

Abstract
The xenobiotic receptors CAR and PXR constitute two important members of the NR1I nuclear receptor family. They function as sensors of toxic byproducts of endogenous metabolism and of exogenous chemicals and enhance their elimination. This unique function of CAR and PXR sets them apart from the steroid hormone receptors. In contrast, the steroid receptors, exemplified by the estrogen receptor (ER) and glucocorticoid receptor (GR), are the sensors that tightly monitor and respond to changes in circulating steroid hormone levels to maintain body homeostasis. This divergence of the chemical- and steroid-sensing functions has evolved to ensure the fidelity of the steroid hormone endocrine regulation while allowing development of metabolic elimination pathways for xenobiotics. The development of the xenobiotic receptors CAR and PXR also reflect the increasing complexity of metabolism in higher organisms, which necessitate novel mechanisms for handling and eliminating metabolic byproducts and foreign compounds from the body. The purpose of this review is to discuss similarities and differences between the xenobiotic receptors CAR and PXR with the prototypical steroid hormone receptors ER and GR. Interesting differences in structure explain in part the divergence in function and activation mechanisms of CAR/PXR from ER/GR. In addition, the physiological roles of CAR and PXR will be reviewed, with discussion of interactions of CAR and PXR with endocrine signaling pathways.
Keywords: Constitutive androstane receptor, pregnane X receptor, estrogen receptor, glucocorticoid receptor, structure-function relationship, physiology
1. Introduction
During the last decade, much progress has been made in dissecting the mechanisms by which foreign compounds elicit alterations in hepatic xeno- and endobiotic metabolism. A regulatory mechanism for these alterations is the binding of a chemical to a member of the nuclear receptor family, followed by induction of enzymes involved in drug metabolism. The NR1I subfamily members, the constitutive active/androstane receptor (CAR) and the pregnane X receptor (PXR), function in this capacity. In addition to regulating drug metabolism, they both play physiological roles in regulating metabolic pathways important for the elimination of cholesterol. Interestingly, no physiological ligands have been definitively identified. This is in contrast to the steroid nuclear receptors, the estrogen and glucocorticoid receptors (ER and GR), which mediate biological responses to known steroid hormones. The purpose of this review is to highlight the significant differences in the characteristics of CAR and PXR receptors from the steroid hormone receptors. Interestingly, some mechanistic similarities in the activation process have been observed between these two classes of nuclear receptors, and these will also be discussed.
2. The xenobiotic receptors: CAR and PXR
2.1 Functions
2.1.1 Xenobiotic and endobiotic metabolism
It is well known that phenobarbital (PB) regulates numerous genes that are involved in Phase I (cytochrome P450), Phase II (conjugation catalyzed by sulfotranserases, glucuronosyltransferases and glutathione S-transferases), and transporter-mediated (i.e. MRPs, OATPs) drug elimination pathways. In the case of PB induction of the xenobiotic-metabolizingcytochromes P450 2B (CYP2B), the mechanism is transcriptional resulting in elevated mRNA levels [1]. CAR expression, summarized by the UniGene database, is primarily in liver and kidney, with some expression detected in heart and GI tissues in the mouse and in brain tissues in humans. Progress in the last ten years has identified PB responsive enhancer modules (PBREM) in the human, mouse, and rat CYP2B gene promoters, to which the identified nuclear receptor constitutive activated/androstane receptor (CAR) was shown to bind (for more detailed review see [2, 3]).Lastly, CAR has been implicated in regulation of circulating thyroid hormone (TH) levels. PB treatment induces TH conjugation pathways in a CAR-dependent manner resulting in diminished serum thyroxine (T4) [4], and fasting-induced reductions in serum triiodothyronine and T4 concentrations TH levels are absent in Car-null mice [5].
In the case of PXR, its role in the induction of Phase I and II enzymes and transporters was elucidated concurrently with CAR. As summarized in the UniGene database, PXR expression is found primarily in the liver with some expression in testis and embryonic tissues in humans, while in the mouse its expression is confined to the GI tract and liver. Interestingly, many early studies (see [6] and references therein) investigating CYP3A induction by glucocorticoids, pregnane compounds (e.g. pregnane 16α-carbonitrile or PCN) and macrolide antibiotics (e.g. rifampicin or RIF) were thought to involve the glucocorticoid receptor.This was partly due to the impact of results obtained from evaluating steroid structure-activity relationships for CYP3A induction, which suggested a non-classical GR pathway. However in the mid-1990s, detailed characterization of promoter elements in the CYP3A23 gene that confers DEX/PCN responsiveness uncovered a novel promoter element. It contained a nuclear receptor half-site, which GR was found not to bind, suggesting a novel nuclear receptor. Confirming this assertion was the subsequent cloning of PXR in 1998 and the definition of the PXR signaling pathway. PXR alone accounted for low-dose chemical induction of CYP3A, as well as the species differences in CYP3A induction by PCN and RIF. Based on these and other finds, PXR is now widely viewed to be the mediator of xenobiotic regulation of CYP3A.
2.1.2 Role in gluconeogenesis and bile acid homeostasis
Both CAR and PXR have been implicated in energy metabolism. CAR has been shown to be linked to PPAR-γ coactivator 1α (PGC-1α), a transcriptional cofactor induced by fasting and regulates energy metabolism [7]. Regulation of gluconeogenesis mediated by the forkhead transcription factor FoxO1 can also be influenced by CAR. CAR was shown to downregulate the binding of FoxO1 to insulin response sequences (IRS), similar to the effect of insulin, and attenuated the ability of FoxO1 to stimulate gluconeogenic genes such as PEPCK[8].This provides a likely explanation of how PB treatment improves insulin sensitivity in non-insulindependent/Type II diabetes [9]. PXR also represses transcriptional activity of FoxO1 on IRS-1 elements [8], implicating PXR as a negative transcriptional regulator of genes involved in glucose metabolism. Further characterization of this novel inhibitory cross-talk is currently ongoing.
PXR has recently been found to play a major role in protection from bile acid toxicity. The basis for PXR being involved in bile acid homeostasis comes from in vivo observations showing that PXR activation is protective against hepatotoxic bile acid [10, 11] and that accumulation of bile acid precursors leads to PXR activation [12]. These studies reveal PXR’s role in regulating conversion of cholesterol into bile acids and detoxifying oxidized cholesterol (“oxysterols”). Evidence further supporting this notion came with the recent report of high mortality associated with hepatitis and hepatocellular injury in PXR-null mice fed a cholesterol- and cholic acid-enriched diet [13]. Cholic acid was included in the diet to block cholesterol catabolism and increase intestinal cholesterol absorption. In addition to liver injury, the PXR target genes Cyp3a11 and Oatp2 were also induced in wild-type mice, but not in PXR-null mice. With respect to CAR’s role in regulating cholesterol levels, pretreatment of PXR-null mice with TCPOBOP did not ameliorate lethality from the cholesterol/cholic acid diet. Thus, PXR serves a protective role against cholesterol toxicity.
Bile acids (BAs) are directly linked to the maintenance of body cholesterol levels, as they are the first products of cholesterol catabolism. BA formation has been shown to be regulated by both CAR and PXR. The rate-limiting enzyme in the BA formation is CYP7A, which is repressed by PXR in response to lithocholic acid [10, 11]; however, this constitutes a BA-sensing mechanism that is secondary to the BA sensor farnesoid X receptor (FXR) [14]. Also shown in mouse, CAR induces enzymes and transporters involved in BA elimination, namely CYP3A11, MRP3, and the sulfotrasferase SULT2A1 [15]. Hence CAR also contributes to maintaining normal cholesterol levels indirectly through regulation of BA homeostasis.
2.2 Ligand promiscuity & species differences
Both PXR and CAR exhibit the ability to bind multiple ligands (Fig. 1), and each receptor’s ligand specificity is species-dependent. For CAR, a variety of compounds bind to it in addition to the classic CYP2B inducer PB. These include androstane steroidal compounds (i.e. 3α,5α-androstanol), 5β-pregnane-3,20-dione, retinoic acids, clotrimazole, chlorpromazine, o,p’-DDT, methoxychlor, and to hydrocarbons such as 2,3,3’,4’,5’,6-hexachlorobiphenyl, 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime (CITCO), and 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene (TCPOBOP) [2, 16-18]. These constitute a structurally-diverse panel of compounds. However, their affinities for CAR vary across species and in some cases their direct binding to CAR remains a matter of debate; TCPOBOP and CITCO are the only compounds shown to specifically bind to mouse CAR (mCAR) and human CAR (hCAR), respectively. Nonetheless, these represent a large variety of chemical structures that are accommodated in the CAR binding pocket. In the case of PXR, ligands include the naturally occurring steroids 5α-pregnane-3,20-dione, progesterone, 17α-hydroxyprogesterone, 17α-hydroxypregnenolone and corticosterone, hyperforin (a component of St. John’s Wort), dexamethasone, the a nti-glucocorticoids PCN and RU486, taxol, and the bisphosphonate ester SR12813 [19]. Selectivity for these compounds also differs across species. For instance, pregnenolone 16α-carbonitrile (PCN) binds to the rodent form of PXR, while rifampicin (RIF) and SR12813 is specific to human PXR [19, 20]. The structural nature of PXR’s ligand binding domain explains in part its broad substrate specificity, as will be discussed later in this review.
Figure 1. Chemical structures of ligands for CAR and PXR, and the nuclear steroid receptors ER and GR.
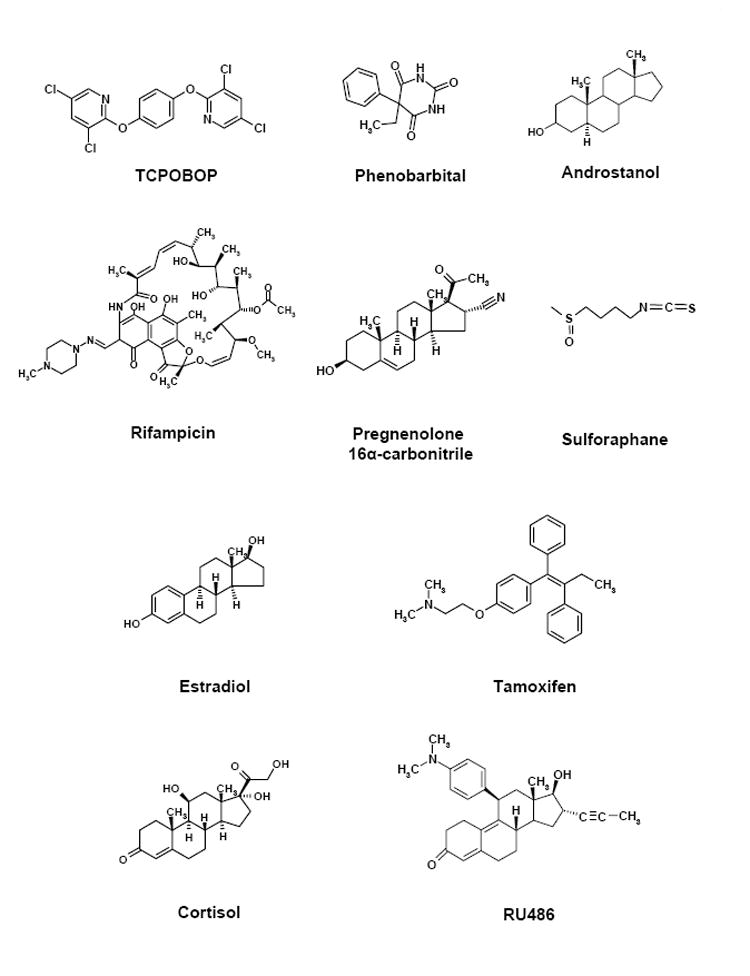
Each horizontal set of compounds represent the prototypical ligands, respectively from top to bottom, for CAR, PXR, ER and GR.
2.3 Mechanisms of activation
2.3.1 CAR
2.3.1.1 Overview
The hallmark mechanism ascribed to PB-induced, CAR-mediated induction of the CYP2B genes is the activation of cytosolic CAR upon PB exposure, resulting in its dissociation from its co-chaperone partnerscytoplasmicCARretentionprotein (CCRP) and HSP90. Translocation of CAR to the nucleus, presumably dependent on the activity of the protein phosphatase PP2A, is followed by association with the retinoid X receptor (RXR) and binding to the PBREM. An additional feature recently uncovered for CAR activation includes its translocation to the membrane [21], which raises the possibility of signaling components important in CAR activation located at the cell membrane. Transcriptional activation occurs upon CAR binding to the PRBREM which contains two nuclear receptor DR4 sites (NR1 and NR2). CAR coactivators identified to date that facilitate transactivation include GRIP1/TIF2 , PGC-1, SRC-1, Sp1, and more recently, ASC-2 [22-26], and SMC-1 in the synergistic activation on the CYP2B6 gene promoter by TCPOBOP and the phosphatase inhibitor okadaic acid [27]. In this signaling paradigm, translocation of CAR into the nucleus serves as an important step to regulate CAR activity. This is an area of active investigation, the highlights of which are discussed in the next section.
2.3.1.2 Nuclear translocation
The steps leading to full ligand-mediated activation of CAR into its transcriptionally competent form has been the focus of investigation in numerous laboratories. To date, individual components of the pathway, namely genes specifically targeted by CAR, promoter elements to which CAR binds (i.e. the PBREM), accessory DNA elements important in transcriptional activation, as well as the physiological role(s) of CAR, have been identified by us and others. However, a detailed understanding of the balance between cytosolic sequestration and translocation, crucial for controlling CAR’s actions on target genes, is lacking.
A general schematic of the CAR pathway and its components, each discussed in more detail below, is shown in Fig. 2. The essential factor regulating CAR’s xenobiotic-induced transcriptional effects is nuclear translocation. Using immunohistochemistry, Takeshi Kawamoto and co-workers found cytosolic staining of CAR in livers of untreated mice and dramatic nuclear staining of CAR in mice within one hour of PB treatment [28]. Nuclear accumulation of fluorescently-tagged CAR in HepG2 cells and primary hepatocytes was also observed using TCPOBOP, and correlated with increased activity of a co-transfected luciferase reporter plasmid. Collectively, these data indicate that CAR transcriptional activity is the consequence of its accumulation in the nucleus.
Figure 2. Summary of signaling pathways for CAR and PXR, and the nuclear steroid receptors ER and GR.
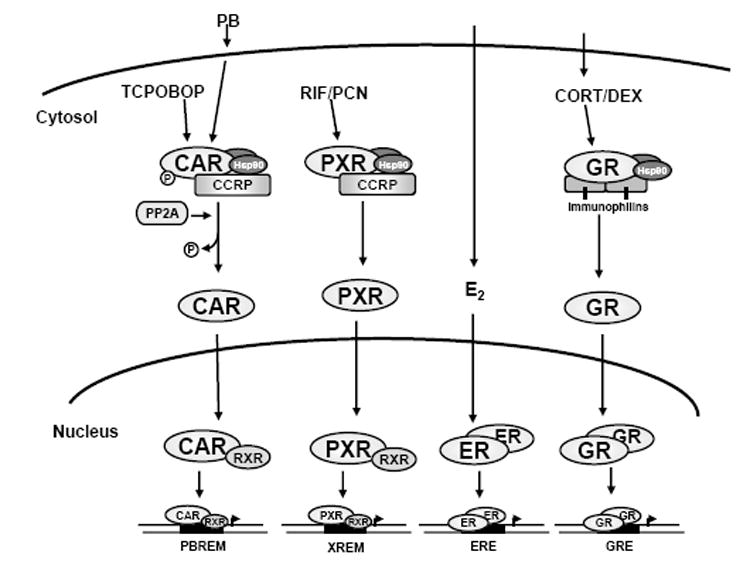
Additional insights of CAR drug-induced nuclear accumulation has been obtained from studies using protein phosphatase inhibitors. One such inhibitor, okadaic acid (OA), blocks the PB induction of rodent CYP2B genes [29], and treatment with OA was shown to prevent nuclear accumulation of CAR [30]. Taken together, these investigations of CAR nuclear translocation revealed that tight control of an otherwise constitutively active receptor is achieved by cytosolic sequestration in vivo. This property appears to apply to hCAR as well, since CPZ treatment induced its nuclear translocation when expressed in mouse liver [31]. Subsequent analysis using fluorescently-tagged hCAR expressed in mouse liver showed that nuclear translocation is AF-2 independent and that PB-inducible translocation is regulated by a 30 amino acid leucine-rich motif in the C-terminal region termed the xenobiotic response sequence (XRS) [31]. Moreover, this translocation activity occurs without a strong nuclear localization sequence (NLS) signal like that present in GR.
Exploring this translocation mechanism further, transient expression of untagged and epitope-tagged versions of CAR (either mouse or human) in transformed human liver cell lines has been attempted. Unfortunately, this strategy is limited by the spontaneous accumulation of both transiently- and stably-expressed CAR in the nucleus. This tendency is somewhat reduced when CAR is expressed transiently compared to stable expression, with cytosolic CAR being detectable under these conditions. This technical difficulty was partially alleviated, however, by the identification of a CAR cytosolic sequestration protein termed CAR cytoplasmic retention protein (CCRP). In vivo, CAR is sequestered in the cytosol in a protein complex with the heatshock chaperone HSP90 [32]. CCRP was found to be a component of this protein complex, and its overexpression in HepG2 indeed stabilized CAR co-expression in the cytosol. This strategy produced a more pronounced nuclear translocation effect by TCPOBOP, mitigating in part the technical limitation of transiently overexpressing CAR in HepG2 cells. Thus, these findings demonstrate the importance of CCRP in the CAR nuclear translocation mechanism.
In addition to CCRP, other proteins regulate CAR nuclear translocation. Follow-up studies of the effect of OA identified phosphatase 2A (PP2A), which is recruited to the HSP90:CAR complex [33]. Initially it was unknown if CAR or HSP90 was targeted for dephosphorylation, until Ser202 of mCAR was identified. When mutated to Asp to mimic phosphorylation, this abrogated PB-induced nuclear accumulation of CAR in vivo, while in contrast mutation to Ala promoted nuclear accumulation [34]. Co-overexpression of PP2A with mCAR also enhanced TCPOBOP-induced CAR nuclear translocation in HepG2 cells. Thus, dephosphorylation of CAR constitutes a crucial signal for its release from the cytosolic complex upon activator treatment.
Other proteins have been suggested to mediate CAR nuclear translocation. Byron Kemper’s group identified the p160 transcription factor GR-interacting protein 1 (GRIP1) as an XRS-binding protein assisting in CAR nuclear accumulation [24, 35]. Generally viewed as a nuclear receptor coregulator of transcriptional activity, overexpression of GRIP1 in mouse liver enhanced PB-induced nuclear accumulation of CAR [24]. This correlated with enhancement of CYP2B-promoter transactivation, thus linking the subcellular localization of CAR to its transcriptional coactivating function. In view of these findings, it will be important to distinguish between GRIP1 functioning as a CAR nuclear retention protein rather than as a protein driving CAR translocation from the cytoplasm to the nucleus. However, questions remain as to the importance of GRIP1 in CAR nuclear translocation, since attempts in our laboratory to recapitulate these effects have been unsuccessful [34]. Interestingly, another protein, PPAR binding protein (PBP)/TRAP220/MED1, has been suggested to play a role in CAR nuclear translocation. Using a mouse PBP knockout model, loss of PBP resulted in the abrogation of PB induced CAR nuclear translocation, implicating PBP in regulating CAR activity [36]. However, as for GRIP1 it remains to be determined whether this is due to PBP either enhancing nuclear import or retaining CAR in the nucleus.
2.3.2 PXR
2.3.2.1 Overview
The features of PXR signaling (Fig. 2) are similar to those for CAR (reviewed in [19, 20]), particularly when comparing the mouse forms of the receptors. As with mCAR, mPXR is sequestered in the cytosol by CCRP [37]. Upon ligand binding, PXR then dissociates and translocates to the nucleus to activate gene transcription as a heterodimer with RXR. Core promoter elements, contained within the defined xenobiotic responsive enhancer module (XREM), consist of DR3 motifs or ER6 motifs in the CYP3A gene promoter depending on species. Recently, numerous transcriptional cofactors reglating PXR activity on target gene promoters have been identified. Given that PXR shares many target genes with CAR, it is not surprising that many of the coregulators found to interact with CAR bind PXR as well (summarized in [38]) These coregulators include members of the p160 family of coregulators, such as SRC-1, TIF2/GRIP1, and ACTR, as well as SUG-1, RIP140, PBP, and PGC-1. Signaling cascades have been implicated in modulating PXR activity, most notably the potentiation of PXR-mediated CYP3A induction by the protein kinase A (PKA) activator forskolin resulting from enhanced recruitment of PGC-1 [39]. Moreover, PXR was shown to be phosphorylated by PKA in vitro, and specific activation of PKA using 8-Br-cAMP enhanced interactions of PBP and SRC1 to PXR. In contrast, protein kinase C (PKC) was shown to repress PXR signaling [40]. Further studies to characterize the role of phosphorylation in PXR translocation will undoubtedly shed new light on the role of rapid-acting kinase pathways in the regulation of PXR. As with CAR, for PXR we will focus on nuclear translocation as it plays an important mechanism forregulating its transcriptional activity.
2.3.2.2 Nuclear Translocation
Compared to CAR, the effect of ligand on PXR’s subcellular localization is not well understood. We recently investigated the localization of mouse PXR in both its resting and ligand-activated state. Like with CAR, mouse PXR was located in the resting state in the cytosol of mouse liver. All functional motifs, namely the nuclear localization sequence or NLS (localized in the C-terminal region of the DBD), the xenobiotic response sequence or XRS, and the transcription activation function 2 (AF-2) domain, were all necessary for PCN-induced nuclear translocation. Like CAR, PXR forms a protein complex with CCRP and HSP90, and CCRP overexpression increased cytosolic levels of PXR [37]. It was also found that transient knockdown of CCRP by siRNA attenuates ligand-induced PXR transcriptional activation, giving additional evidence of the importance of cytosolic sequestration in regulating PXR transcriptional activity [37]. Thus, nuclear translocation is an important step in the activation of PXR as it is for CAR (Fig. 2). However, the precise molecular mechanisms involved in this process remain to be elucidated.
2.4 Structural aspects of CAR and PXR
In order to function as a xenobiotic sensor, receptors serving this role require broad ligand specificity. We propose this to be a defining characteristic of the xenobiotic receptors. CAR and PXR both possess unique hydrophobic ligand pockets that are larger and more flexible than those of the estrogen and glucocorticoid receptors. Both flexibility and larger ligand pocket volume can largely account for the diversity and range of size of CAR and PXR ligands. In addition, the overall structure of CAR and PXR differs when compared to steroid receptors, most notably poor conservation of the amino-terminal domain (Fig. 3). The structural elements underlying these unique trademarks of CAR and PXR will be highlighted in the next two sections.
Figure 3. Contrasting the functional domain architecture of CAR and PXR to that of the steroid receptors.

ERα, representing the steroid receptors, is shown together with human and mouse forms of CAR and PXR. The percent identities in the DBDs and LBDs are shown relative to hCAR.
2.4.1 CAR
Using different approaches to evaluate the structural elements of CAR underlying its constitutive and ligand-dependent activities, several important findings have been uncovered. Investigating the mechanism by which CAR’s helix-12/AF-2 (aa 350-357) mediated the repressive effects of androstanol on the transcriptional activity of mouse but not human CAR [41], Thr350 in mouse CAR was mutated to Met, the equivalent residue at this position in hCAR [42]. This mutation abolished this repression of androstanol on mCAR. However, this mCAR point mutant did not lose its ability to interact with SRC-1 as its activity was regulated by SRC-1. Evaluating further the role of Thr350 in regulating CAR activity, a hydrogen bond between Thr350 with Thr176 was identified to play a role to enhance stability of the AF-2 conformation favoring interactions with coregulators [43]. Maintenance of this hydrogen bond in the original Thr350Met mutant explains its interaction and thus activation by SRC-1. Three other residues of mouse CAR (K187, L352 and E355) were also shown to play a role in the binding of the coregulators, SRC-1 and SMRT, and when mutated causes loss of coregulator binding and strongly reduces ligand-dependent and -independent activities of CAR [44]. The significance of these studies is that they provided important insights into the structural determinants required for CAR’s AF-2 interaction with coregulators, and were also among the first to hypothesize rigidity in CAR’s structure that constrains it in the active conformation resulting in constitutive activity. Perhaps not surprisingly, this notion was confirmed from CAR X-ray crystallography studies (see below)
Lys205 was also found to be important for mouse CAR activity, adding a fourth residue that contributes to helix 12/AF-2 stabilization in accordance with homology modeling of CAR based on the PXR crystal structure [44]. Additional work also identified C357 in AF-2 function; however, mutation of equivalent residues in human CAR revealed a subtle species difference. Specifically, C357 contributes more to AF-2 stabilization in mouse CAR than the equivalent C347 of human CAR; however, a second residue (Ile330) is utilized by human CAR to enhance the stabilization of helix 12 [45]. More detailed analysis by the same research group delineated additional residues in helix 3, 5, 11, and 12 contributing to basal activity, and ligand selectivity by helix 3, 5, and 7 [46].
Recent X-ray crystallography studies have complemented the mutagenesis studies and in some respects mirrors their findings, particularly in regards to conformation of the C-terminal helix 12/AF-2. This segment of the nuclear receptor (such as ERα) assumes the “active” conformation that facilitates interaction with a coactivator LXXLL binding surface upon ligand binding. This conformational stabilization of the AF-2 includes capping the ligand binding pocket and/or surface exposure for co-regulator interactions. Three studies of the CAR LBD crystal structures, simultaneously published in 2004, revealed unique properties of its helix 12/AF-2 that explain both its ligand-dependent and –independent activities.
In regards to structural elements that influence conformation of the AF-2, an α-helical turn composed of four residues named Helix X (H-X) is present between a relatively shortened helix 10 and the AF-2 (helix 12) in human CAR [47]. However, unique in CAR is a shorter linker between H-X and the AF-2 helix that constrains the AF-2 in a more rigid conformation that is permissive to interaction with RXR. Stabilization of this conformation is further enhanced by interaction of the AF-2 C-terminal carboxyl group with helix 4 [47]. This confirmed what was originally predicted in homology models of mouse CAR [44].
The role of ligand in AF-2 stabilization has also been highlighted from the CAR LBD crystal structures. The hCAR ligand pocket itself is smaller than that of human VDR and PXR (870 Å3 and 1290-1540 Å3, respectively, versus 675 Å3 for CAR), which accounts for the relatively small size of CAR ligands compared to those found for hPXR (i.e. taxol and rifampicin) [18]. Interactions of the pocket with ligand are also different for CAR compared to other nuclear receptors. For instance, 5β-pregnanedione orientation within the hCAR binding pocket forms a single hydrogen bond at the D-ring end of the steroid (the C20 carbonyl group) [47], while 17β-estradiol binding to ERα involves hydrogen bonds at both the A and D ring ends of the steroid [48, 49]. Also, CAR has a structural barrier dividing the ligand pocket from the AF-2 [47]. However, the potent mouse CAR activator, TCPOBOP, can penetrate this barrier, based on the X-ray crystal structure of mouse CAR with bound TCPOBOP [50]. The crystal structure reveals that TCPOBOP penetrates the barrier to form multiple hydrophobic interactions with residues of the AF-2 domain, including Thr350, which was previously identified to be necessary for activation. Thus these direct interactions may provide a molecular basis for the ability of TCPOBOP to potently activate mouse CAR [50].
In contrast, how human CAR is activated by CITCO is not fully understood from the crystal structure data [47]. But in the case of androstanol, its effect on AF-2 conformation is different and might explain its activity as an inverse agonist for CAR [51]. It does not contact the AF-2 and disrupts the C-terminus of the AF-2 interacting with helix 4 that otherwise favors the active form of CAR. This presumably prevents stable interaction with a co-regulator. Consequently, it can now be proposed that for CAR, destabilization of helix 12/AF-2 is crucial to repress its constitutive activity. This can occur with certain chemical ligands, such as androstanol, and perhaps might be mitigated through protein-protein interactions with CAR-binding proteins.
The ligand-binding and activation model established from the crystal structure data confirms that which was proposed by homology modeling of CAR using human VDR and PXR crystal structure data. The modeling strategies and results for CAR [44, 52] proved to be highly accurate and hence proved useful for analyzing amino acid residues involved in ligand recognition [53, 54]. It will be interesting to see what new CAR ligands will be uncovered from homology modeling strategies, and more importantly, whether such strategies will ultimately identify CAR’s endogenous ligand(s).
2.4.2 PXR
The X-ray crystal structure of PXR reveals that its ligand binding pocket, at 1150 Å3, is approximately twice the volume of that of CAR. This may be a one structural feature explaining PXR’s ligand promiscuity, which is further enhanced by a higher degree of protein flexibility than CAR [55, 56]. Unique to PXR is a flexible loop (aa 309-321 in human PXR) replacing the α6 helix and that lies next to the ligand binding pocket to accommodate ligands of various sizes [56-58]. In addition, there is an insertion of two β-strands, in the region of aa 210 to 226 (β1, aa 210-217; β1’, aa 221-226), that results in the formation a five-strand antiparallel β-sheet. This structure, which also incorporates a four-residue turn and a pseudohelical segment, replaces the α2 helix present in both GR and ER and actually forms the bottom surface of a smooth and spherical ligand binding pocket.
The structure of the pocket itself has been resolved with different ligands that include hyperforin, rifampicin, and the cholesterol-lowering drug SR12813 [56-58]. Based on current crystal structures with these compounds, the ligand pocket can expand from approximately 1200 Å3 to more than 1600 Å3, a property not yet ascribed to CAR. Twenty-eight residues line the ligand binding pocket, of which six commonly interact with these ligands (Met243, Ser247, Gln285, Trp299, His407, Phe420) [38]. Rifampicin, owing to its larger bulk, interacts with two additional residues (Ser208, Arg410) [57]. In all, the ligand binding pocket is predominantly hydrophobic, with a salt bridge that neutralizes the charged character of certain residues. An interesting feature is an expandable pore that is formed by the aforementioned 309-321 loop [56], which opens to accommodate large ligands and thereby provides additional hydrophobic residues. This was confirmed upon crystallization of the PXR LBD with bound rifampicin, and the flexibility of the ligand binding pocket is apparent when comparing it to apo form of the LBD (Fig. 4). Interestingly, both rifampicin and the LBD acquire a disordered conformation, with the 4-methyl-1-piperazinyl ring of rifampicin and several loops in the five-strand antiparallel β-sheet showing no clear electron densities indicating mobility in these molecular structures [57]. In this same study, the species difference in responsiveness to rifampicin (activator of human PXR) versus pregnenolone 16α-carbonitrile (PCN; activator of mouse PXR) was attributed at least in part to a single residue, Phe305 in mouse and Leu308 in human PXRs. In view of the crystal structure findings, the flexibility of PXR’s ligand binding pocket explains the broad range in sizes of ligands that activate it, ranging from simpler bile acids to the more complex macrolide antibiotic compounds exemplified by rifampicin.
Figure 4. Comparison of the PXR LBD with bound rifampicin to the apo-PXR LBD.
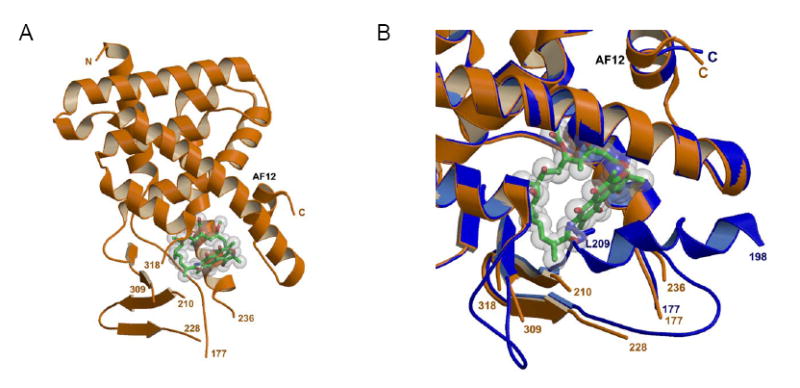
(A) Crystal structure of the Ligand binding domain of human PXR in complex with Rifampicin (green) (pdb idcode 1ILG). (B) Crystal structure of the ligand binding domain of hPXR (orange) in complex with Rifampicin (green) (pdb idcode 1ILG) superimposed with the apo form of the LBD (blue) (pdb idcode 1SKX). Two loops become disordered upon binding Rifampicin (residues 229-235 and 310-317). The position of Leucine residue 209 in the apo form is displaced by Rifampicin in the halo form resulting in residues 198-209 also becoming disordered.
PXR possesses an additional unique structural feature that impacts on its ligand activation and the acquisition of its AF-2 to a conformation that favors interaction with coregulators [59]. A novel α2 helix is also part of the β1-β1’ insert mentioned above. The α2, found to be flexible and loosely structured, is thought to function in ligand entry to and exit from the binding pocket by opening an access pathway for ligand [56, 58, 59]. Indeed, in some PXR crystal structures a channel 3 Å wide and 9 Å lies adjacent to α2. This channeling mechanism attributable to the α2 appears unique for PXR. In summary, the unique α2 seems to serve a dual function to facilitate binding of large ligands in the pocket and as a guide for the association-dissociation of ligands in and out of PXR.
In contrast to CAR, while it is still unclear the degree to which a ligand directly contributes to the proper positioning of PXR’s AF-2 to favor interactions with coregulators, a recent crystal structure of the PXR LBD in a complex with SR12813 and a single SRC-1 LXXLL domain provides an novel insight [59]. The results suggest that coregulator and ligand binding work in concert to stabilize the active LBD, rather than stabilization by direct contacts with a properly-oriented ligand. It was found that SRC-1 binding fixes SR12813 in a single, distinct orientation, while in the absence of SRC-1, SR12813 was observed to adopt three binding modes [55, 59]. Thus, rather than ligand directly contributing to AF-2 stabilization, thereby favoring AF-2 interact with coregulators as seen with CAR, coregulator interactions with the AF-2 defines ligand orientation in the PXR LBD and when eventually assumed the AF-2 is stabilized. That SR12813 and SRC-1 binding to the LBD together exerted an additive effect on overall LBD stabilization gives further credence to this idea [59]. However, amino acid substitutions introduced in full-length PXR that had been speculated to affect LBD stability did not correlate with changes in basal transcriptional activity [57]. This raises new questions of the role of AF-2 stabilization in PXR transcriptional activity, and whether coregulators are selective for stabilized PXR AF-2 domains. However, the data suggests another possibility: if coregulator works in concert with ligand to stabilize the LBD, then the ligand-induced transcriptional response would depend on which coregulator binds to PXR’s LBD/AF-2. If so, this would allow for promoter context (i.e. the combination of coregulators associated with a target gene promoter) to also be important in determining the transcriptional activity of PXR. Support for this notion comes from greater PXR activation by pregnenolone on a CYP3A4 PXR response element (PXRE) than on a PXRE from the multidrug resistance protein 1 (MDR1) gene, that correlates with recruitment of SRC-1 and not AIB-1 [60].
3. Comparison of CAR/PXR to ER/GR
3.1 Structure
Both the ER and GR are classical steroid receptors. The estrogen receptors α and β mediate the physiological effects of estrogen on reproductive development and homeostasis, while the glucocorticoid receptor (GR) serves as a master regulator for transcriptional responses to glucocorticoid hormones (i.e. cortisol) released under physiological stress [61]. Both ERs contain an amino-terminal ligand-independent activation function 1 (AF-1) domain, which may be regulated by growth factor signaling such as MAPK [62]. It has the DNA-binding domain (DBD), hinge domain, and LBD regions, and finally, the carboxy-terminal AF-2 and an F domain whose activity is regulated by estrogen binding [63]. GR possesses the same modular organization as ERα, except that its amino terminal A/B (AF-1) domain is extended, conferring constitutive (or ligand-independent) transcriptional activity. Other functional contributions of GR’s AF-1 have been described elsewhere [64]. The F domain found in ERα is not described to be present in GR.
This classic domain organization of ER and GR serves as a useful starting point to highlight what distinguishes them from CAR and PXR. First, the A/B and F domains present in ERα are absent in both CAR and PXR; the elongated A/B (AF-1) domain of GR is also lacking in CAR and PXR. Hence, CAR and PXR lack AF-1 function. The amino-terminal domains (A/B domain) among members of the nuclear receptor family are in fact poorly conserved [18] and so the functions of their amino termini, if any, are not precisely known.
The second notable structural difference between ER/GR and CAR/PXR is the size of the ligand binding pocket. Based on crystal structures for ERα and ERβ [49, 65, 66], their ligand pocket volumes are 450 Å3 and 390 Å3, respectively, approximately 40% smaller than that of CAR (at 675 Å3) and roughly one-third that of PXR (~1200 Å3). In the case of GR, the ligand binding pocket volume is larger than the ERs, at 578-599 Å3[67], approximating that for CAR, but is about half the volume of PXR’s LBD. The large volume of PXR’s ligand binding pocket is particularly interesting. Coupled with the inherent flexibility of its structure, PXR has the capability to bind a diverse array of compounds that can vary widely both in size and chemical structure. In contrast, ER and GR have more limited ligand selectivity for steroidal compounds resembling estradiol and corticsol. This is important to prevent ER and GR from being activated by structurally-divergent endogenous and exogenous compounds, and their high selectivity requires that drugs targeting ER and GR possess structural elements resembling to estradiol and cortisol (e.g. tamoxifin and dexamethasone)
The third important structural difference between the receptors is the conformational orientations of the AF-2 domain upon ligand binding. For the ERs, upon estradiol binding the AF-2 positions itself into the hydrophobic pocket to “cap” the access channel. Thus the AF-2 of ER serves a dual function: to encase estradiol into the binding pocket and to interact with coactivators, such as GRIP1, via their LXXLL motifs [66]. The GR ligand pocket also envelopes the ligand, however unique to GR there remains a hollow side pocket formed by rearrangement of helix 6 and 7 [67]. Like with ERα, the GR LBD structure also shows that the AF-2/helix 12 closes onto the binding pocket when ligand is present. At the same time, the AF-2 assumes a conformation favoring interactions with coregulators, a notion supported by, for instance, the identification an additional charge clamp unique to GR that confers selectivity for the coactivator GRIP1 [67]. For CAR and PXR, the AF-2 functions to interact with coactivators but not to cap the access channel.
A surprising observation from the GR crystal structure identifies a unique similarity between GR and PXR. The GR LBD dimerizes forming an intermolecular β-sheet; the interface of the dimer was subsequently found to be important for GR transctiptional activity [67]. Similar structure has very recently been observed for the PXR LBD, found to homodimerize in an antiparallel orientation to form a novel tryptophan-zipper at the dimer interface [68]. The functional significance of receptor dimerization remains to be determined, as it is unknown whether dimerization occurs in the resting state, upon translocation to the nucleus, or upon DNA binding. Nevertheless, work is likely ongoing that might shed further light on this issue and other questions undoubtedly raised by the observation of GR and PXR dimerization.
In summary, the domain architecture of CAR and PXR differs significantly from ER and GR in that CAR and PXR lack the A/B domain hence AF-1 activities are absent. For both CAR and PXR, their ligand binding pockets are larger that those of ER and GR which accounts for the greater structural diversity of CAR and PXR ligands. And lastly, the tight encapsulation of ligand, particularly the capping of the ligand access channel, found for both ER and GR, is not observed in current CAR and PXR crystal structures.
3.2 Intracellular trafficking
Functionally speaking, there are commonalities between ER/GR and CAR/PXR (Table 1). ERα and GR activity, like CAR and PXR, is regulated in part by interactions with components of the cellular chaperone machinery, such as HSP90. However it should be noted that there is some controversy over the role of HSP chaperones in regulating subcellular distribution of the ERs. They do not appear to be involved in cytoplasmic retention of ER per se, but have been found to interact with ER to regulate in part ligand-dependent and -independent ER transcriptional activity [69-75].
TABLE I.
a) Modifications associated with ligand-induced receptor redistribution
| Receptor | Ligand | Modification | Site | Enzyme | Redistribution | Proposed mechanism | Ref. |
|---|---|---|---|---|---|---|---|
| ERα | Estradiol | phosphorylation | Ser118 | cdk7 | cytosol →nuclear | modulates AF1 | [102] |
| ICI-182,780 | phosphorylation | Ser118 | ??? | nuclear→ extranuclear/ insoluble fraction | AF2-independent; release from HSP90 | [83] | |
| ??? | phosphorylation | Thr311 | p38/MAPK | cytosol →nuclear | favor interactions with coactivator | [84] | |
| estradiol & ligand-indep. | phosphorylation | Ser118 | ??? | membrane →nuclear | ??? | [103] | |
| GR | DEX | dhosphorylation | Ser226 | JNK | nuclear → cytosol | ??? | [104] |
| dephosphorylation | ??? | PP1, PP2A | nuclear → cytosol | ??? | [105] | ||
| CAR | TCPOBOP, PB | dephosphorylation | S202 | PP2A | cytosol → nuclear | release from CCRP-Hsp90 complex | [34] |
| PXR | --- | forskolin-induced phosphorylation | ??? | PKA | unknown | potentiation of transcription | [39] |
| --- | phosphorylation | ??? | PKC | unknown | transcriptional repression | [40] | |
| dephosphorylation | ??? | PP1/PP2A | unknown | transcriptional repression | [40] | ||
| b) Modifications regulating receptor levels | |||||||
|
| |||||||
| Receptor | Modification | Protein(s) involved | Consequence | Mechanism | Ref | ||
|
| |||||||
| ERα | ubiquitination | CHIP, E4-A6 | receptor degradation | ligand-induced | [106-109] | ||
| GR | ubiquitination | CHIP | receptor degradation | ligand induced | [110, 111] | ||
| c) Proteins associated with subcellular localization | |||||||
|
| |||||||
| Receptor | Protein | Role | Ref | ||||
|
| |||||||
| ERα | GPR30 | estrogen signaling;: recruitment of ERα to membrane? | [78-80] | ||||
| HSP90 | regulation of transcription | [75] | |||||
| other (cytoplasmic and/or nuclear retention?) | [112] | ||||||
| GR | HSP90 | cytoplasmic retention; nuclear retention | [113] | ||||
| Immunophilins (FKBP51, FKBP52) | cytoplasmic retention; nuclear transport | [114] | |||||
| Gβ | nuclear translocation | [115] | |||||
| importin 7 | nuclear import | [116] | |||||
| importin-α/importin-β | nuclear import | [116] | |||||
| calreticulin | nuclear export | [117, 118] | |||||
| CCRP/Tpr2 | cytoplasmic retention | [81] | |||||
| 14-3-3σ | cytoplasmic retention | [119] | |||||
| β2 adrenergic receptors | nuclear translocation | [120] | |||||
| Ran/TC4 GTPase | nuclear translocation | [121] | |||||
| CAR | CCRP/Tpr2 | cytoplasmic retention | [32] | ||||
| ??? | cell membrane recruitment | [21] | |||||
| PXR | CCRP/Tpr2 | cytoplasmic retention | [37] | ||||
In the classic model for GR (Fig. 2), unliganded receptor resides in the cytoplasm in a complex with Hsp90, and ligand binding causes GR to acquire a nuclear-localized form [61, 76]. This model applies to CAR and PXR. Although the ERs are generally considered nuclearlocalized receptors (Fig. 2), signaling systems at the cell membrane have been observed to explain non-genomic actions of estradiol that suggests the controversial notion of ERα being recruited to membranes [77]. GPR30, a G-protein coupled receptor, was identified to mediate rapid estradiol-dependent signaling at the plasma membrane [78] and endoplasmic reticulum [79]. The controversy of membrane recruitment by GPR30 stems from identification of membrane-derived E2 binding proteins as ERα and that rapid responses to E2 were absent in GPR30-positive, ERα-negative cells [80]. Whether GPR30 requires interaction with ERα to respond to E2, or that ERα functions downstream of GPR30 to mediate signaling, remains to be determined. Nevertheless, while a specific membrane protein that binds CAR remains to be identified, we have observed PB-dependent localization of CAR at the cell membrane [21].
Interestingly, GR appears to interact with CCRP (also known as Tpr2), the same cochaperone that retains CAR and PXR in the cytosol. CCRP contains seven tetratricopeptide (Tpr) motifs that function in chaperone substrate recognition. It also possesses a DnaJ domain that stimulates ATPase hydrolysis and protein binding by Hsp70 [81]. This activity of CCRP, coupled with its ability to induce ATP-independent dissociation of Hsp90 from chaperonesubstrate complexes, made it an attractive candidate to regulate GR activation. Indeed, DEXinduced GR transactivation was attenuated by 60% by CCRP co-expression; partial depletion of CCRP also attenuated GR transactivation [81]. While an examination of whether these effects correlated with GR redistribution was not performed in this study, CCRP was shown to inhibit Hsp90-dependent GR refolding to almost the same extent as geldanamycin, suggesting that CCRP opposes Hsp70 to Hsp90 protein transfer. Hence, CCRP may serve to promote GR retention in the cytosol by preventing release of GR from the Hsp70-Hsp90 complex. This mechanism could also explain CCRP’s cytoplasmic retention of both CAR and PXR.
3.3 Protein Phosphorylation
Another common feature between CAR/PXR and ER/GR is the role of phosphorylation for receptor activity. For ER, phosphorylation appears to be important for regulating receptor nucleocytoplasmic shuttling and nuclear functioning [82, 83]. For instance, p38/MAPK has been identified to phosphorylate ER, leading to ER extranuclear localization [84]. More has been documented for the regulation of GR activity by phosphorylation. Seven phosphorylation sites have been identified for mouse GR [85], five of which are conserved among rodent and human GRs [64]. All are located in the N-terminal A/B domain in or near the AF-1 region of GR. The effects of phosphorylation on GR activity is ascribed to alterations in coactivator and/or corepressor interactions that vary the transcriptional actions of GR (Table I). However, phosphorylation at only one site is not effective at modulating GR transcription. Rather, as shown with mouse GR on an MMTV-driven reporter construct, multiple phosphorylations on GR (hyperphosphorylation) are necessary to affect GR transcriptional activation [86]. A potential role for protein phosphatases in nucleocytoplasmic shuttling of GR has also been implicated based on the inhibition of this translocation activity by OA [87], suggesting dephosphorylation contributes to GR activation.
As described above for CAR, phosphorylation at Ser202 plays a more important role in cytosolic retention, and dephosphorylation of this residue results in its nuclear translocation. Protein phosphatase 2A (PP2A) is believed to remove the phosphate group from CAR; however, which kinase(s) phosphorylate CAR remains unknown. Studies characterizing CAR membrane localization and of signaling pathways that impact on CAR activity are currently ongoing. For PXR, evidence has bee found for its regulation by PKA- and PKC-mediated phosphorylation [39, 40]. Other modifications of ER and GR, and those of PXR and CAR, are summarized in Table I.
4. Indirect role of endocrine factors in CAR/PXR signaling
4.1 Steroid hormones
Glucocorticoids consistently show stimulatory effects on CAR activity; however, this effect is mutual as CAR potentiates GR signaling. Thus, GR and CAR can synergize to induce target genes, as documented with CYP isoforms [88]. For particular CYPs, such as CYP3A, PXR can also contribute to GR-dependent regulation of these genes [89]. This forms a somewhat complex regulatory loop, where the interrelationships between CAR, PXR and GR can be sequential, synergistic, or occurring in tandem [88]. The influence of glucocorticoids on the activity of CAR and PXR necessitates consideration of physiological stress as factor contributing to the physiological response to xenobiotics. Owing to the particular role of PXR in regulating CYP3A4, it will be important to more precisely define the physiological relationships among GR, CAR, and PXR.
Sex hormones might regulate CAR activity in addition to glucocorticoids, which might explain the sexual dimorphism in CYP2B1 induction by PB [90, 91]. A recent study in our laboratory revealed higher inducibility of hepatic CYP2B1 mRNA in male versus female Wistar-Kyoto rats coincided with elevated CAR protein levels in males [92]. In addition, CAR and gonadal hormone signaling components (i.e. ER) can interact by other mechanisms. CAR has been shown to repress ER transcriptional activation [93], proposed to occur by squelching p160 coactivators to limit ER-mediated transctivation. It remains to be determined whether an interaction between gonadal hormones and CAR is physiologically relevant, but it might be important for regulating enzyme systems involved in xenobiotic metabolism.
Lastly, while circulating TH levels are regulated by CAR, TH does not appear to regulate CAR activity, as it has been shown that TH does not alter PB-induced transcriptional activation using a PBRU-containing reporter construct [94]. Hence, cross-talk between TH and CAR signaling pathways is not mutual, in contrast to cross-talk observed between ERα and CAR.
4.2 Growth factors
Evidence is accumulating of the role of growth factors in regulating CAR signaling. Studies investigating the repressive effect of epidermal growth factor (EGF) on PB induction of CYP2B1 expression [95] using rat primary hepatocyte culturesrevealed that a putative PBresponsive CYP2B1 promoter enhancer region (at -2230 to -2170 ) mediating EGF repression of PB-induced reporter activity contained a section equivalent to the 51-bp PBREM of the mouse Cyp2b10 gene [96]. In this same study, growth hormone (GH) also gave similar repression of PB-induced transcriptional activation using the same CYP2B1 promoter reporter construct; the repressive actions of both EGF and GH was also shown on NR1 protein binding by gel shift analysis. Similarly, interleukin-1β (IL-1β) represses PB induction of CYPs 3A4, 2B6, and 2C9 in human hepatocytes, but by a mechanism where IL-1β suppresses hepatocyte CAR expression [97]. Reductions in CAR expression also appears to be the cause of the repressive actions of a novel hepatotrophic growth factor, augmenter of liver regeneration (ALR), on PB-induced CYP2B6 induction in human hepatocytes [98]. However in contrast, this study showed that ALR has no effects on PXR expression; but nevertheless, ALR repressed RIF-induced CYP3A4 expression. Another report investigating growth factor regulation of PXR showed interleukin-6 (IL-6) negatively regulating PXR expression, and also the expression of CAR, in human hepatocytes [99]. These expression changes correlated with reduced RIF- and PB-mediated induction of CYPs 2B6, 2C8, 2C9, and 3A4. These findings collectively implicate growth factors as negative regulators of both CAR and PXR.
CAR, however, can impact on growth factor signaling as well. Gadd45β, an early response gene during liver proliferation induced by TCPOBOP, can be more greatly induced in the absence of upstream tumor necrosis factor (TNFα) signaling [100]. This induction, however, was absent in Car-null mice. These observations implicate CAR regulation of TNFα-induced signaling; Since TNF activates nuclear factor (NF)-κB, the enhanced proliferative response in TNFα receptor-null mice suggested an inhibitory effect of CAR on NF- κB. It will be interesting to see what molecular mechanism underlies this novel inhibitory cross-talk between CAR and TNFα-NF-κB signaling.
5. Concluding Remarks and Future Directions
CAR and PXR are xenochemical sensing nuclear receptors each with structural features that define their functional characteristics that make them distinct from the classical steroid receptors ER and GR. CAR is a constitutively active receptor due in part to its rigid LBD structure that maintains its helix-12/AF-2 in a conformation favoring interactions with its heterodimerization partner RXR. Interaction of the ligand with the AF-2 of CAR appears to be important, and blockage of contact between a ligand and AF-2 by a helical barrier presumably reduces the ability of some but not all ligands to activate CAR. However, this model is based solely on interaction of TCPOBOP with the AF-2 of mCAR, and discovery of novel CAR ligands that can contact the AF-2 is needed to assess this model further.
PXR is a receptor capable of binding ligands of various sizes, owing to the flexibility of its LBD. Its ligand pocket is fairly large and can expand upon binding of large and bulky molecules. The active conformation of PXR’s AF-2 is determined partly by ligand and partly by coactivator binding. However, acquisition of the ligand into the proper conformation for full receptor activation is finally determined by coactivator interaction with the LBD. While this is based solely on PXR crystal structures with bound SR12183, this raises the question of whether promoter context, and particularly which coactivator is present at the target gene promoter, determines the level of transcriptional response induced by a PXR ligand. Further structural characterization of interactions between PXR and other ligands will undoubtedly give additional insights into cooperativity of coactivator binding and ligand binding to positioning PXR’s LBD into its active conformation.
Current efforts to understand the role(s) and functioning of CAR and PXR are revealing novel mechanisms of regulation of these important nuclear receptors. Components of their signaling cascade include cell membrane signaling molecules, cell chaperones, protein kinases and/or protein phosphatases, and accessory factors including transcriptional coregulators. It will be interesting whether novel modes of protein modification in addition to phosphorylation are involved in the regulation of CAR and PXR. One such modification is ubiquitination, a pathway identified to regulate activities of the steroid receptors such as GR and ER (Table I). If CAR and PXR are found to be targets of ubiquitin-dependent regulation, this would add another layer of regulation for these receptors. Preliminary observations of ubiquitin-proteasomal regulation of PXR have already been noted [101]. Indeed, this is an active area of research that will ultimately aid in our understanding of the physiological roles of CAR and PXR and for the design of therapeutic drugs that modulate these receptors to treat metabolic disorders.
Acknowledgments
We would like to thank Lars C. Pedersen (Structure and Function Group, Laboratory of Structural Biology, NIEHS) for his assistance in generating the PXR crystal structure figure. We also would like to thank Drs. Bonnie J. Deroo (Laboratory of Reproductive and Developmental Toxicology, NIEHS) and Andrew Wallace (Department of Environmental and Molecular Toxicology, North Carolina State University) for their comments on the manuscript.
Footnotes
This research was supported [in part] by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kodama S, Negishi M. Phenobarbital confers its diverse effects by activating the orphan nuclear receptor CAR. Drug Metab Rev. 2006;38:75–87. doi: 10.1080/03602530600569851. [DOI] [PubMed] [Google Scholar]
- 2.Sueyoshi T, Negishi M. Phenobarbital response elements of cytochrome P450 genes and nuclear receptors. Annu Rev Pharmacol Toxicol. 2001;41:123–43. doi: 10.1146/annurev.pharmtox.41.1.123. [DOI] [PubMed] [Google Scholar]
- 3.Swales K, Kakizaki S, Yamamoto Y, Inoue K, Kobayashi K, Negishi M. Novel CAR-mediated mechanism for synergistic activation of two distinct elements within the human cytochrome P450 2B6 gene in HepG2 cells. J Biol Chem. 2005;280:3458–66. doi: 10.1074/jbc.M411318200. [DOI] [PubMed] [Google Scholar]
- 4.Qatanani M, Zhang J, Moore DD. Role of the constitutive androstane receptor in xenobiotic-induced thyroid hormone metabolism. Endocrinology. 2005;146:995–1002. doi: 10.1210/en.2004-1350. [DOI] [PubMed] [Google Scholar]
- 5.Maglich JM, Watson J, McMillen PJ, Goodwin B, Willson TM, Moore JT. The nuclear receptor CAR is a regulator of thyroid hormone metabolism during caloric restriction. J Biol Chem. 2004;279:19832–8. doi: 10.1074/jbc.M313601200. [DOI] [PubMed] [Google Scholar]
- 6.Quattrochi LC, Guzelian PS. CYP3A regulation: from pharmacology to nuclear receptors. Drug Metab Dispos. 2001;29:615–22. [PubMed] [Google Scholar]
- 7.Miao J, Fang S, Bae Y, Kemper JK. Functional inhibitory cross-talk between constitutive androstane receptor and hepatic nuclear factor-4 in hepatic lipid/glucose metabolism is mediated by competition for binding to the DR1 motif and to the common coactivators, GRIP-1 and PGC-1α. J Biol Chem. 2006;281:14537–46. doi: 10.1074/jbc.M510713200. [DOI] [PubMed] [Google Scholar]
- 8.Kodama S, Koike C, Negishi M, Yamamoto Y. Nuclear receptors CAR and PXR cross talk with FOXO1 to regulate genes that encode drug-metabolizing and gluconeogenic enzymes. Mol Cell Biol. 2004;24:7931–40. doi: 10.1128/MCB.24.18.7931-7940.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lahtela JT, Arranto AJ, Sotaniemi EA. Enzyme inducers improve insulin sensitivity in non-insulin-dependent diabetic subjects. Diabetes. 1985;34:911–6. doi: 10.2337/diab.34.9.911. [DOI] [PubMed] [Google Scholar]
- 10.Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A, Liu Y, Klaassen CD, Brown KK, Reinhard J, Willson TM, Koller BH, Kliewer SA. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci U S A. 2001;98:3369–74. doi: 10.1073/pnas.051551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie W, Radominska-Pandya A, Shi Y, Simon CM, Nelson MC, Ong ES, Waxman DJ, Evans RM. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc Natl Acad Sci U S A. 2001;98:3375–80. doi: 10.1073/pnas.051014398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goodwin B, Gauthier KC, Umetani M, Watson MA, Lochansky MI, Collins JL, Leitersdorf E, Mangelsdorf DJ, Kliewer SA, Repa JJ. Identification of bile acid precursors as endogenous ligands for the nuclear xenobiotic pregnane X receptor. Proc Natl Acad Sci U S A. 2003;100:223–8. doi: 10.1073/pnas.0237082100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sonoda J, Chong LW, Downes M, Barish GD, Coulter S, Liddle C, Lee CH, Evans RM. Pregnane X receptor prevents hepatorenal toxicity from cholesterol metabolites. Proc Natl Acad Sci U S A. 2005;102:2198–203. doi: 10.1073/pnas.0409481102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goodwin B, Kliewer SA. Nuclear receptors. I. Nuclear receptors and bile acid homeostasis. Am J Physiol Gastrointest Liver Physiol. 2002;282:G926–31. doi: 10.1152/ajpgi.00044.2002. [DOI] [PubMed] [Google Scholar]
- 15.Zhang J, Huang W, Qatanani M, Evans RM, Moore DD. The constitutive androstane receptor and pregnane X receptor function coordinately to prevent bile acid-induced hepatotoxicity. J Biol Chem. 2004;279:49517–22. doi: 10.1074/jbc.M409041200. [DOI] [PubMed] [Google Scholar]
- 16.Maglich JM, Parks DJ, Moore LB, Collins JL, Goodwin B, Billin AN, Stoltz CA, Kliewer SA, Lambert MH, Willson TM, Moore JT. Identification of a novel human constitutive androstane receptor (CAR) agonist and its use in the identification of CAR target genes. J Biol Chem. 2003;278:17277–83. doi: 10.1074/jbc.M300138200. [DOI] [PubMed] [Google Scholar]
- 17.Tzameli I, Pissios P, Schuetz EG, Moore DD. The xenobiotic compound 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene is an agonist ligand for the nuclear receptor CAR. Mol Cell Biol. 2000;20:2951–8. doi: 10.1128/mcb.20.9.2951-2958.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Willson TM, Kliewer SA. PXR, CAR and drug metabolism. Nat Rev Drug Discov. 2002;1:259–66. doi: 10.1038/nrd753. [DOI] [PubMed] [Google Scholar]
- 19.Goodwin B, Redinbo MR, Kliewer SA. Regulation of CYP3A gene transcription by the pregnane X receptor. Annu Rev Pharmacol Toxicol. 2002;42:1–23. doi: 10.1146/annurev.pharmtox.42.111901.111051. [DOI] [PubMed] [Google Scholar]
- 20.Kliewer SA, Goodwin B, Willson TM. The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism. Endocr Rev. 2002;23:687–702. doi: 10.1210/er.2001-0038. [DOI] [PubMed] [Google Scholar]
- 21.Koike C, Moore R, Negishi M. Localization of the nuclear receptor CAR at the cell membrane of mouse liver. FEBS Lett. 2005;579:6733–6. doi: 10.1016/j.febslet.2005.10.070. [DOI] [PubMed] [Google Scholar]
- 22.Choi E, Lee S, Yeom SY, Kim GH, Lee JW, Kim SW. Characterization of activating signal cointegrator-2 as a novel transcriptional coactivator of the xenobiotic nuclear receptor constitutive androstane receptor. Mol Endocrinol. 2005;519:1711–9. doi: 10.1210/me.2005-0066. [DOI] [PubMed] [Google Scholar]
- 23.Kim HJ, Lee SK, Na SY, Choi HS, Lee JW. Molecular cloning of xSRC-3, a novel transcription coactivator from Xenopus, that is related to AIB1, p/CIP, and TIF2. Mol Endocrinol. 1998;512:1038–47. doi: 10.1210/mend.12.7.0139. [DOI] [PubMed] [Google Scholar]
- 24.Min G, Kemper JK, Kemper B. Glucocorticoid receptor-interacting protein 1 mediates ligand-independent nuclear translocation and activation of constitutive androstane receptor in vivo. J Biol Chem. 2002;277:26356–63. doi: 10.1074/jbc.M200051200. [DOI] [PubMed] [Google Scholar]
- 25.Muangmoonchai R, Smirlis D, Wong SC, Edwards M, Phillips IR, Shephard EA. Xenobiotic induction of cytochrome P450 2B1 (CYP2B1) is mediated by the orphan nuclear receptor constitutive androstane receptor (CAR) and requires steroid co-activator 1 (SRC-1) and the transcription factor Sp1. Biochem J. 2001;355:71–8. doi: 10.1042/0264-6021:3550071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shiraki T, Sakai N, Kanaya E, Jingami H. Activation of orphan nuclear constitutive androstane receptor requires subnuclear targeting by peroxisome proliferator-activated receptor γ coactivator-1α. A possible link between xenobiotic response and nutritional state. J Biol Chem. 2003;278:11344–50. doi: 10.1074/jbc.M212859200. [DOI] [PubMed] [Google Scholar]
- 27.Inoue K, Borchers CH, Negishi M. Cohesin protein SMC1 represses the nuclear receptor CAR-mediated synergistic activation of a human P450 gene by xenobiotics. Biochem J. 2006;398:125–33. doi: 10.1042/BJ20060109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawamoto T, Sueyoshi T, Zelko I, Moore R, Washburn K, Negishi M. Phenobarbitalresponsive nuclear translocation of the receptor CAR in induction of the CYP2B gene. Mol Cell Biol. 1999;19:6318–22. doi: 10.1128/mcb.19.9.6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sidhu JS, Omiecinski CJ. An okadaic acid-sensitive pathway involved in the phenobarbital-mediated induction of CYP2B gene expression in primary rat hepatocyte cultures. J Pharmacol Exp Ther. 1997;282:1122–9. [PubMed] [Google Scholar]
- 30.Honkakoski P, Negishi M. Protein serine/threonine phosphatase inhibitors suppress phenobarbital-induced Cyp2b10 gene transcription in mouse primary hepatocytes. Biochem J. 1998;330:889–95. doi: 10.1042/bj3300889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zelko I, Sueyoshi T, Kawamoto T, Moore R, Negishi M. The peptide near the C terminus regulates receptor CAR nuclear translocation induced by xenochemicals in mouse liver. Mol Cell Biol. 2001;21:2838–46. doi: 10.1128/MCB.21.8.2838-2846.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kobayashi K, Sueyoshi T, Inoue K, Moore R, Negishi M. Cytoplasmic accumulation of the nuclear receptor CAR by a tetratricopeptide repeat protein in HepG2 cells. Mol Pharmacol. 2003;64:1069–75. doi: 10.1124/mol.64.5.1069. [DOI] [PubMed] [Google Scholar]
- 33.Yoshinari K, Kobayashi K, Moore R, Kawamoto T, Negishi M. Identification of the nuclear receptor CAR:HSP90 complex in mouse liver and recruitment of protein phosphatase 2A in response to phenobarbital. FEBS Lett. 2003;548:17–20. doi: 10.1016/s0014-5793(03)00720-8. [DOI] [PubMed] [Google Scholar]
- 34.Hosseinpour F, Moore R, Negishi M, Sueyoshi T. Serine 202 regulates the nuclear translocation of constitutive active/androstane receptor. Mol Pharmacol. 2006;69:1095–102. doi: 10.1124/mol.105.019505. [DOI] [PubMed] [Google Scholar]
- 35.Xia J, Kemper B. Structural determinants of constitutive androstane receptor required for its glucocorticoid receptor interacting protein-1-mediated nuclear accumulation. J Biol Chem. 2005;280:7285–93. doi: 10.1074/jbc.M409696200. [DOI] [PubMed] [Google Scholar]
- 36.Jia Y, Guo GL, Surapureddi S, Sarkar J, Qi C, Guo D, Xia J, Kashireddi P, Yu S, Cho YW, Rao MS, Kemper B, Ge K, Gonzalez FJ, Reddy JK. Transcription coactivator peroxisome proliferator-activated receptor-binding protein/mediator 1 deficiency abrogates acetaminophen hepatotoxicity. Proc Natl Acad Sci U S A. 2005;102:12531–6. doi: 10.1073/pnas.0506000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Squires EJ, Sueyoshi T, Negishi M. Cytoplasmic localization of pregnane X receptor and ligand-dependent nuclear translocation in mouse liver. J Biol Chem. 2004;279:49307–14. doi: 10.1074/jbc.M407281200. [DOI] [PubMed] [Google Scholar]
- 38.Orans J, Teotico DG, Redinbo MR. The nuclear xenobiotic receptor pregnane X receptor: recent insights and new challenges. Mol Endocrinol. 2005;19:2891–900. doi: 10.1210/me.2005-0156. [DOI] [PubMed] [Google Scholar]
- 39.Ding X, Staudinger JL. Induction of drug metabolism by forskolin: the role of the pregnane X receptor and the protein kinase A signal transduction pathway . J Pharmacol Exp Ther. 2005;312:849–56. doi: 10.1124/jpet.104.076331. [DOI] [PubMed] [Google Scholar]
- 40.Ding X, Staudinger JL. Repression of PXR-meCYP3A gene expression by protein kinase C. Biochem Pharmacol. 2005;69:867–73. doi: 10.1016/j.bcp.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 41.Kawamoto T, Kakizaki S, Yoshinari K, Negishi M. Estrogen activation of the nuclear orphan receptor CAR (constitutive active receptor) in induction of the mouse Cyp2b10 gene. Mol Endocrinol. 2000;14:1897–905. doi: 10.1210/mend.14.11.0547. [DOI] [PubMed] [Google Scholar]
- 42.Ueda A, Kakizaki S, Negishi M, Sueyoshi T. Residue threonine 350 confers steroid hormone responsiveness to the mouse nuclear orphan receptor CAR. Mol Pharmacol. 2002;61:1284–8. doi: 10.1124/mol.61.6.1284. [DOI] [PubMed] [Google Scholar]
- 43.Ueda A, Matsui K, Yamamoto Y, Pedersen LC, Sueyoshi T, Negishi M. Thr176 regulates the activity of the mouse nuclear receptor CAR and is conserved in the NR1I subfamily members PXR and VDR. Biochem J. 2005;388:623–30. doi: 10.1042/BJ20041572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dussault I, Lin M, Hollister K, Fan M, Termini J, Sherman MA, Forman BM. A structural model of the constitutive androstane receptor defines novel interactions that mediate ligand-independent activity. Mol Cell Biol. 2002;22:5270–80. doi: 10.1128/MCB.22.15.5270-5280.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frank C, Molnar F, Matilainen M, Lempiainen H, Carlberg C. Agonist-dependent and agonist-independent transactivations of the human constitutive androstane receptor are modulated by specific amino acid pairs. J Biol Chem. 2004;279:33558–66. doi: 10.1074/jbc.M403946200. [DOI] [PubMed] [Google Scholar]
- 46.Jyrkkarinne J, Windshugel B, Makinen J, Ylisirnio M, Perakyla M, Poso A, Sippl W, Honkakoski P. Amino acids important for ligand specificity of the human constitutive androstane receptor. J Biol Chem. 2005;280:5960–71. doi: 10.1074/jbc.M411241200. [DOI] [PubMed] [Google Scholar]
- 47.Xu RX, Lambert MH, Wisely BB, Warren EN, Weinert EE, Waitt GM, Williams JD, Collins JL, Moore LB, Willson TM, Moore JT. A structural basis for constitutive activity in the human CAR/RXRalpha heterodimer. Mol Cell. 2004;16:919–28. doi: 10.1016/j.molcel.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 48.Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–8. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 49.Tanenbaum DM, Wang Y, Williams SP, Sigler PB. Crystallographic comparison of the estrogen and progesterone receptor’s ligand binding domains. Proc Natl Acad Sci U S A. 1998;95:5998–6003. doi: 10.1073/pnas.95.11.5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suino K, Peng L, Reynolds R, Li Y, Cha JY, Repa JJ, Kliewer SA, Xu HE. The nuclear xenobiotic receptor CAR: structural determinants of constitutive activation and heterodimerization. Mol Cell. 2004;16:893–905. doi: 10.1016/j.molcel.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 51.Shan L, Vincent J, Brunzelle JS, Dussault I, Lin M, Ianculescu I, Sherman MA, Forman BM, Fernandez EJ. Structure of the murine constitutive androstane receptor complexed to androstenol: a molecular basis for inverse agonism. Mol Cell. 2004;16:907–17. doi: 10.1016/j.molcel.2004.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xiao L, Cui X, Madison V, White RE, Cheng KC. Insights from a three-dimensional model into ligand binding to constitutive active receptor. Drug Metab Dispos. 2002;30:951–6. doi: 10.1124/dmd.30.9.951. [DOI] [PubMed] [Google Scholar]
- 53.Poso A, Honkakoski P. Ligand recognition by drug-activated nuclear receptors PXR and CAR: structural, site-directed mutagenesis and molecular modeling studies. Mini Rev Med Chem. 2006;6:937–47. doi: 10.2174/138955706777935008. [DOI] [PubMed] [Google Scholar]
- 54.Windshugel B, Jyrkkarinne J, Vanamo J, Poso A, Honkakoski P, Sippl W. Comparison of homology models and X-ray structures of the nuclear receptor CAR: Assessing the structural basis of constitutive activity. J Mol Graph Model. 2006 doi: 10.1016/j.jmgm.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 55.Watkins RE, Noble SM, Redinbo MR. Structural insights into the promiscuity and function of the human pregnane X receptor. Curr Opin Drug Discov Devel. 2002;5:150–8. [PubMed] [Google Scholar]
- 56.Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP, Willson TM, Kliewer SA, Redinbo MR. The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science. 2001;292:2329–33. doi: 10.1126/science.1060762. [DOI] [PubMed] [Google Scholar]
- 57.Chrencik JE, Orans J, Moore LB, Xue Y, Peng L, Collins JL, Wisely GB, Lambert MH, Kliewer SA, Redinbo MR. Structural disorder in the complex of human pregnane X receptor and the macrolide antibiotic rifampicin. Mol Endocrinol. 2005;19:1125–34. doi: 10.1210/me.2004-0346. [DOI] [PubMed] [Google Scholar]
- 58.Watkins RE, Maglich JM, Moore LB, Wisely GB, Noble SM, Davis-Searles PR, Lambert MH, Kliewer SA, Redinbo MR. 2.1 Å crystal structure of human PXR in complex with the St. John’s wort compound hyperforin. Biochemistry. 2003;42:1430–8. doi: 10.1021/bi0268753. [DOI] [PubMed] [Google Scholar]
- 59.Watkins RE, Davis-Searles PR, Lambert MH, Redinbo MR. Coactivator binding promotes the specific interaction between ligand and the pregnane X receptor. J Mol Biol. 2003;331:815–28. doi: 10.1016/s0022-2836(03)00795-2. [DOI] [PubMed] [Google Scholar]
- 60.Masuyama H, Suwaki N, Tateishi Y, Nakatsukasa H, Segawa T, Hiramatsu Y. The pregnane X receptor regulates gene expression in a ligand- and promoter-selective fashion. Mol Endocrinol. 2005;19:1170–80. doi: 10.1210/me.2004-0434. [DOI] [PubMed] [Google Scholar]
- 61.Zhou J, Cidlowski JA. The human glucocorticoid receptor: one gene, multiple proteins and diverse responses. Steroids. 2005;70:407–17. doi: 10.1016/j.steroids.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 62.Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, Metzger D, Chambon P. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270:1491–4. doi: 10.1126/science.270.5241.1491. [DOI] [PubMed] [Google Scholar]
- 63.Kumar V, Green S, Stack G, Berry M, Jin JR, Chambon P. Functional domains of the human estrogen receptor. Cell. 1987;51:941–51. doi: 10.1016/0092-8674(87)90581-2. [DOI] [PubMed] [Google Scholar]
- 64.Kumar R, Thompson EB. Gene regulation by the glucocorticoid receptor: structure:function relationship. J Steroid Biochem Mol Biol. 2005;94:383–94. doi: 10.1016/j.jsbmb.2004.12.046. [DOI] [PubMed] [Google Scholar]
- 65.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–37. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 66.Shiau AK, Barstad D, Radek JT, Meyers MJ, Nettles KW, Katzenellenbogen BS, Katzenellenbogen JA, Agard DA, Greene GL. Structural characterization of a subtype-selective ligand reveals a novel mode of estrogen receptor antagonism. Nat Struct Biol. 2002;9:359–64. doi: 10.1038/nsb787. [DOI] [PubMed] [Google Scholar]
- 67.Bledsoe RK, Stewart EL, Pearce KH. Structure and function of the glucocorticoid receptor ligand binding domain. Vitam Horm. 2004;68:49–91. doi: 10.1016/S0083-6729(04)68002-2. [DOI] [PubMed] [Google Scholar]
- 68.Noble SM, Carnahan VE, Moore LB, Luntz T, Wang H, Ittoop OR, Stimmel JB, Davis-Searles PR, Watkins RE, Wisely GB, LeCluyse E, Tripathy A, McDonnell DP, Redinbo MR. Human PXR forms a tryptophan zipper-mediated homodimer. Biochemistry. 2006;45:8579–89. doi: 10.1021/bi0602821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oxelmark E, Roth JM, Brooks PC, Braunstein SE, Schneider RJ, Garabedian MJ. The cochaperone p23 differentially regulates estrogen receptor target genes and promotes tumor cell adhesion and invasion. Mol Cell Biol. 2006;26:5205–13. doi: 10.1128/MCB.00009-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ogawa S, Oishi H, Mezaki Y, Kouzu-Fujita M, Matsuyama R, Nakagomi M, Mori E, Murayama E, Nagasawa H, Kitagawa H, Yanagisawa J, Yano T, Kato S. Repressive domain of unliganded human estrogen receptor α associates with Hsc70. Genes Cells. 2005;10:1095–102. doi: 10.1111/j.1365-2443.2005.00904.x. [DOI] [PubMed] [Google Scholar]
- 71.Oxelmark E, Knoblauch R, Arnal S, Su LF, Schapira M, Garabedian MJ. Genetic dissection of p23, an Hsp90 cochaperone, reveals a distinct surface involved in estrogen receptor signaling. J Biol Chem. 2003;278:36547–55. doi: 10.1074/jbc.M305960200. [DOI] [PubMed] [Google Scholar]
- 72.Schlegel A, Wang C, Katzenellenbogen BS, Pestell RG, Lisanti MP. Caveolin-1 potentiates estrogen receptor α (ERα) signaling. caveolin-1 drives ligand-independent nuclear translocation and activation of ERα. J Biol Chem. 1999;274:33551–6. doi: 10.1074/jbc.274.47.33551. [DOI] [PubMed] [Google Scholar]
- 73.Knoblauch R, Garabedian MJ. Role for Hsp90-associated cochaperone p23 in estrogen receptor signal transduction. Mol Cell Biol. 1999;19:3748–59. doi: 10.1128/mcb.19.5.3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Devin-Leclerc J, Meng X, Delahaye F, Leclerc P, Baulieu EE, Catelli MG. Interaction and dissociation by ligands of estrogen receptor and Hsp90: the antiestrogen RU 58668 induces a protein synthesis-dependent clustering of the receptor in the cytoplasm. Mol Endocrinol. 1998;12:842–54. doi: 10.1210/mend.12.6.0121. [DOI] [PubMed] [Google Scholar]
- 75.Inano K, Curtis SW, Korach KS, Omata S, Horigome T. Heat shock protein 90 strongly stimulates the binding of purified estrogen receptor to its responsive element. J Biochem (Tokyo) 1994;116:759–66. doi: 10.1093/oxfordjournals.jbchem.a124593. [DOI] [PubMed] [Google Scholar]
- 76.Schaaf MJ, Cidlowski JA. Molecular mechanisms of glucocorticoid action and resistance. J Steroid Biochem Mol Biol. 2002;83:37–48. doi: 10.1016/s0960-0760(02)00263-7. [DOI] [PubMed] [Google Scholar]
- 77.Evinger AJ, 3rd, Levin ER. Requirements for estrogen receptor alpha membrane localization and function. Steroids. 2005;70:361–3. doi: 10.1016/j.steroids.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 78.Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. 2005;146:624–32. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- 79.Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–30. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- 80.Pedram A, Razandi M, Levin ER. Nature of functional estrogen receptors at the plasma membrane. Mol Endocrinol. 2006;20:1996–2009. doi: 10.1210/me.2005-0525. [DOI] [PubMed] [Google Scholar]
- 81.Brychzy A, Rein T, Winklhofer KF, Hartl FU, Young JC, Obermann WM. Cofactor Tpr2 combines two TPR domains and a J domain to regulate the Hsp70/Hsp90 chaperone system. Embo J. 2003;22:3613–23. doi: 10.1093/emboj/cdg362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Likhite VS, Stossi F, Kim K, Katzenellenbogen BS, Katzenellenbogen JA. Kinase-specific phosphorylation of the estrogen receptor changes receptor interactions with ligand, DNA, and coregulators associated with alterations in estrogen and tamoxifen activity. Mol Endocrinol. 2006 doi: 10.1210/me.2006-0068. [DOI] [PubMed] [Google Scholar]
- 83.Lipfert L, Fisher JE, Wei N, Scafonas A, Su Q, Yudkovitz J, Chen F, Warrier S, Birzin ET, Kim S, Chen HY, Tan Q, Schmidt A, Dininno F, Rohrer SP, Hammond ML, Rodan GA, Freedman LP, Reszka AA. Antagonist-induced, activation function-2-independent estrogen receptor α phosphorylation. Mol Endocrinol. 2006;20:516–33. doi: 10.1210/me.2005-0190. [DOI] [PubMed] [Google Scholar]
- 84.Lee H, Bai W. Regulation of estrogen receptor nuclear export by ligand-induced and p38-mediated receptor phosphorylation. Mol Cell Biol. 2002;22:5835–45. doi: 10.1128/MCB.22.16.5835-5845.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bodwell JE, Orti E, Coull JM, Pappin DJ, Smith LI, Swift F. Identification of phosphorylated sites in the mouse glucocorticoid receptor. J Biol Chem. 1991;266:7549–55. [PubMed] [Google Scholar]
- 86.Almlof T, Wright AP, Gustafsson JA. Role of acidic and phosphorylated residues in gene activation by the glucocorticoid receptor. J Biol Chem. 1995;270:17535–40. doi: 10.1074/jbc.270.29.17535. [DOI] [PubMed] [Google Scholar]
- 87.Galigniana MD, Housley PR, DeFranco DB, Pratt WB. Inhibition of glucocorticoid receptor nucleocytoplasmic shuttling by okadaic acid requires intact cytoskeleton. J Biol Chem. 1999;274:16222–7. doi: 10.1074/jbc.274.23.16222. [DOI] [PubMed] [Google Scholar]
- 88.Pascussi JM, Gerbal-Chaloin S, Drocourt L, Maurel P, Vilarem MJ. The expression of CYP2B6, CYP2C9 and CYP3A4 genes: a tangle of networks of nuclear and steroid receptors. Biochim Biophys Acta. 2003;1619:243–53. doi: 10.1016/s0304-4165(02)00483-x. [DOI] [PubMed] [Google Scholar]
- 89.Pascussi JM, Drocourt L, Gerbal-Chaloin S, Fabre JM, Maurel P, Vilarem MJ. Dual effect of dexamethasone on CYP3A4 gene expression in human hepatocytes. Sequential role of glucocorticoid receptor and pregnane X receptor. Eur J Biochem. 2001;268:6346–58. doi: 10.1046/j.0014-2956.2001.02540.x. [DOI] [PubMed] [Google Scholar]
- 90.Agrawal AK, Shapiro BH. Phenobarbital induction of hepatic CYP2B1 and CYP2B2: pretranscriptional and post-transcriptional effects of gender, adult age, and phenobarbital dose. Mol Pharmacol. 1996;49:523–31. [PubMed] [Google Scholar]
- 91.Chang TKH, Anderson MD, Bandiera SM, Bellward GD. Effect of ovariectomy and androgen on phenobarbital induction of hepatic CYP2B1 and CYP2B2 in Sprague-Dawley rats. Drug Metab Dispos. 1997;25:994–1000. [PubMed] [Google Scholar]
- 92.Yoshinari K, Sueyoshi T, Moore R, Negishi M. Nuclear receptor CAR as a regulatory factor for the sexually dimorphic induction of CYB2B1 gene by phenobarbital in rat livers. Mol Pharmacol. 2001;59:278–84. doi: 10.1124/mol.59.2.278. [DOI] [PubMed] [Google Scholar]
- 93.Min G, Kim H, Bae Y, Petz L, Kemper JK. Inhibitory cross-talk between estrogen receptor (ER) and constitutively activated androstane receptor (CAR). CAR inhibits ER-mediated signaling pathway by squelching p160 coactivators. J Biol Chem. 2002;277:34626–33. doi: 10.1074/jbc.M205239200. [DOI] [PubMed] [Google Scholar]
- 94.Ganem LG, Trottier E, Anderson A, Jefcoate CR. Phenobarbital induction of CYP2B1/2 in primary hepatocytes: endocrine regulation and evidence for a single pathway for multiple inducers. Toxicol Appl Pharmacol. 1999;155:32–42. doi: 10.1006/taap.1998.8599. [DOI] [PubMed] [Google Scholar]
- 95.Kietzmann T, Hirsch-Ernst KI, Kahl GF, Jungermann K. Mimicry in primary rat hepatocyte cultures of the in vivo perivenous induction by phenobarbital of cytochrome P-450 2B1 mRNA: role of epidermal growth factor and perivenous oxygen tension. Mol Pharmacol. 1999;56:46–53. doi: 10.1124/mol.56.1.46. [DOI] [PubMed] [Google Scholar]
- 96.Bauer D, Wolfram N, Kahl GF, Hirsch-Ernst KI. Transcriptional regulation of CYP2B1 induction in primary rat hepatocyte cultures: repression by epidermal growth factor is mediated via a distal enhancer region. Mol Pharmacol. 2004;65:172–80. doi: 10.1124/mol.65.1.172. [DOI] [PubMed] [Google Scholar]
- 97.Assenat E, Gerbal-Chaloin S, Larrey D, Saric J, Fabre JM, Maurel P, Vilarem MJ, Pascussi JM. Interleukin 1beta inhibits CAR-induced expression of hepatic genes involved in drug and bilirubin clearance. Hepatology. 2004;40:951–60. doi: 10.1002/hep.20387. [DOI] [PubMed] [Google Scholar]
- 98.Thasler WE, Dayoub R, Muhlbauer M, Hellerbrand C, Singer T, Grabe A, Jauch KW, Schlitt HJ, Weiss TS. Repression of cytochrome P450 activity in human hepatocytes in vitro by a novel hepatotrophic factor, augmenter of liver regeneration. J Pharmacol Exp Ther. 2006;316:822–9. doi: 10.1124/jpet.105.094201. [DOI] [PubMed] [Google Scholar]
- 99.Pascussi JM, Gerbal-Chaloin S, Pichard-Garcia L, Daujat M, Fabre JM, Maurel P, Vilarem MJ. Interleukin-6 negatively regulates the expression of pregnane X receptor and constitutively activated receptor in primary human hepatocytes. Biochem Biophys Res Commun. 2000;274:707–13. doi: 10.1006/bbrc.2000.3219. [DOI] [PubMed] [Google Scholar]
- 100.Columbano A, Ledda-Columbano GM, Pibiri M, Cossu C, Menegazzi M, Moore DD, Huang W, Tian J, Locker J. Gadd45β is induced through a CAR-dependent, TNF-independent pathway in murine liver hyperplasia. Hepatology. 2005;42:1118–26. doi: 10.1002/hep.20883. [DOI] [PubMed] [Google Scholar]
- 101.Masuyama H, Inoshita H, Hiramatsu Y, Kudo T. Ligands have various potential effects on the degradation of pregnane X receptor by proteasome. Endocrinology. 2002;143:55–61. doi: 10.1210/endo.143.1.8578. [DOI] [PubMed] [Google Scholar]
- 102.Chen D, Riedl T, Washbrook E, Pace PE, Coombes RC, Egly JM, Ali S. Activation of estrogen receptor α by S118 phosphorylation involves a ligand-dependent interaction with TFIIH and participation of CDK7. Mol Cell. 2000;6:127–37. [PubMed] [Google Scholar]
- 103.Lu Q, Ebling H, Mittler J, Baur WE, Karas RH. MAP kinase mediates growth factor-induced nuclear translocation of estrogen receptor α. FEBS Lett. 2002;516:1–8. doi: 10.1016/s0014-5793(02)02432-8. [DOI] [PubMed] [Google Scholar]
- 104.Itoh M, Adachi M, Yasui H, Takekawa M, Tanaka H, Imai K. Nuclear export of glucocorticoid receptor is enhanced by c-Jun N-terminal kinase-mediated phosphorylation. Mol Endocrinol. 2002;16:2382–92. doi: 10.1210/me.2002-0144. [DOI] [PubMed] [Google Scholar]
- 105.DeFranco DB, Qi M, Borror KC, Garabedian MJ, Brautigan DL. Protein phosphatase types 1 and/or 2A regulate nucleocytoplasmic shuttling of glucocorticoid receptors. Mol Endocrinol. 1991;5:1215–28. doi: 10.1210/mend-5-9-1215. [DOI] [PubMed] [Google Scholar]
- 106.Fan M, Park A, Nephew KP. CHIP (carboxyl terminus of Hsc70-interacting protein) promotes basal and geldanamycin-induced degradation of estrogen receptor-alpha. Mol Endocrinol. 2005;19:2901–14. doi: 10.1210/me.2005-0111. [DOI] [PubMed] [Google Scholar]
- 107.Reid G, Hubner MR, Metivier R, Brand H, Denger S, Manu D, Beaudouin J, Ellenberg J, Gannon F. Cyclic, proteasome-mediated turnover of unliganded and liganded ERα on responsive promoters is an integral feature of estrogen signaling. Mol Cell. 2003;11:695–707. doi: 10.1016/s1097-2765(03)00090-x. [DOI] [PubMed] [Google Scholar]
- 108.Tateishi Y, Kawabe Y, Chiba T, Murata S, Ichikawa K, Murayama A, Tanaka K, Baba T, Kato S, Yanagisawa J. Ligand-dependent switching of ubiquitin-proteasome pathways for estrogen receptor. EMBO J. 2004;23:4813–23. doi: 10.1038/sj.emboj.7600472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tateishi Y, Sonoo R, Sekiya YI, Sunahara N, Kawano M, Wayama M, Hirota R, Kawabe YI, Kato S, Kimura K, Yanagisawa J. Turning off estrogen receptor β-mediated transcription requires estrogen-dependent receptor proteolysis. Mol Cell Biol. 2006 doi: 10.1128/MCB.00713-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Connell P, Ballinger CA, Jiang J, Wu Y, Thompson LJ, Hohfeld J, Patterson C. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat Cell Biol. 2001;3:93–6. doi: 10.1038/35050618. [DOI] [PubMed] [Google Scholar]
- 111.Wallace AD, Cidlowski JA. Proteasome-mediated glucocorticoid receptor degradation restricts transcriptional signaling by glucocorticoids. J Biol Chem. 2001;276:42714–21. doi: 10.1074/jbc.M106033200. [DOI] [PubMed] [Google Scholar]
- 112.Binart N, Lombes M, Baulieu EE. Distinct functions of the 90 kDa heat-shock protein (hsp90) in oestrogen and mineralocorticosteroid receptor activity: effects of hsp90 deletion mutants. Biochem J. 1995;311:797–804. doi: 10.1042/bj3110797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pratt WB, Galigniana MD, Harrell JM, DeFranco DB. Role of hsp90 and the hsp90-binding immunophilins in signalling protein movement. Cell Signal. 2004;16:857–72. doi: 10.1016/j.cellsig.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 114.Davies TH, Ning YM, Sanchez ER. A new first step in activation of steroid receptors: hormone-induced switching of FKBP51 and FKBP52 immunophilins. J Biol Chem. 2002;277:4597–600. doi: 10.1074/jbc.C100531200. [DOI] [PubMed] [Google Scholar]
- 115.Kino T, Tiulpakov A, Ichijo T, Chheng L, Kozasa T, Chrousos GP. G protein beta interacts with the glucocorticoid receptor and suppresses its transcriptional activity in the nucleus. J Cell Biol. 2005;169:885–96. doi: 10.1083/jcb.200409150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Freedman ND, Yamamoto KR. Importin 7 and importin α/importin β are nuclear import receptors for the glucocorticoid receptor. Mol Biol Cell. 2004;15:2276–86. doi: 10.1091/mbc.E03-11-0839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Holaska JM, Black BE, Love DC, Hanover JA, Leszyk J, Paschal BM. Calreticulin is a receptor for nuclear export. J Cell Biol. 2001;152:127–40. doi: 10.1083/jcb.152.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Walther RF, Lamprecht C, Ridsdale A, Groulx I, Lee S, Lefebvre YA, Hache RJ. Nuclear export of the glucocorticoid receptor is accelerated by cell fusion-dependent release of calreticulin. J Biol Chem. 2003;278:37858–64. doi: 10.1074/jbc.M306356200. [DOI] [PubMed] [Google Scholar]
- 119.Kino T, Souvatzoglou E, De Martino MU, Tsopanomihalu M, Wan Y, Chrousos GP. Protein 14-3-3σ interacts with and favors cytoplasmic subcellular localization of the glucocorticoid receptor, acting as a negative regulator of the glucocorticoid signaling pathway. J Biol Chem. 2003;278:25651–6. doi: 10.1074/jbc.M302818200. [DOI] [PubMed] [Google Scholar]
- 120.Eickelberg O, Roth M, Lorx R, Bruce V, Rudiger J, Johnson M, Block LH. Ligand-independent activation of the glucocorticoid receptor by β2-adrenergic receptor agonists in primary human lung fibroblasts and vascular smooth muscle cells. J Biol Chem. 1999;274:1005–10. doi: 10.1074/jbc.274.2.1005. [DOI] [PubMed] [Google Scholar]
- 121.Carey KL, Richards SA, Lounsbury KM, Macara IG. Evidence using a green fluorescent protein-glucocorticoid receptor chimera that the Ran/TC4 GTPase mediates an essential function independent of nuclear protein import. J Cell Biol. 1996;133:985–96. doi: 10.1083/jcb.133.5.985. [DOI] [PMC free article] [PubMed] [Google Scholar]