors of cellular lipid raft structure inhibits UPEC invasion and key cellular components so far implicated in bacterial invasion of BECs are typically localized with lipid raft structure (Duncan et al., 2004). One such lipid raft mediator of bacterial invasion is the Rho GTPase member, Rac-1, whose activation is critical because it enhances accumulation of actin filaments at sites of bacterial entry (Duncan et al., 2004; Martinez and Hultgren, 2002).
During infection of the urinary tract, both TLR4 mediated signaling and lipid raft mediated bacterial invasion of BECs are believed to occur. Here, we reveal the existence of crosstalk between the two pathways, where TLR4 signaling through increased production of the secondary messenger, cAMP, negatively regulates lipid raft mediated bacterial invasion.
RESULTS
Increased invasion of mouse bladders by Gram negative bacteria when TLR4 signaling is abrogated
One of the earliest in vivo examples revealing the critical role of PRRs in host defense was the observation made over 20 years ago that TLR4 mutant C3H/HeJ mice, in contrast to isogenic control C3H/HeN mice, were unable to clear experimental UTIs (Hagberg et al., 1984; Shahin et al., 1987). This observation was ascribed to the inability of TLR4 mutant mice to mount a local cytokine and neutrophil response (Hagberg et al., 1984; Shahin et al., 1987). Here, we examined if in addition to their inability to mount an adequate inflammatory response these TLR4 mutant mice exhibited increased susceptibility to bacterial invasion. A small volume (50 μl) of saline containing 1×108 of a type 1 fimbriated uropathogenic E. coli strain CI5 was introduced via catheter into the bladders of a group of anesthetized TLR4 mutant and isogenic control mice. One hour later, the bladders were emptied and instilled with gentamicin for 30 min to kill all extracellular bacteria. Thereafter, the mice were sacrificed and the bladders were removed, rinsed and the intracellular numbers of bacteria were determined by standard colony counts of bladder homogenates (Hagberg et al., 1984). The values obtained from each bladder homogenate represent the number of intracellular bacteria. Since the incubation time was limited to only 1 hr, the contribution of recruited immune cells to bacterial clearance was minimal. We found that the number of intracellular E. coli in the bladders of TLR4 mutant mice were at least 12 fold higher than the controls (Fig. 1A). Immunomicroscopy of bladder cross sections of both groups of mice confirmed bacterial association with the superficial epithelium of the bladders of TLR4 mutant but not in control mice (Fig. 1B and 1C). Employing immunoprobes for TLR4, we sought to confirm earlier claims of the presence of TLR4 in bladder cells (Backhed et al., 2001; Samuelsson et al., 2004). Whereas no discernable staining was observed when an isotype antibody was employed (Fig. 1D), the relatively large basal level of TLR4 expression on superficial BECs in control mice was striking (Fig. 1E). Interestingly, neither the distribution nor expression levels of TLR4 appeared to change following bacterial infection (Fig. 1F). To demonstrate the specificity of this TLR4 mediated response for LPS, we examined the invasive capacity of another LPS producing bacteria, a type 1 fimbriated uropathogenic Klebsiella pneumoniae strain 1236, as well as a non LPS producer, a clinical isolate Staphylococcus aureus strain 54. Whereas no significant difference between the TLR4 mutant and control mice in staphylococcal invasion was detected, an 11-fold difference in invasion was observed with K. pneumoniae (Fig. 1G and 1H). These observations reveal that compared to controls, TLR4 mutant mice are highly susceptible to invasion by type 1 fimbriated Gram negative enteric bacteria. Thus, TLR4 markedly suppresses the invasion of BECs by type 1 fimbriated enterobacteria. To further support this conclusion, we compared the ability of a LPS modified type 1 fimbriated K-12 E. coli strain MLK1067 (msbB mutant) and its parent strain W3110 (Clementz et al., 1997) to invade bladders of wild type mice. The msbB mutant E. coli MLK1067 produces a penta-acylated lipid A that is poorly recognized by the TLR4 signaling complex and as a consequence this strain fails to trigger TLR4-LPS signaling in host cells (Coats et al., 2005). Predictably, unlike the parent strain, the msbB mutant will not activate TLR4 and therefore a significantly greater invasion of mouse bladders should result. Indeed, we found a 6.3-fold greater invasion of mouse bladders by the mutant MLK1067 strain compared to the parent strain (Fig. 1I). Consistent with this idea, when we compared bladder invasion by wild-type and msbB mutant E. coli in the TLR4 mutant mice, no significant difference in invasion between the two strains was seen (Fig. 1I). Cumulatively, these observations provide definitive evidence that TLR4-LPS signaling plays a key role in reducing bacterial invasion of bladder cells.
Figure 1. Increased invasion of mouse bladders by Gram negative bacteria when TLR4 signaling is abrogated.
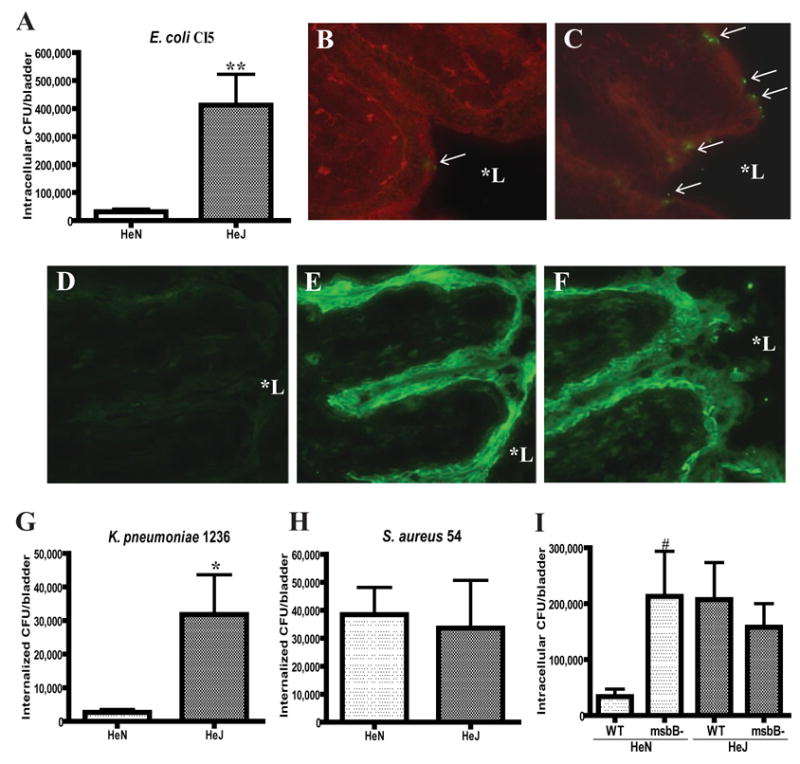
A, TLR4 mutant C3H/HeJ (HeJ) and isogenic control C3H/HeN (HeN) mice were inoculated transurethrally with 1×108 of E. coli CI5 (A). Bacterial invasion after 1 hr was assessed by gentamicin protection assays followed by colony counts of bladder homogenates. B–C, Fluorescent images of bladder sections of HeN (B) and HeJ (C) mice 1 hr following instillation of 1×108 E. coli CI5. Green, E. coli; Red, Wheat Germ Agglutinin (WGA). D–F, TLR4 immunostaining images of bladder sections of HeN mice before (D and E) and 1 hr after (F) instillation of 1×108 E. coli CI5. E and F were stained with TLR4-specific antibody whereas D was stained with isotype control antibody. G–H, TLR4 mutant HeJ and control HeN mice were inoculated transurethrally with 1×108 of type 1 fimbriated K. pneumoniae 1236 (G), or S. aureus 54 (H). Bacterial invasion after 1 hr was assessed as before. I, TLR4 mutant HeJ and control HeN mice were inoculated transurethrally with 1×108 of type 1 fimbriated msbB mutant MLK1067 (msbB-) or corresponding wild-type (WT) E. coli W3110. Bacterial invasion after 1 hr was assessed as before. In A, G, H, and I, **P <0.0001; * P<0.01, relative to values of HeN; #P<0.05, relative to values of wild-type E. coli W3110 infected HeN. Bars represent the mean + S. D.. In B–F, magnification was 200×. *L stands for lumen.
Recapitulation of in vivo observations using a human BEC line
To elucidate the molecular basis for TLR4 mediated abrogation of bacterial invasion, we initiated studies employing a well established in vitro model, the 5637 human BEC line. It has previously been demonstrated that interactions of this cell line with Gram negative bacteria closely mimics in vivo behavior of BECs (Duncan et al., 2004; Schilling et al., 2001). We sought to recapitulate our in vivo observations of the modulatory role of TLR4 on E. coli invasion. First, we compared invasion of BECs by wild type E. coli W3110 and its msbB mutant derivative, and observed a 9-fold greater invasion by the mutant E. coli compared to the parent E. coli strain (Fig. 2A) which is consistent with the in vivo data. Next, using RNA interference techniques, we generated BECs where expression of TLR4 was knocked down (KD). Densitometric quantitation of message levels in the TLR4 KD BECs, revealed that the expression of TLR4 was reduced by 49% (Fig. 2B). We sought to compare invasion of control (transfected with control vector) BECs and TLR4 KD BECs by the type 1 fimbriae expressing E. coli ORN103(pSH2). This K-12 laboratory E. coli strain expressing recombinant type 1 fimbriae rather than a UPEC strain was employed for most of our subsequent studies (Orndorff and Falkow, 1984). Typically, in addition to type 1 fimbriae, UPEC express several other virulent factors which could potentially confound our studies. As expected, we found that E. coli ORN103(pSH2) invaded TLR4 KD BECs in significantly higher numbers than control transfected or non-transfected BECs (Fig. 2C). It is noteworthy that the adherence of E. coli to both control BECs and TLR4 KD BECs were identical (Fig. 2D). It is also noteworthy that invasion of TLR4 KD BECs by E. coli ORN103(pSH2) was also FimH dependent because limited invasion of TLR4 KD BECs was observed when the isogenic FimH mutant E. coli ORN103(pUT2002) (Minion et al., 1986) was tested (data not shown). Thus, our in vitro data recapitulate our in vivo observations implicating TLR4 as a negative modulator of E. coli invasion into BECs.
Figure 2. TLR4-LPS signaling abrogates in vitro invasion of BECs by type 1 fimbriated Enterobacteria.
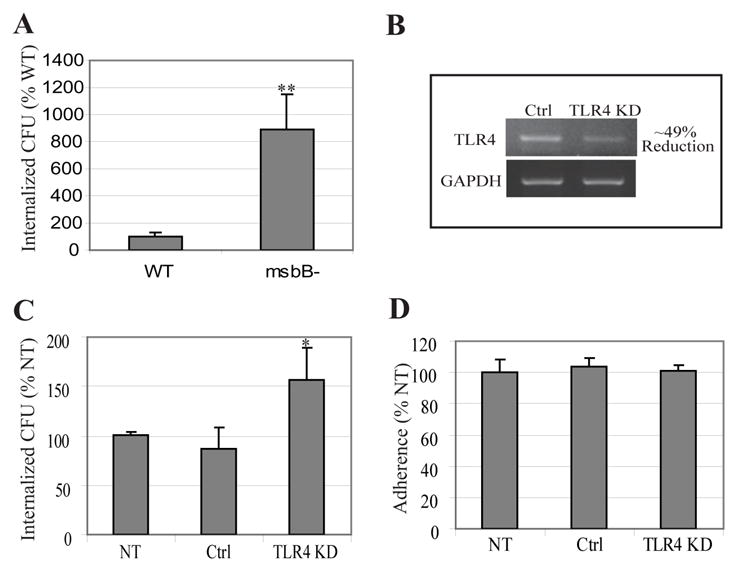
A, Bacterial invasion following exposure of BECs to wild-type E. coli W3110 (WT) or msbB mutant E. coli MLK1067 (msbB-). **P< 0.001, relative to values of wild-type E. coli infected BECs. B, RT-PCR of control-transfected BECs (Ctrl) and TLR4 knockdown BECs (TLR4 KD). Glyseraldehyde-3-phosphate dehydrogenase (GAPDH) was employed as a loading control. C–D, Bacterial invasion (C) or bacterial adherence (D) following exposure of non-transfected BECs (NT), control-transfected BECs (Ctrl), and TLR4 KD BECs to type 1 fimbriated E. coli ORN103(pSH2). *P< 0.03, relative to control values. Bars represent the mean + S. D. in A, C and D.
TLR4 KD BECs exhibit an enhanced Rac-1 response to E. coli
To gain an understanding of how TLR4 signaling was negatively impacting bacterial invasion in BECs, we sought to compare the level of Rac-1 activation in TLR4 KD BECs and control BECs following exposure to type 1 fimbriated E. coli ORN103(pSH2). We and others have previously implicated Rac-1, a critical mediator of actin cytosketal dynamics and a component of lipid rafts, in the invasion of BECs by E. coli (Duncan et al., 2004; Martinez and Hultgren, 2002). Shown in Fig. 3A is a confirmatory experiment where invasion by type 1 fimbriated E. coli ORN103(pSH2) of control BECs, BECs overexpressing constitutively activated Rac1 and BECs overexpressing a dominant negative form of Rac-1 was examined. Compared to control BECs, bacterial invasion of BECs where Rac1 was constitutively activated was markedly higher. Conversely, bacterial invasion of BECs with the dominant negative form of Rac-1 was significantly lower than the controls (Fig. 3A). Both of these observations reiterate the idea that Rac-1 activation is essential to the invasion of BECs by type 1 fimbriated E. coli. To investigate the activation states of Rac-1 in TLR4 KD BECs, non-transfected BECs and control transfected BECs, we isolated activated Rac-1 from each of the BEC lysates before and 30 min following exposure to E. coli ORN103(pSH2). The assay we employed to detect the activated form of Rac-1 was a pull down assay employing a fusion protein comprising of GST-fused to PAK which specifically binds active GTP bound Rac-1 (Manser et al., 1994). An appreciable increase in activated Rac-1 was detected in each of the cell types 30 min following exposure to bacteria, although the amounts of activated Rac-1 BECs in TLR4 KD BECs was markedly higher than in any of the control BECs (Fig. 3B). Indeed, the level of activated Rac-1 in each cell type (Fig. 3B) correlated closely with the extent of bacterial invasion seen (Fig. 2). These observations suggest that TLR4 mediated suppression of bacterial invasion is through inhibition of Rac-1 activation. It is noteworthy that even before exposure to bacteria, the levels of activated Rac-1 in TLR4 KD BECs was relatively high (Fig. 3B) suggesting that TLR4 may be constitutively suppressing Rac-1 in quiescent cells.
Figure 3. TLR4 KD BECs exhibit an enhanced Rac-1 response to E. coli which correlates with enhanced bacterial invasion.
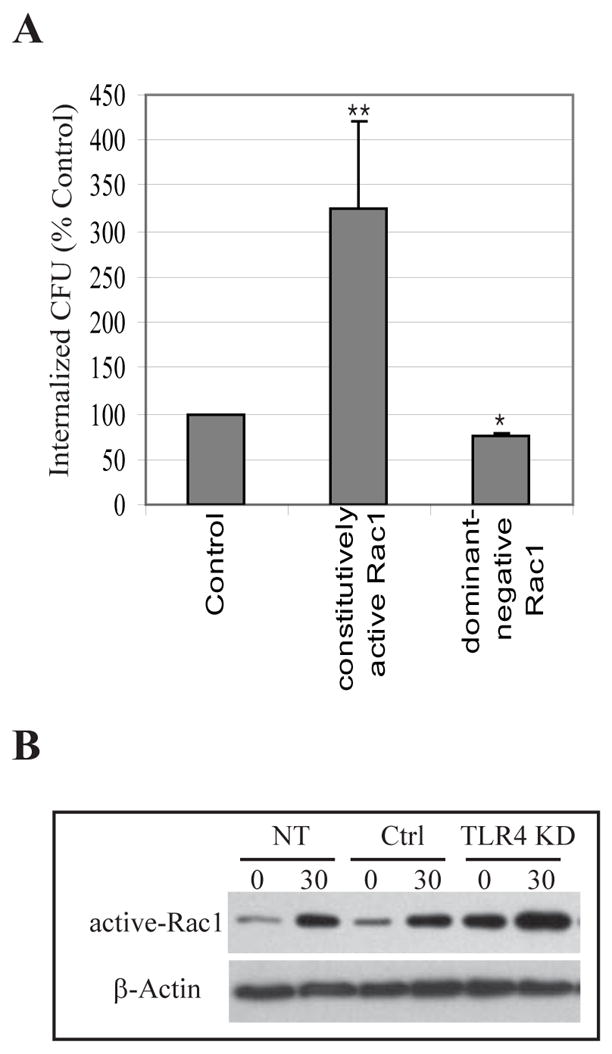
A, Invasion of control-transfected BECs, BECs overexpressing constritutively active Rac1, or BECs with dominant-negative Rac1 by type 1 fimbriated E. coli ORN103(pSH2). **P< 0.01; *P<0.05, relative to control values. Bars represent the mean + S. D.. B, GTP-bound Rac1 levels of non-transfected (NT), control-transfected (Ctrl), and TLR4 KD BECs before (0) and 30 min after (30) exposure to E. coli. An actin-specific Western blot was used as a loading control.
TLR4 KD BECs evoke a reduced cAMP response to E. coli
Since TLR4 has not previously been reported to suppress activation of Rac-1, we reasoned that the mechanism would involve one or more inhibitory intermediaries. Because secondary messengers can have profound and sometimes inhibitory effects on multiple cellular functions, we examined the possibility of cAMP as the candidate inhibitory substrate. cAMP has previously been shown to negatively regulating Rac-1 activity and subsequent Rac-1 mediated reorganization of actin cytoskeleton (Nagasawa et al., 2005). Rac-1 inactivation by cAMP has been shown to specifically involve protein kinase A (PKA), whose catalytic domains are activated by cAMP binding (Howe, 2004; O’Connor and Mercurio, 2001; Waschke et al., 2004). Before determining whether cAMP was involved in suppressing Rac-1 activation it was important to demonstrate cAMP production by BECs following exposure to type 1 fimbriated E. coli ORN103(pSH2). We examined if this was the case and if so, whether TLR4 KD BECs evoked a diminished cAMP response. We compared the levels of intracellular cAMP in control and TLR4 KD BECs before and 1 h after exposure to E. coli ORN103(pSH2). We observed a marked increase in intracellular cAMP in BECs following exposure to bacteria (Fig. 4A). This bacteria-induced increase in intracellular cAMP was significantly reduced in TLR4 KD BECs (Fig. 4A) indicating that TLR4 was mediating this cAMP response. A similar observation was made if the E. coli was replaced by purified LPS (Fig. 4A) which is consistent with the notion that the bacterial component responsible for activating TLR4 was indeed LPS. It should be noticed that bacteria induced increase in intracellular cAMP was seen as early as 15 min after exposure, but the 1 h time point was selected so as to maximize difference in cAMP levels under various conditions. To further investigate the relationship between intracellular cAMP and E. coli invasion of BECs, we compared bacterial entry into non-transfected, control-transfected and TLR4 KD BECs following treatment with 50 μM forskolin (Fsk), a broad spectrum activator of adenylyl cyclases, or 1 mM dibutyryl cAMP (dbcAMP), a membrane permeable cAMP analog. In all cases, Fsk and dbcAMP treatments markedly reduced bacterial uptake indicating that increasing intracellular cAMP levels had a powerful effect in blocking E. coli invasion (Fig. 4B). We next investigated if the Fsk mediated increase in intracellular cAMP would also negatively impact Rac-1 activation. We examined for bacterial induced Rac-1 activation in untreated and Fsk treated BECs. As shown in Fig. 4C, Fsk treated BECs failed to evoke any Rac-1 activation in response to bacteria which is consistent with the finding that Fsk significantly reduces bacterial invasion in BECs (Fig. 4B). In additional experiments, we compared the effects of two membrane-permeable cAMP analogs, dbcAMP and 8-CPT-cAMP on bacterial invasion. Whereas the former analog activates two downstream effectors PKA and the exchange protein directly activated by cAMP (Epac), the latter activates only Epac. We found that dbcAMP but not 8-CPT-cAMP significantly inhibited bacterial invasion (Fig. 4D) indicating that PKA but not Epac was involved in the uptake of bacteria. Consistent with this notion, treatment of BECs with a PKA specific inhibitor peptide (PKI) or another cell permeable cAMP analog, 6-Bnz-cAMP which specifically activates PKA revealed negative impacts of cAMP-PKA on E. coli invasion (Fig. 4D). Thus, the enhanced levels of bacterial invasion seen in TLR4 KD BECs compared to control BECs are directly attributable to their limited cAMP response to bacteria. Our observations cumulatively support the notion that TLR4 mediated increase in intracellular cAMP and its downstream effector, PKA, are largely responsible for suppressing Rac-1 mediated bacterial invasion.
Figure 4. TLR4 KD BECs evoke a reduced cAMP response to E. coli which correlates with increased bacterial invasion.
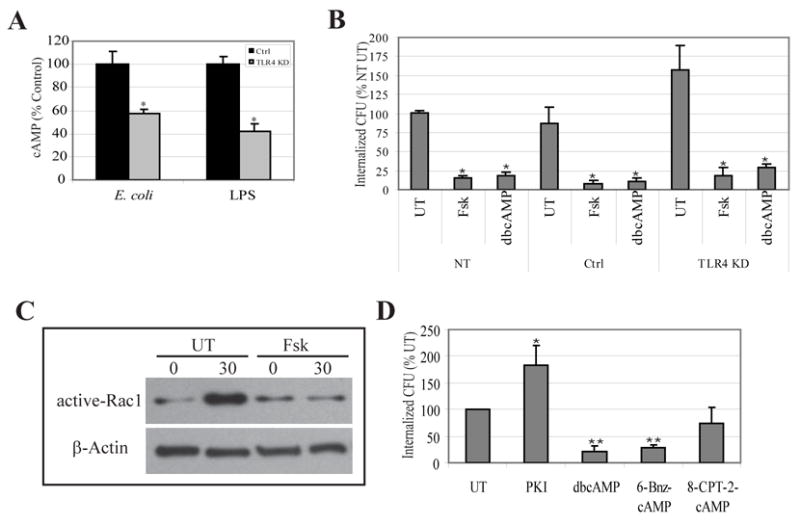
A, Intracellular cAMP levels of control-transfected BECs (Ctrl) and TLR4 KD BECs after exposure to type 1 fimbriated E. coli ORN103(pSH2) or E. coli LPS. *P<0.05 relative to E. coli (EC) or LPS treated control. B, E. coli invasion of non-transfected (NT), control-transfected (Ctrl), and TLR4 KD BECs left untreated (UT) or following treatment with 50 μM forskolin (Fsk) or 1 mM dibutryl cAMP (dbcAMP). *P<0.001 relative to values of respective UT BECs. C, GTP-bound Rac1 levels in untreated (UT) and forskolin-treated (Fsk) BECs before (0) or after 30 min (30) exposure to E. coli. An actin-specific Western blot was used as a loading control. D, E. coli invasion of untreated (UT) BECs or BECs following 30 min treatment with 0.2 μM PKA inhibitor peptide (PKI), 1 mM dibutyryl cAMP (dbcAMP), 1 mM 6-Bnz-cAMP, or 1 mM 8-CPT-2-cAMP. **P< 0.001; *P< 0.01, relative to UT values. Bars represent the mean + S. D. in A, B, and D.
The BEC cAMP response to E. coli is mediated by Adenylyl cyclase 3
Next, we sought to determine how TLR4 was increasing intracellular cAMP levels in BECs. Because there are currently 10 known isoforms of mammalian adenylyl cyclases (ACs) (Sunahara and Taussig, 2002) it was of interest to identify the specific AC in BECs responsible for the TLR4 mediated cAMP response. First, we sought to determine which AC isoforms were actually expressed in BECs. RT-PCR was performed on total cellular RNA, using primers specific for each known AC isoform and only mRNA for AC isoforms 3, 4, 6, and 7 was detectable in BECs (Song et al., 2007) (data not shown). We confirmed that the other AC isotype-specific primers used were functional by undertaking RT-PCR on total RNA from human embryonic kidney (HEK) cells, positive control cells, where all ACs except AC4 and 8 were expressed (Ludwig and Seuwen, 2002) (data not shown). RNAi was utilized to minimize the expression of each AC, which was verified by AC isotype-specific RT-PCR (Fig. 5A). To see which of the 4 ACs were involved in suppressing E. coli invasion, we exposed the respective KD BECs to E. coli ORN103(pSH2) and examined for cAMP production and bacterial invasion. We observed an increase in intracellular cAMP in all the KD cells except AC3 KD BECs (Fig. 5B). Similarly, the level of invasion into the various BECs was comparable with that of the control except AC3 KD BECs following E. coli exposure (Fig. 5C). Indeed, the level of bacterial invasion in AC3 KD BECs was over 3 fold higher than in controls. Consistent with these findings, there was a high level of Rac-1 activation in AC-3 KD BECs following exposure to E. coli ORN103(pSH2) relative to controls (Fig. 5D). Thus, AC3 is the BEC AC isoform linked to the intracellular cAMP response following E. coli exposure. This means that AC3 is also the isoform which is responsible for suppressing invasion of human BECs by E. coli. The high level of Rac-1 activation seen in quiescent AC-3 KD BECs is reminiscent of the situation in TLR4 KD BECs and suggests that the suppressive effects of TLR4 on Rac-1 activation in quiescent BECs is through AC-3 derived cAMP.
Figure 5. The BEC cAMP response to E. coli and its subsequent effect on bacterial invasion is mediated by Adenylyl cyclase 3.
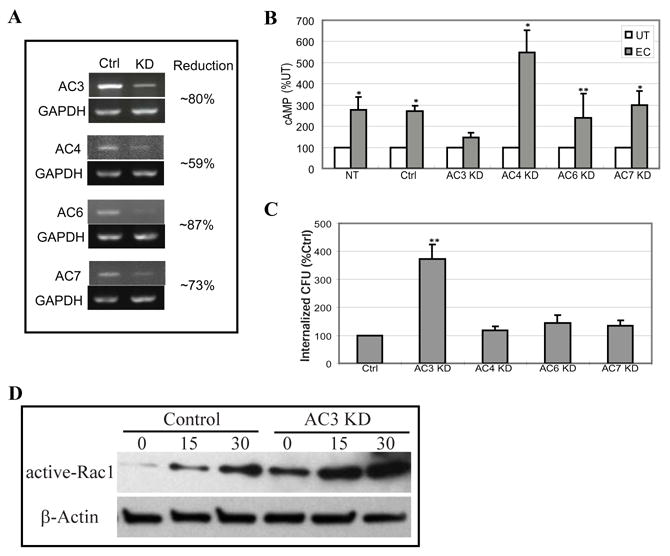
A, RT-PCR of control-transfected BECs (Ctrl) and AC-3, -4, -6, or -7 KD BECs. GAPDH-specific RT-PCR was used as a loading control. B, Intracellular cAMP levels in non-transfected BECs (NT), control-transfected BECs (Ctrl), or AC-3, -4, -6 or -7 KD BECs before (UT) or after exposure to E. coli ORN103(pSH2) (EC). *P<0.005 and **P<0.02 relative to respective UT values. C, E. coli invasion of control-transfected BECs (Ctrl), or AC-3, -4, -6, or -7 KD BECs. **P<0.0001 relative to control values. Bars represent the mean + S. D. in B and C. D, Active-Rac1 levels of control and AC3 KD BECs before and at 15 or 30 min after exposure to E. coli. An actin-specific Western blot was used as a loading control.
Application of a booster of intracellular cAMP in the urinary tracts of TLR4 mutant mice reduces bacterial invasion
In our in vivo studies, we had observed that TLR4 mutant mice were highly susceptible to invasion by uropathogenic E. coli and K. pneumoniae (Fig. 1A and 1B). Our in vitro studies suggest the existence of a cAMP dependent innate mechanism in BECs for countering bacterial invasion. We sought to establish a link between intracellular cAMP in BECs and susceptibility to bacterial invasion in vivo. Our in vitro studies have suggested that TLR4 constitutively regulates cAMP levels in BEC in a positive fashion, if so, intracellular levels of cAMP in TLR4 mice would predictably be lower than that seen in control mice. We compared the basal cAMP levels in superficial BECs of bladders in TLR4 mutant and control mice and as predicted, intracellular cAMP levels in TLR4 mutant mice were found to be significantly lower than in control BECs (Fig. 6A). Thus, the increased susceptibility of TLR4 mutant mice to enterobacterial invasion could be attributable to low levels of intracellular cAMP in BECs. If the increased susceptibility of TLR4 mutant mice to bacterial invasion is attributable to their inability mount an appropriate cAMP response, then application of the cAMP booster, Fsk, should compensate for the cAMP deficiency and reduce susceptibility to bacterial invasion. Indeed, when bacterial invasion and intracellular cAMP levels in BECs of TLR4 mutant mice were examined following combined intraperitoneal/intravesicular Fsk treatment, the levels were comparable to that seen in the wild type mice (Fig. 6A and 6B), indicating that the increased susceptibility could be reversed merely by treating the urinary tract with a booster of intracellular cAMP levels. Application of Fsk to TLR4 mutant mice also reduced invasion by type 1 fimbriated K. pneumoniae 1236 but not invasion by S. aureus 54 (Fig. 6C and 6D).
Figure 6. Reduced intracellular levels of cAMP in BECs of TLR4 mutant mice and use of Fsk to enhance resistance of BECs to invasion by type 1 fimbriated Enterobacteria.
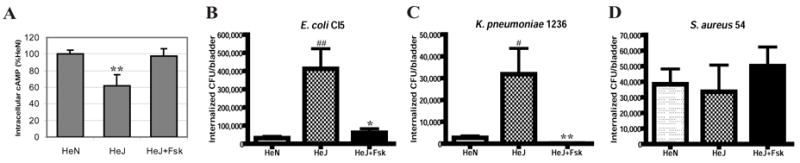
A, Intracellular cAMP in superficial BECs obtained from control (HeN), TLR4 mutant (HeJ) mice, and TLR4 mutant mice treated with Fsk (HeJ+Fsk) (n=3–5).**P<0.01, relative to HeN values as well as relative to HeJ+Fsk values. B–D, Invasion of bladders of control (HeN) mice, TLR4 mutant (HeJ) mice and TLR4 mutant (HeJ) mice pretreated with Fsk by 1×108 of E. coli CI5 (B), K. pneumoniae 1236 (C), or S. aureus 54 (D). When indicated, the mice were pretreated for 1 h with forskolin via intravesicular catheter instillation and intraperitoneal (IP) injection. Bars represent the mean + S. D. in A–D. ##P<0.001; #P<0.01, relative to HeN values. ** P<0.01; * P<0.03, relative to HeJ values.
DISCUSSION
Invasion of host cells is the singular most important mechanism that pathogens employ to avoid rapid clearance by the host’s immune system. Here, we report a novel host cell adaptation for resisting this powerful microbial trait. The mechanism in BECs for resisting invasion by type 1 fimbriated E. coli involves TLR4, an immune surveillance molecule, which is well known for mobilizing a wide range of innate immune responses against Gram negative bacteria including secretion of critical proinflammatory mediators and antimicrobial peptides (Saemann et al., 2005; Samuelsson et al., 2004; Schilling et al., 2003; Shahin et al., 1987). Blocking invasion of type 1 fimbriated enterobacteria represents a novel al beit surprising role for TLR4 considering that TLRs in macrophages have been implicated in promoting bacterial uptake and subsequent phagosome maturation (Blander and Medzhitov, 2004, 2006; Ozinsky et al., 2000; Underhill and Gantner, 2004).
Invasion of BECs by type 1 fimbriated UPEC involves a distinct but poorly defined endocytic pathway. What is currently known regarding UPEC invasion of BECs is that cellular lipid raft components are involved and that the internalized bacteria are retained within compartments resembling fusiform vesicles of BECs (Bishop et al., 2007; Duncan et al., 2004). A number of lipid raft components have been shown to be necessary for invasion of BECs by UPEC including Uroplakin 1a, the FimH receptor, caveolin-1, a scaffolding protein, and Rac-1, an inducer of cytoskeletal remodeling (Duncan et al., 2004; Martinez and Hultgren, 2002). Interestingly, our studies reveal that Rac-1 is also a molecular target of TLR4 signaling and that Rac-1 represents the site where TLR4 signaling and lipid raft mediated phagocytosis intersect. Shown in Fig. 7 is a diagrammatic depiction of how TLR4 signaling converges with the bacterial invasion pathway.
Figure 7. Diagrammatic depiction of the parallel signaling reactions occurring in BECs during infection by type 1 fimbriated enterobacteria.
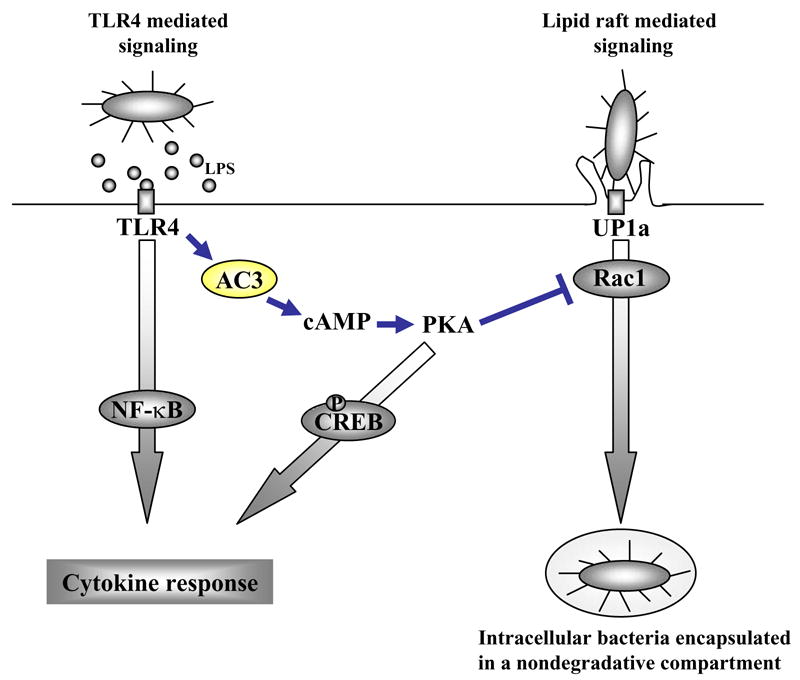
Highlighted in the figure is the TLR4 initiated and AC-3, cAMP, and PKA dependent signaling pathway that dissects with and abrogates the lipid raft mediated endocytic pathway.
The connection between TLR4 and Rac-1 was revealed from the findings that the level of Rac-1 activation in TLR4 KD BECs was markedly higher relative to control BECs even in the steady state (Fig. 3B). Although the levels of activated Rac-1 correspondingly increased in each cell type following exposure to type 1 fimbriated E. coli, the amounts in TLR4 KD BECs was strikingly higher compared to that in control BECs (Fig. 3B). Thus, TLR4 was negatively impacting Rac-1 activation, a necessary step for actin remodeling and bacterial invasion (Duncan et al., 2004; Martinez and Hultgren, 2002). TLR4 mediated suppression of Rac-1 appears to be the underlying basis for why bacterial invasion in TLR4 KD BECs is several fold higher than in control BECs (Fig. 2 and 3). Since previous reports in other cell types have shown that TLR4 regulation of Rac-1 activity is of a positive nature (Schmeck et al., 2006; Wissel et al., 2005; Zhang et al., 2005), our finding is consistent with the notion that TLR4 signaling circuitry in BECs is more complex than in other cell types.
Since TLR4 has not previously been reported to negatively impact activation of Rac-1, TLR4 mediated suppression of Rac-1 activity presumably involves a distinct set of ancillary molecules. Our studies point to a second messenger, cAMP and its down stream element, PKA, as critical intermediaries in the TLR4 circuitry responsible for suppressing Rac-1 activation and subsequent bacterial invasion. We showed that following ligation of TLR4, cAMP, derived primarily from AC-3, played a critical role in impeding bacterial invasion. That AC-3 generated cAMP was critical for abrogating bacterial invasion was evident from the finding that both Rac-1 activation and bacterial invasion was markedly enhanced in the AC-3 KD BECs compared to control BECs. Indeed, the levels of Rac-1 activation and bacterial invasion in AC-3 KD BECs were comparable to the strikingly high levels observed previously in TLR4 KD BECs. Interestingly, in addition to regulating cAMP and Rac-1 activity during Gram negative infections, TLR4 appears to play a constitutive role in regulating these molecules even in quiescent BECs.
Since inhibitors of the transcriptional factor NF-κB such as MG-132 had limited effect on either intracellular production of cAMP or bacterial invasion (data not shown), these events appeared independent of the traditional TLR4 signaling pathway. Recently, it was reported that TLR4 activation in BECs resulted in signaling via the traditional NF-κB pathway as well as a second and a “more rapid” pathway involving AC-3 derived cAMP and the transcriptional factor, CREB (Song et al., 2007) (Fig. 7). That this TLR4 initiated and cAMP dependent pathway blocks bacterial invasion reveals an important and previously unsuspected physiological function for this pathway.
Presumably, the invasion-abrogating defenses in BECs lining the lumen are triggered when TLR4 molecules make contact with LPS shed from bacteria, an event likely to be initiated as soon as bacteria enter the bladder. If the contaminating bacteria are not able to overcome cellular actions that impede penetration, they will be rapidly eliminated by the flushing forces of urine. That BECs are invaded by type 1 fimbriated E. coli in spite of its TLR4 mediated resistance argue that expression of type 1 fimbriae could be a trait specifically evolved by UPEC to invade BECs. Indeed a survey of UPEC isolates has revealed that over 80% of UPEC expressed type 1 fimbriae (Hagberg et al., 1981; Langermann et al., 1997). The discovery of the central role of cAMP in modulating cellular defenses to bacterial invasion is also of particular interest because intracellular cAMP levels are readily regulatable in vivo with a variety of small molecule modulators (Elmslie, 2004; Insel and Ostrom, 2003; Pressman, 1976; Shafer et al., 1998), raising the possibility of boosting bladder defenses for therapeutic purposes. We have demonstrated that adminstering TLR4 mutant mice with the cAMP inducer, forskolin, can markedly boost both intracellular cAMP levels and resistance of BECs to bacterial invasion to levels typically found in wild type cells. Recently, it was reported that administering forkolin in the urinary tracts of UPEC-infected mice reduced bladder infections by triggering the exocytosis of UPEC from fusiform vesicles of infected BECs (Bishop et al., 2007). Our findings suggest that the protective properties ascribed to forskolin in the urinary tract could also be attributable to reducing bacterial invasion of BECs.
With the growing realization that most pathogens seek refuge in host cells at some stage in the infectious process, much of the research has focused on elucidating the various strategies employed by pathogens to penetrate and subsequently survive within cells. The intrinsic capacity of certain host cells to completely or partially abrogate microbial entry has gone largely unrecognized. Our studies demonstrating the intrinsic properties of BECs to counter bacterial invasion could partly explain the remarkable intractability of the urinary tract to infection in spite of frequent contamination by gut flora.
MATERIALS AND METHODS
In vivo invasion assay
C3H/HeN and C3H/HeJ female mice were obtained from NCI and The Jackson Laboratory. 8–10-week-old female mice were anesthetized with sodium pentobarbital and inoculated transurethrally with 1×108 of uropathogenic E. coli CI5 by urethral catheterization. After 1 hr, 0.1 mg of gentamicin was applied for 30 min to kill all extracellular bacteria, and bladders were washed briefly using 100 μl of PBS. Bladders were aseptically removed and homogenized in 0.1% Triton X-100 in PBS. Homogenate dilutions were plated on LB plates and incubated overnight at 37°C for colony counts. n=10–20 per each experimental group. When indicated, a group of mice were pretreated intraperitoneally with 10 mg/kg forskolin and transurethrally with 100 μM forskolin for 1 hr. All experiments were done according to protocols approved by the Duke Division of Laboratory Animal Resources and the Duke University Institutional Animal Care and Use Committee.
Immunofluorescence of bladder sections
8–10-week-old female mice were anesthetized and inoculated transurethrally with 1×108 of E. coli CI5. After 1 h, bladders were aseptically removed, embeded with OCT solution, and kept at −80 °C until use. Frozen sections of uninfected and infected bladders were cut using standard methods. Each section was fixed in 4% paraformaldehyde for 10 min at RT and in precooled ethanol:acetic acid (2:1) for an additional 5 min at − 20 °C, blocked in PBS with 3% BSA for 30 min at RT, and incubated with primary antibodies diluted in PBS with 3% BSA for 30 min at RT. Sections were incubated for 30 min at RT with Alexa 488 conjugated secondary antibodies diluted in PBS with 1% BSA. To visualize epithelial cells, sections were incubated with wheat germ agglutinin (WGA) for 30 min at RT when the secondary antibodies were applied. Coverslips were mounted using Prolong Gold anti-fade reagent (Molecular Probes) and viewed. For TLR4 staining, sections were fixed as before, blocked in PBS containing 1% goat serum, and incubated with 1:100 diluted rat anti-mouse TLR4 antibody (R&D Systems) in blocking solution overnight at 4 °C. Same amounts of Rat IgG were used for the control staining. Sections were incubated with FITC-conjugated secondary antibody for 30 min at RT, mounted, and viewed.
Bacteria and Bladder epithelial cells
E. coli ORN103(pSH2) (Orndorff and Falkow, 1984), E. coli ORN103(pUT2002) (Minion et al., 1986), uropathogenic E. coli strain CI5 (Abraham et al., 1985; Orndorff and Falkow, 1984; Thankavel et al., 1997), E. coli MLK1067 (insertional inactivation mutant of msbB1) (Clementz et al., 1997), E. coli W3110 (corresponding wild-type strain for MLK1067) (Clementz et al., 1997), uropathogenic Klebsiella pneumoniae strain 1236 (a clinical isolate from Duke University Medical Center), and a Gram positive clinical isolate Staphylococcus aureus strain 54 (a clinical isolate from DUMC) were utilized in this study. Bacteria were grown overnight in Luria-Bertani (LB) broth or in Brain Heart Infusion (BHI) broth prior to use. 80 μg/ml of chloramphenicol was added for E. coli ORN103(pSH2) and E. coli MLK1067. The human bladder epithelial cell line 5637 (ATCC HTB-9) was grown in RPMI 1640 (Invitrogen) containing 10% fetal bovine serum (HyClone) and incubated at 37° C with 5% CO2.
In vitro bacterial Invasion and Adherence Assays
5637 human BECs were seeded onto 96-well plates at a density of 4×104 cells/well and incubated overnight. The cells were infected with 100 MOI bacteria for 1 h. The medium was replaced with fresh culture medium containing 100 μg/ml of the membrane-impermeable antibiotic gentamicin (Invitrogen) to kill extracellular bacteria and incubated at 37 °C for additional 1 hour. Each well was washed three times with PBS. In order to lyse the cells, 100 μl of 0.1% Triton X-100 in PBS was added to each well and incubated for 15 min. Cells were scraped, diluted, and plated onto LB agar plates containing 80 μg/ml of chloramphenicol. Colonies were counted to quantify the number of invading bacteria. To test the effect of various drugs on bacterial invasion, 50 μM forskolin (Sigma), 1 mM dibutyryl cAMP (Sigma), 1 mM N6- Benzoyladenosine- 3′, 5′-cyclic monophosphate (6-Bnz-cAMP, Sigma), 1 mM 8-(4-Chlorophenylthio)-2′-O-methyladenosine 3′,5′-cyclic monophosphate (8-CPT-2-cAMP, Sigma), or 0.2 μM Protein Kinase A inhibitor fragment (PKI, Sigma) in serum-free medium was added to the cells for 30 min prior to infection. The viability of the cells was not affected by any of the treatments used as determined by trypan blue exclusion. For the MTT adherence assay, cells were plated 96-well plates, incubated overnight, and fixed for 15 min with 3% paraformaldehyde. The monolayers were washed three times with sterile PBS and pretreated for 1 h at RT with blocking buffer (3% bovine serum albumin in PBS). 100 μl of E. coli strain ORN103(pSH2) (A600 =~1.0) in PBS, was incubated with cells for 1 h at 37 °C. Nonadherent bacteria were removed by washing the cell monolayers three times with PBS. Fifty microliters of LB was applied to each monolayer and incubated for 15 min at 37 °C. Fifty microliters of 2 mg/ml MTT (Sigma) in PBS was added, and the plates were incubated for 15 min at 37 °C to allow reduction of MTT to formazan by live bacteria. Next, 150 μl of mixture of isopropyl alcohol and hydrochloric acid (24:1) was added to solubilize the formazan, and the absorbance was measured at 450 nm using a Tecan Sunrise remote microplate reader.
Creation of TLR4 knockdowns using RNA Interference
RNA interference vectors were generated using pQCXIN retroviral vector (BD Biosciences). Briefly, pQCXIN was digested by BamHI and EcoRI and then was religated to generate pQCXIN1. Human U6 small nuclear RNA promoter was PCR-amplified from pTZ U6 + 1 (gift from John Rossi, Beckman Research Institute of the City of Hope, Duarte, CA) with added BglII site (5′ ends), BamHI, and XbaI sites (3′ ends). The PCR product was cloned to the BglII and XbaI sites of pQCXIN1 to generate pQCXIN-U6. The following oligonucleotides were ordered from Integrated DNA Technologies, Inc.: TLR4a, 5′-GATCCGTTCCGATTAGCATACTTAGTTCAAGAGACTAAGTATGCTAATCGG AACTTTTTTT-3′, and TLR4b, 5′-CTAGAAAAAAAGTTCCGATTAGCATACTTAGTCTCTTGAACTAAGTATGCT AATCGGAACG-3′. The boldface and underlined sequences are forward and reverse sequences, respectively, which correspond to nucleotides 1026-1044 of the human TLR4 gene (GenBank™ accession number U88880). The oligos were annealed to form double-stranded DNA and cloned into the BamHI and XbaI sites of pQCXIN-U6 to generate pSi-TLR4. The Amphopack-293 Cell Line (BD Biosciences) was used to produce the viral particles. Production of viral particles, infection of target cell line (5637), and selection of viral infected cells were performed as recommended by the vendor of the pQCXIN vector (BD Biosciences). The geneticin-resistant stable-transfected cell lines were named TLR4 KD BECs. Knockdowns were verified by RT-PCR using the specific primers listed below.
RNA isolation and reverse transcription-polymerase chain reaction (RT-PCR)
Total cellular RNA was isolated using RNeasy purification system (Qiagen). Two μg of total RNA was reverse transcribed and amplified with gene-specific primers using the RT-PCR System kit (Bio-rad). The primer sequences for the genes were as follows: 5′-CGATTCCATTGCTTCTTG-3′ (sense) and 5′-GCTCAGGTCCAGGTTCTT-3′ (antisense) for TLR4 and 5′-ATCCCATCACCATCTTCCAG-3′ (sense) and 5′-CCTGCTTCACCACCTTCTTG-3′ (antisense) for GAPDH. The primer sequences for the AC genes and expected product sizes were as summarized in Table 1 (Supplementary information). We confirmed that the AC isotype-specific primers were functional by undertaking RT-PCR on total RNA from HEK cells (a positive control cell, where all ACs except AC4 and 8 were expressed) (Ludwig and Seuwen, 2002).
Creation of BECs overexpressing constitutively activated, or dominant-negative Rac1
Rac1 was PCR amplified and inserted into the pLEGFP-C1 (Clontech) to produce GFP-Rac1. The GFP-Rac1 construct was then used to generate the dominant-active GFP-Rac1 (Q61L), or the dominant-negative GFP-Rac1 (T17N) by using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) following the vendor’s instruction.
Rac1 activity assay
For Rac1 activity assay, we used a glutathione-S-transferase (GST)-PAK-CD (p21-activated kinase (PAK) CRIB domain; which interacts with activated form of Rac1) fusion protein to selectively isolate activated form of Rac1 from human BECs. Multiple steps were involved in this approach; cloning and production of GST-PAK-CD fusion protein followed by pull down of activated form of Rac1 from BEC extracts employing the GST-PAK fusion protein.
Cloning and Production of GST-PAK-CD fusion protein
We first generated a plasmid containing GST-PAK-CD as described previously (Sander et al., 1998). Briefly, a DNA fragment encoding amino acids 56-141 from human PAK1B (GenBank accession number AF071884) was amplified by standard PCR and inserted into pGEX-4T-1 (Amersham Pharmacia Biotech) to produce pGST-PAK-CD. This plasmid was transformed into E. coli BL21. To purify GST-PAK-CD protein, E. coli harboring pGST-PAK-CD were grown overnight in LB broth with 100 μg/ml of ampicillin. On the following day, 1 ml of grown bacteria was added into 20 ml LB broth with ampicillin, and incubated for additional ~2 hours (A600=0.6–0.8). Expression of GST-PAK-CD protein was induced by addition of 0.1 mM IPTG for another 4 hours. Cells were harvested, resuspended in lysis buffer (0.1 M NaCl, 10 mM Tris-Cl, pH 8.0, 1 mM EDTA, pH 8.0, 100 μg/ml lysozyme, 5 mM DTT, protease inhibitor cocktail, 1.5% Sarkosyl), and then passed in 27-G needle 10 times. The supernatant was taken and incubated with 50% Glutathione sepharose beads slurry (Amersham Pharmacia Biotech) for 30 min at 4°C. Fusion protein bound the beads washed and directly used for a pull-down assay described as follows.
Pull down and quantitation of activated Rac1
5637 human BECs were seeded onto 10-cm culture dish at a density of 4.2 × 106 cells/plate (~60% confluency) and incubated overnight. The medium was replaced with fresh culture medium with 0.2% FBS and incubated at 37 °C for additional 5 hrs. The cells were inoculated with E. coli ORN103(pSH2) (100 MOI), and incubated at 37 °C for experimental time frames. The cells were lysed in a RIPA buffer (Upstate Biotechnology) containing 1 mM PMSF and a 1:100 dilution of mammalian protease inhibitor cocktail (Sigma). The cell suspension was passed 20 times through a 27-gauge needle, and centrifuged for 5 min with the precipitates then being discarded. Protein concentrations were determined using the Bradford reagent (Bio-Rad) with bovine serum albumin as a standard. To pull down activated form of Rac1 from BEC extracts, 200 μg BEC extracts were mixed with purified GST-PAK-CD protein bound the beads. The mixture was incubated for 1 hr at 4°C with shaking, and pellets were collected by centrifugation and washed four times with 1 ml of lysis buffer. After final wash, 50 μl of 2 x Sample buffer (Bio-Rad) was added and the samples were boiled for 5 min. Western blotting for Rac1 (BD Biosciences) was performed using 10 μl of the samples.
Measurement of intracellular cAMP levels
5637 human BECs were seeded onto 6-well plates and grown overnight. The cells were uninfected or infected with 100 MOI E. coli ORN103(pSH2), or treated with 100 μg/ml E. coli LPS (Sigma) for indicated time points. The cells were washed four times with PBS to remove culture media, and lysed in 250 μl of 0.1 M HCl for 10 min. After centrifugation, the supernatant was directly used for the cAMP assay. Intracellular concentrations of cAMP were determined using a cAMP enzyme immunoassay kit (Sigma) according to the manufacturer’s instructions. In order to measure intracellular cAMP levels of BECs obtained from control HeN and TLR4 mutant HeJ mice, 8–10 week old female mice were anesthetized and sacrificed, and bladders were aseptically removed, bisected, turned epithelium-side out, lysed in 300 μl of 0.1 M HCl for 20 min. Supernatant from centrifugation was directly used for the assay as before.
Statistics
Two tailed Student’s T-tests were preformed in order to determine the statistical significance of experimental changes from control values.
Supplementary Material
Acknowledgments
We thank Meta Kuehn (Department of Biochemistry, Duke University Medical Center) for critical reading of this manuscript and Christian Raetz (Department of Biochemistry, Duke University Medical Center) for the gift of the msbB mutant E. coli and corresponding wild-type E. coli. This work was supported in part with research grants from the National Institutes of Health (AI 056101, AI 150021, and DK 050814).
Footnotes
JS performed the experiments in this work. GL generated the E. coli harboring pGST-PAK-CD and E. coli harboring the dominant-positive GFP-Rac1 and the dominant-negative GFP-Rac1. BLB and MJD initiated the laboratory use of the in vivo and the in vitro models of E. coli infection of BECs. JS and SNA wrote the paper; the other authors read and commented on the manuscript.
COMPETING INTERESTS
The authors have declared that no competing interests exist.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abraham SN, Babu JP, Giampapa CS, Hasty DL, Simpson WA, Beachey EH. Protection against Escherichia coli-induced urinary tract infections with hybridoma antibodies directed against type 1 fimbriae or complementary D-mannose receptors. Infect Immun. 1985;48:625–628. doi: 10.1128/iai.48.3.625-628.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham SN, Sun D, Dale JB, Beachey EH. Conservation of the D-mannose-adhesion protein among type 1 fimbriated members of the family Enterobacteriaceae. Nature. 1988;336:682–684. doi: 10.1038/336682a0. [DOI] [PubMed] [Google Scholar]
- Apodaca G. The uroepithelium: not just a passive barrier. Traffic. 2004;5:117–128. doi: 10.1046/j.1600-0854.2003.00156.x. [DOI] [PubMed] [Google Scholar]
- Backhed F, Soderhall M, Ekman P, Normark S, Richter-Dahlfors A. Induction of innate immune responses by Escherichia coli and purified lipopolysaccharide correlate with organ- and cell-specific expression of Toll-like receptors within the human urinary tract. Cell Microbiol. 2001;3:153–158. doi: 10.1046/j.1462-5822.2001.00101.x. [DOI] [PubMed] [Google Scholar]
- Bishop BL, Duncan MJ, Song J, Li G, Zaas D, Abraham SN. Cyclic AMP-regulated exocytosis of Escherichia coli from infected bladder epithelial cells. Nat Med. 2007;13:625–630. doi: 10.1038/nm1572. [DOI] [PubMed] [Google Scholar]
- Blander JM, Medzhitov R. Regulation of phagosome maturation by signals from toll-like receptors. Science. 2004;304:1014–1018. doi: 10.1126/science.1096158. [DOI] [PubMed] [Google Scholar]
- Blander JM, Medzhitov R. On regulation of phagosome maturation and antigen presentation. Nat Immunol. 2006;7:1029–1035. doi: 10.1038/ni1006-1029. [DOI] [PubMed] [Google Scholar]
- Clementz T, Zhou Z, Raetz CR. Function of the Escherichia coli msbB gene, a multicopy suppressor of htrB knockouts, in the acylation of lipid A. Acylation by MsbB follows laurate incorporation by HtrB. J Biol Chem. 1997;272:10353–10360. doi: 10.1074/jbc.272.16.10353. [DOI] [PubMed] [Google Scholar]
- Coats SR, Pham TT, Bainbridge BW, Reife RA, Darveau RP. MD-2 mediates the ability of tetra-acylated and penta-acylated lipopolysaccharides to antagonize Escherichia coli lipopolysaccharide at the TLR4 signaling complex. J Immunol. 2005;175:4490–4498. doi: 10.4049/jimmunol.175.7.4490. [DOI] [PubMed] [Google Scholar]
- Duncan MJ, Li G, Shin JS, Carson JL, Abraham SN. Bacterial penetration of bladder epithelium through lipid rafts. J Biol Chem. 2004;279:18944–18951. doi: 10.1074/jbc.M400769200. [DOI] [PubMed] [Google Scholar]
- Elmslie KS. Calcium channel blockers in the treatment of disease. J Neurosci Res. 2004;75:733–741. doi: 10.1002/jnr.10872. [DOI] [PubMed] [Google Scholar]
- Fischer H, Yamamoto M, Akira S, Beutler B, Svanborg C. Mechanism of pathogen-specific TLR4 activation in the mucosa: fimbriae, recognition receptors and adaptor protein selection. Eur J Immunol. 2006;36:267–277. doi: 10.1002/eji.200535149. [DOI] [PubMed] [Google Scholar]
- Hagberg L, Hull R, Hull S, Falkow S, Freter R, Svanborg Eden C. Contribution of adhesion to bacterial persistence in the mouse urinary tract. Infect Immun. 1983;40:265–272. doi: 10.1128/iai.40.1.265-272.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg L, Hull R, Hull S, McGhee JR, Michalek SM, Svanborg EC. Difference in susceptibility to gram-negative urinary tract infection between C3H/HeJ and C3H/HeN mice. Infect Immun. 1984;46:839–844. doi: 10.1128/iai.46.3.839-844.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg L, Jodal U, Korhonen TK, Lidin-Janson G, Lindberg U, Svanborg Eden C. Adhesion, hemagglutination, and virulence of Escherichia coli causing urinary tract infections. Infect Immun. 1981;31:564–570. doi: 10.1128/iai.31.2.564-570.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe AK. Regulation of actin-based cell migration by cAMP/PKA. Biochim Biophys Acta. 2004;1692:159–174. doi: 10.1016/j.bbamcr.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Insel PA, Ostrom RS. Forskolin as a tool for examining adenylyl cyclase expression, regulation, and G protein signaling. Cell Mol Neurobiol. 2003;23:305–314. doi: 10.1023/a:1023684503883. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. TLR signaling. Cell Death Differ. 2006;13:816–825. doi: 10.1038/sj.cdd.4401850. [DOI] [PubMed] [Google Scholar]
- Langermann S, Palaszynski S, Barnhart M, Auguste G, Pinkner JS, Burlein J, Barren P, Koenig S, Leath S, Jones CH, et al. Prevention of mucosal Escherichia coli infection by FimH-adhesin-based systemic vaccination. Science. 1997;276:607–611. doi: 10.1126/science.276.5312.607. [DOI] [PubMed] [Google Scholar]
- Ludwig MG, Seuwen K. Characterization of the human adenylyl cyclase gene family: cDNA, gene structure, and tissue distribution of the nine isoforms. J Recept Signal Transduct Res. 2002;22:79–110. doi: 10.1081/rrs-120014589. [DOI] [PubMed] [Google Scholar]
- Manser E, Leung T, Salihuddin H, Zhao ZS, Lim L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature. 1994;367:40–46. doi: 10.1038/367040a0. [DOI] [PubMed] [Google Scholar]
- Martinez JJ, Hultgren SJ. Requirement of Rho-family GTPases in the invasion of Type 1-piliated uropathogenic Escherichia coli. Cell Microbiol. 2002;4:19–28. doi: 10.1046/j.1462-5822.2002.00166.x. [DOI] [PubMed] [Google Scholar]
- Minion FC, Abraham SN, Beachey EH, Goguen JD. The genetic determinant of adhesive function in type 1 fimbriae of Escherichia coli is distinct from the gene encoding the fimbrial subunit. J Bacteriol. 1986;165:1033–1036. doi: 10.1128/jb.165.3.1033-1036.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvey MA, Schilling JD, Martinez JJ, Hultgren SJ. Bad bugs and beleaguered bladders: interplay between uropathogenic Escherichia coli and innate host defenses. Proc Natl Acad Sci U S A. 2000;97:8829–8835. doi: 10.1073/pnas.97.16.8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasawa SY, Takuwa N, Sugimoto N, Mabuchi H, Takuwa Y. Inhibition of Rac activation as a mechanism for negative regulation of actin cytoskeletal reorganization and cell motility by cAMP. Biochem J. 2005;385:737–744. doi: 10.1042/BJ20041060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor KL, Mercurio AM. Protein kinase A regulates Rac and is required for the growth factor-stimulated migration of carcinoma cells. J Biol Chem. 2001;276:47895–47900. doi: 10.1074/jbc.M107235200. [DOI] [PubMed] [Google Scholar]
- Orndorff PE, Falkow S. Identification and characterization of a gene product that regulates type 1 piliation in Escherichia coli. J Bacteriol. 1984;160:61–66. doi: 10.1128/jb.160.1.61-66.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, Schroeder L, Aderem A. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc Natl Acad Sci U S A. 2000;97:13766–13771. doi: 10.1073/pnas.250476497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pressman BC. Biological applications of ionophores. Annu Rev Biochem. 1976;45:501–530. doi: 10.1146/annurev.bi.45.070176.002441. [DOI] [PubMed] [Google Scholar]
- Reid G, Sobel JD. Bacterial adherence in the pathogenesis of urinary tract infection: a review. Rev Infect Dis. 1987;9:470–487. doi: 10.1093/clinids/9.3.470. [DOI] [PubMed] [Google Scholar]
- Saemann MD, Weichhart T, Horl WH, Zlabinger GJ. Tamm-Horsfall protein: a multilayered defence molecule against urinary tract infection. Eur J Clin Invest. 2005;35:227–235. doi: 10.1111/j.1365-2362.2005.01483.x. [DOI] [PubMed] [Google Scholar]
- Samuelsson P, Hang L, Wullt B, Irjala H, Svanborg C. Toll-like receptor 4 expression and cytokine responses in the human urinary tract mucosa. Infect Immun. 2004;72:3179–3186. doi: 10.1128/IAI.72.6.3179-3186.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander EE, van Delft S, ten Klooster JP, Reid T, van der Kammen RA, Michiels F, Collard JG. Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell-cell adhesion or cell migration and is regulated by phosphatidylinositol 3-kinase. J Cell Biol. 1998;143:1385–1398. doi: 10.1083/jcb.143.5.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling JD, Martin SM, Hunstad DA, Patel KP, Mulvey MA, Justice SS, Lorenz RG, Hultgren SJ. CD14- and Toll-like receptor-dependent activation of bladder epithelial cells by lipopolysaccharide and type 1 piliated Escherichia coli. Infect Immun. 2003;71:1470–1480. doi: 10.1128/IAI.71.3.1470-1480.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling JD, Mulvey MA, Vincent CD, Lorenz RG, Hultgren SJ. Bacterial invasion augments epithelial cytokine responses to Escherichia coli through a lipopolysaccharide-dependent mechanism. J Immunol. 2001;166:1148–1155. doi: 10.4049/jimmunol.166.2.1148. [DOI] [PubMed] [Google Scholar]
- Schmeck B, Huber S, Moog K, Zahlten J, Hocke AC, Opitz B, Hammerschmidt S, Mitchell TJ, Kracht M, Rosseau S, et al. Pneumococci induced TLR- and Rac1-dependent NF-kappaB-recruitment to the IL-8 promoter in lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2006;290:L730–L737. doi: 10.1152/ajplung.00271.2005. [DOI] [PubMed] [Google Scholar]
- Shafer SH, Phelps SH, Williams CL. Reduced DNA synthesis and cell viability in small cell lung carcinoma by treatment with cyclic AMP phosphodiesterase inhibitors. Biochem Pharmacol. 1998;56:1229–1236. doi: 10.1016/s0006-2952(98)00260-3. [DOI] [PubMed] [Google Scholar]
- Shahin RD, Engberg I, Hagberg L, Svanborg EC. Neutrophil recruitment and bacterial clearance correlated with LPS responsiveness in local gram-negative infection. J Immunol. 1987;138:3475–3480. [PubMed] [Google Scholar]
- Song J, Duncan MJ, Li G, Chan C, Grady R, Stapleton A, Abraham SN. A Novel TLR4-Mediated Signaling Pathway Leading to IL-6 Responses in Human Bladder Epithelial Cells. PLoS Pathogens. 2007;3:e60. doi: 10.1371/journal.ppat.0030060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunahara RK, Taussig R. Isoforms of mammalian adenylyl cyclase: multiplicities of signaling. Mol Interv. 2002;2:168–184. doi: 10.1124/mi.2.3.168. [DOI] [PubMed] [Google Scholar]
- Svanborg C, Bergsten G, Fischer H, Godaly G, Gustafsson M, Karpman D, Lundstedt AC, Ragnarsdottir B, Svensson M, Wullt B. Uropathogenic Escherichia coli as a model of host-parasite interaction. Curr Opin Microbiol. 2006;9:33–39. doi: 10.1016/j.mib.2005.12.012. [DOI] [PubMed] [Google Scholar]
- Thankavel K, Madison B, Ikeda T, Malaviya R, Shah AH, Arumugam PM, Abraham SN. Localization of a domain in the FimH adhesin of Escherichia coli type 1 fimbriae capable of receptor recognition and use of a domain-specific antibody to confer protection against experimental urinary tract infection. J Clin Invest. 1997;100:1123–1136. doi: 10.1172/JCI119623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truschel ST, Wang E, Ruiz WG, Leung SM, Rojas R, Lavelle J, Zeidel M, Stoffer D, Apodaca G. Stretch-regulated exocytosis/endocytosis in bladder umbrella cells. Mol Biol Cell. 2002;13:830–846. doi: 10.1091/mbc.01-09-0435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underhill DM, Gantner B. Integration of Toll-like receptor and phagocytic signaling for tailored immunity. Microbes Infect. 2004;6:1368–1373. doi: 10.1016/j.micinf.2004.08.016. [DOI] [PubMed] [Google Scholar]
- Waschke J, Drenckhahn D, Adamson RH, Barth H, Curry FE. cAMP protects endothelial barrier functions by preventing Rac-1 inhibition. Am J Physiol Heart Circ Physiol. 2004;287:H2427–2433. doi: 10.1152/ajpheart.00556.2004. [DOI] [PubMed] [Google Scholar]
- Wissel H, Schulz C, Koehne P, Richter E, Maass M, Rudiger M. Chlamydophila pneumoniae induces expression of toll-like receptor 4 and release of TNF-alpha and MIP-2 via an NF-kappaB pathway in rat type II pneumocytes. Respir Res. 2005;6:51. doi: 10.1186/1465-9921-6-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Glogauer M, Zhu F, Kim TH, Chiu B, Inman RD. Innate immunity and arthritis: neutrophil Rac and toll-like receptor 4 expression define outcomes in infection-triggered arthritis. Arthritis Rheum. 2005;52:1297–1304. doi: 10.1002/art.20984. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.