

Abstract
Serum retinol binding protein 4 (RBP4) was recently described as a new adipokine that reduced peripheral and hepatic insulin sensitivity and increased hepatic gluconeogenesis. The RBP4 gene maps to 10q23-24, near a region linked to T2DM in Caucasian and Mexican American populations. Hence, sequence variants that alter RBP4 expression or function could increase T2DM susceptibility and reduce insulin sensitivity. We screened the 6 exons, flanking intronic sequence, and 5’ and 3’ flanking sequences in 48 Caucasian and 48 African American subjects. We identified 21 SNPs, of which 8 were unique to the African American population. Additional public database SNPs were chosen for regions not screened. We selected SNPs for typing based on frequency, linkage disequilibrium, and location in a putative functional or conserved region. We typed 10 SNPs in 191 Caucasians with T2DM and a family history of T2DM, and 188 euglycemic controls with no family history of diabetes. We similarly typed 14 variants in 182 controls and 353 diabetic individuals of African American ancestry. No single variant was associated with type 2 diabetes in either population (p>0.15 in African Americans, p>0.09 in Caucasians), but a haplotype of 8 common SNPs in Caucasians was significantly increased in type 2 diabetics compared with controls (0.137 vs 0.076, p=0.008). Furthermore, SNPs -804 and +9476 were associated with reduced insulin secretion, (p=0.01 and 0.001, respectively), and SNP +390 with reduced insulin sensitivity (p=0.0005) in Caucasians. Our data suggest that noncoding SNPs may increase diabetes susceptibility in Caucasians and may contribute to insulin resistance and reduced insulin secretion.
Keywords: insulin resistance, type 2 diabetes, single nucleotide polymorphism, linkage disequilibrium, allelic association
Introduction
Insulin resistance and progressive β-cell failure are key factors in the pathogenesis of type 2 diabetes mellitus (T2DM) [1]. Genome scans and association studies suggest a complex genetic etiology for diabetes susceptibility [2]. Most glucose after a meal is taken up by muscle by way of insulin-mediated recruitment of GLUT4 transporters to the cell surface [3]. However, the markedly increased risk and prevalence of T2DM with accumulating adiposity is inconsistent with the small role that adipocytes play in post-prandial glucose uptake. Such arguments suggest that adipocytes influence T2DM pathogenesis beyond their minor role in glucose disposal. Indeed, adipocytes secrete a plethora of cytokine-like products that alter insulin sensitivity, including tumor necrosis factor α, resistin, leptin, interleukin 6, and adiponectin [4].
Recent studies have suggested an even broader role for adipocytes in the control of whole body glucose metabolism. Mice with an adipose-specific knockout of GLUT4 developed both muscle and hepatic insulin resistance along with glucose intolerance [5]. Because muscle insulin sensitivity was normal ex vivo, a circulating adipocyte factor was proposed [5]. Subsequently, these investigators demonstrated elevated expression of retinol binding protein 4 (RBP4) in adipocytes from the same adipocyte-Glut4-/- mice [6]. Serum levels of RBP4 were increased in adipocyte-Glut4-/- animals. When RBP4 levels were reduced by a synthetic retinoid (fenretinide) or by RBP4 deletion, peripheral insulin sensitivity increased [6]. Furthermore, increased serum RBP4 stimulated hepatic gluconeogenesis through stimulation of phosphoenolpyruvate carboxykinase [6]. These results suggested that adipocyte-derived RBP4 may be a key factor regulating peripheral tissue response to insulin-stimulated GLUT4 action.
Additional evidence for a primary role of RBP4 in humans was reported recently by Graham et al [7]. In three populations studied, RBP4 levels correlated with total adiposity (body mass index, BMI), abdominal obesity, and fasting insulin levels, and were inversely proportionate to glucose disposal using euglycemic clamp studies [7]. RBP4 levels were increased in subjects with impaired glucose tolerance or diabetes relative to euglycemic controls, and RBP4 serum levels fell in individuals responding to exercise training with improved insulin sensitivity. Finally, serum RBP4 levels correlated inversely with insulin sensitivity among individuals with a family history of T2DM. Together with mouse data [6], these studies support a primary role of RBP4 in insulin action in humans and suggest that genetic variation in RBP4 might alter risk for T2DM [8].
The RBP4 gene is located on chromosome 10q24 in humans near a region linked to elevated fasting blood glucose and 20-year mean blood glucose levels in European Caucasians [9] and to T2DM in Mexican-American subjects [10]. We hypothesized that genetic variation in RBP4 resulting in over-expression of the gene product contributes to the reported increase in serum RBP4 levels, insulin resistance, and T2DM. We extensively screened the RBP4 gene for sequence variants in African American and Caucasian subjects and evaluated the role of these variants by testing the association with T2DM, and by examining the impact on prediabetic intermediate traits (insulin sensitivity and insulin secretion) in nondiabetic individuals.
Materials and Methods
Experimental Subjects
The study populations are summarized in Table 1. Caucasian individuals were ascertained in Utah for Northern European ancestry, as described previously [11;12]. Additional Caucasian and African-American individuals were ascertained from Arkansas for similar criteria [12]. Briefly, all individuals with T2DM had at least one other first-degree relative with type 2 diabetes. Nondiabetic control individuals had no known family history of diabetes and had either a normal 75-g oral glucose tolerance test or a fasting glucose level below 5.6 mmol/l (100 mg/dl). All subjects provided written informed consent under protocols approved by either the University of Utah or the University of Arkansas for Medical Sciences Institutional Review Boards.
Table 1.
Summary of Study Populations
| Population | Male/Female | Body Mass Index (kg/m2) | Age (years) |
|---|---|---|---|
| Caucasian Case/Control | Control 73/115
Case 134/57 |
29.3 (19.8, 43.2) | 56.9 (14.1) |
| African American Case/Control | Control 90/92
Case 189/164 |
31.2 (20.0, 48.8) | 50.0 (13.9) |
| Utah Caucasian Family Based Metabolic Study | 50/72 | 27.5 (18.3, 41.3) | 39.3 (10.5) |
| Arkansas Caucasian Population based metabolic study | 73/135 | 30.8 (18.2, 41.9) | 36.4 (9.0) |
| African American Population based metabolic study | 48/71 | 30.1 (19.9, 45.8) | 37.5 (8.9) |
Means are shown as arithmetic means for normal variables and geometric means for skewed variables (BMI). Age is shown as mean (SD); body mass index is shown as mean (95% CI) transferred to the linear scale from the ln-transformation.
Sequence variation was determined in 48 unrelated Caucasian and 48 unrelated African American volunteers, including 36 subjects with T2DM requiring therapy and 12 glucose-tolerant control subjects from each population. Case-control association studies with T2DM were conducted in 191 Caucasian subjects with T2DM and 188 Caucasian control subjects, and in 353 African American subjects with T2DM and 182 nondiabetic African American subjects. Association of RBP4 SNPs with insulin sensitivity and insulin secretion was tested in three populations totaling 449 individuals (Table 1). The first population included 122 nondiabetic members of families ascertained in Utah for at least 2 diabetic siblings who were studied using the tolbutamide-modified intravenous glucose tolerance test [13], of which 27 individuals had impaired glucose tolerance. A second Caucasian population of 208 subjects was ascertained in Arkansas for normal glucose tolerance but with variable family history. Finally, we studied 119 African American individuals who were similarly ascertained in Arkansas. The two Arkansas populations underwent either tolbutamide modified intravenous glucose tolerance tests (65 African American subjects; 100 Caucasian subjects), or because tolbutamide became unavailable during this ongoing study, an insulin modified test using 0.04 units/kg of insulin (54 African American and 108 Caucasian subjects).
Variant detection
We designed primers from the human genome sequence (AL356214) and alignment with the human RBP4 mRNA sequence (NM_006744) to cover each of the predicted 6 exons, including the untranslated exon 1, introns 1-3, 100-250 bp of intronic sequence flanking exons 4, 5 and 6, and 1.0 - 1.2 kb of 5’ and 3’ flanking sequence (Figure 1). Initial screening for sequence variants was by denaturing high-performance liquid chromatography (DHPLC) using a Transgenomic WAVE HT DNA Fragment Analysis System (Transgenomic, Inc, Omaha, NE). Amplification primers were designed using Oligo 6.0 (Molecular Biology Insights, Cascade, CO) and Wavemaker Software, Version 4.0 (Transgenomic, Inc). Altered migration was confirmed and characterized by bidirectional sequence analysis using a CEQ8000 (Beckman-Coulter, Fullerton, CA) capillary sequencer or a LI-COR GR-4200 (LI-COR Biotech, Lincoln, NE) sequencer according to manufacturer’s protocols. Additional variants in introns 4 and 5, which were not completely screened, were selected for evaluation from available HapMap data [14].
Figure 1. Map of RBP4 gene with genotyped variants.
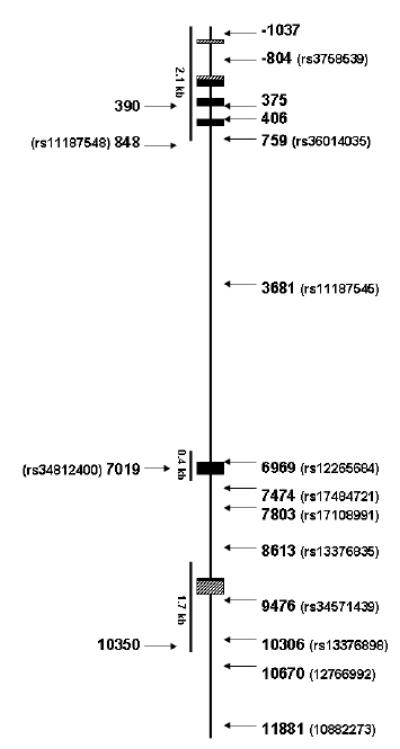
Boxes represent exons, the horizontal line represents the introns and flanking sequence. Translated sequences are shown in black and the untranslated exons are indicated by diagonal patterned boxes. Arrowheads represent approximate location of each single nucleotide polymorphism (SNP). Regions screened are shown as lines below the figure; other single nucleotide variants were obtained from public databases.
Genetic Analysis
Single nucleotide polymorphisms (SNPs) were typed by Pyrosequencing (PSQ96, Biotage AB, Uppsula, Sweden) using manufacturer protocols, except that a universal sequence was appended to one sequence-specific primer and amplification was performed in the presence of the universal biotinylated primer. Additionally, at least 68 unrelated, nondiabetic individual samples were tested from each ethnic group to determine linkage disequilibrium (LD). All SNPs were in Hardy-Weinberg equilibrium (p>0.2). Variants for typing in the full case-control association study were selected in part based on pairwise linkage disequilibrium for r2 < 0.9 ([15]. Markers for typing in individuals who were assessed for insulin sensitivity (SI) were chosen from the combined case-control sample for a minor allele frequency over 10% and pairwise selection of haplotype tagging SNPs using TAGGER (parameter r2=0.8, Haploview v 3.2; [16]). Selection of SNPs was performed separately for African American and Caucasian populations.
Statistical Analysis
Allelic association was tested separately for each ethnic group using the Fisher Exact Test. Marker phase and linkage disequilibrium were determined from unrelated samples using the Expectation-Maximization algorithm in combined case and control populations using programs implemented in Haploview v 3.2 [16]. Haplotype block structure was determined using both solid spline and confidence interval block definitions [17]. Haplotype associations were tested using HaploView 3.2 and adjusted p values determined by permutation tests. Insulin sensitivity (SI) was calculated using either the MinMod program from intravenous glucose tolerance test data [18], or for subjects studied in Arkansas, the MinMod Millenium program [19]. Acute insulin response to glucose (AIRG) was determined from the mean 2 min – 10 min excursion over baseline in the Utah study, or as the area under curve taken from the MinMod Millenium output for Arkansas studies. Disposition index (DI) was calculated as SI*AIRG [20;21]. Association of SNPs with quantitative metabolic traits was determined using the general linear regression models in SPSS for Windows v 12 (SPSS, Inc, Chicago, IL). Skewed variables (BMI, SI, AIRG, DI) were ln-transformed prior to analysis. Fixed factors and covariates included age, gender, BMI, diagnosis (Utah study), and protocol (Arkansas studies). To correct for relatedness of subjects, family membership was included as a random variable in the Utah study. All marginal means were compared after inclusion of covariates. P values are presented based on comparison of marginal means and are reported without correction for multiple hypothesis testing. Significance thresholds were determined for each population based on the estimation of the effective number of SNPs proposed by Nyholt based on the correlation matrix [22].
Results
We identified 21 SNPs by screening in both ethnic groups (Figure 1, Table 2; and Online Supplement, Table 6S), of which 13 were present in the public database (dbSNP) [23]. No nonsynonymous SNP was observed, and 8 SNPs were observed only in African Americans. We examined an additional 6 SNPs from the HapMap project located in introns 4 and 5. Of the 21 SNPs identified by screening, 7 had minor allele frequencies <5% and hence were not typed due to low power to detect an association (Online Supplement, Table 6S). An additional 2 SNPs (Figure 1, -9727 and -180) could not be typed as a result of homopolymeric sequences surrounding the SNP. We assessed linkage disequilibrium for 17 SNPs in African Americans and 14 SNPs in Caucasians (Online Supplemental Data, Figures 2S and 3S). Based on strong pairwise linkage disequilibrium (r2>0.9), we excluded an additional 3 SNPs in African Americans and 4 SNPs in Caucasians from further typing. Results of case control allelic association studies are shown in Table 1, and genotypic counts are shown in Online Supplement, Table 7S. No individual SNP showed an allelic association with T2DM in either population (Table 2); the strongest association gave an uncorrected p value of 0.095 (Table 2; Online Supplement, Table 7S).
Table 2.
Summary of Single Nucleotide Polymorphisms and Allele Frequencies
| Position | RS Number | Variant | Location | African- American Case/Control Frequency | Caucasian Case/Control Frequency |
|---|---|---|---|---|---|
| -1037 | novel | G/A | 5′FLANKING | 0.063/0.072 | ND |
| -804 | rs3758539 | G/A | 5′FLANKING | 0.072/0.069 | 0.168/0.185 |
| 375 | novel | C/T | INTRON 3 | 0.013/0.025 | 0.021/0.027 |
| 390 | novel | C/G | INTRON 3 | 0.078/0.080 | 0.076/0.066 |
| 406 | novel | T/C | INTRON 3 | 1 | 0.071/0.058 |
| 759 | rs36014035 | T/G | INTRON 4 | 0.419/0.429 | 0.404/0.361 |
| 848 | rs11187548 | G/A | INTRON 4 | 0.140/0.141 | ND |
| 3681 | rs11187545 | T/C | INTRON 4 | 0.121/0.0127 | 1 |
| 6969 | rs12265684 | G/C | INTRON 4 | 0.333/0.345 | 0.190/0.198 |
| 7019 | rs34812400 | ACA(Thr)127/ACG(Thr) | EXON 5 (S) | 0.126/0.124 | ND |
| 7803 | rs17108991 | G/T | INTRON 5 | 0.204/0.214 | 1 |
| 9476 | rs34571439 | T/G | 3′FLANKING | 0.359/0.390 | 0.203/0.196 |
| 10306 | rs13376898 | G/C | 3′FLANKING | 0.062/0.047 | ND |
| 10350 | novel | G/A | 3′FLANKING | 2 (<3%) | 0.057/0.044 |
| 10670 | rs12766992 | G/A | 3′FLANKING | 0.077/0.091 | 0.192/0.200 |
| 11881 | rs10882273 | C/T | 3′FLANKING | 0.409/0.364 | 0.402/0.3403 |
Not typed in full set because of strong linkage disequilibrium in initial study;
not typed in full set because of low frequency in initial study;
p=0.095 uncorrected.
To test the hypothesis that a haplotype harboring a SNP that was not tested or could not be typed might contribute to T2DM, we constructed haplotypes for each population using all SNPs with a minor allele frequency over 5% (Table 2). In Caucasians, visual inspection and block structure based on solid spline of linkage disequilibrium placed all 8 common SNPs in a single block (-804, 390, 406, 759, 6969, 9476, 10670, and 11881) with 5 haplotypes (see Online Supplement, Figure S1). A single haplotype was associated with T2DM (p=0.008; permuted p, 0.04 on 100,000 permutations) with a frequency of 13.7% in cases and 7.6% in controls (Table3). No other haplotype was significantly more common in cases than controls. In contrast, no haplotype, regardless of block definition (see Online Supplement, Figure 3S), was more common in African American cases than controls (data not shown).
Table 3.
Haplotype Frequencies for RBP4 SNPs in Caucasian Cases and Controls
| Haplotype | Case Freq | Control Freq | Chi Sq. | p value |
|---|---|---|---|---|
| GGTTGTGT | 0.578 | 0.631 | 2.178 | 0.14 |
| AGTGCGAC | 0.166 | 0.172 | 0.054 | 0.8168 |
| GGTGGTGC | 0.137 | 0.076 | 7.039 | 0.008 |
| GCCGGTGC | 0.069 | 0.057 | 0.427 | 0.5134 |
| GGTGCGAC | 0.018 | 0.019 | 0.005 | 0.9456 |
Haplotypes were estimated from Haploview in Caucasian cases and controls for SNPs -804, 390, 406, 759, 6969, 9476, 10670, and 11881 (Table 1). Uncommon SNPs were not included in the analysis. P values are shown without correction; the corrected p value by permutation was 0.039 for haplotype GGTGGTGC (100,000 permutations).
Based on the evidence that circulating RBP4 protein levels correlate with insulin resistance [7], we tested the role of RBP4 sequence variation in SI, AIRG, and DI. We selected 5 RBP4 SNPs in Caucasians (-804, +390, +759, +9476, and + 10350), and 7 RBP4 SNPs in African Americans (+759, +848, +3681, +7019, +7803, +9476, and +11881) for a minor allele frequency >0.05 and pairwise linkage disequilibrium of r2 >0.8. Results are summarized in Table 4, and marginal means for nominally significant associations are shown in Table 5. SNP +390 was significantly associated with reduced SI (3.32 to 1.69; p=0.0005). The correlated value, DI (SI x AIRG), was also significantly decreased (1230 to 695; p=0.005). Using the Nyholt method [22] to correct for multiple testing while considering the strong linkage disequilibrium among SNPs, and ignoring the correlation between DI and both SI; and AIRG, we determined an appropriate threshold for significance (3 traits and 4.3 effective SNPs) at 0.004. Hence, the association with SI remained significant, and the association with DI was borderline significant. The association of AIRG with SNP +9476 in the Utah Population (210 vs 131; p=0.001) was the only other significant association based on this somewhat conservative threshold. The latter association appears to be supported by similar data in Arkansas Caucasians (402 vs 331; p=0.04 uncorrected).
Table 4.
Summary of RBP4 Association with Intravenous Glucose Tolerance Test Measures
| Population | SNP | N | Genotype Counts | SI | AIRG | DI |
|---|---|---|---|---|---|---|
| Af. Am. | +759 | 119 | 30/57/32 | 0.51 | 0.49 | 0.96 |
| Af. Am. | +848 | 118 | 103/15/0 | 0.62 | 0.77 | 0.97 |
| Af. Am. | +3681 | 118 | 87/31/0 | 0.97 | 0.026 | 0.081 |
| Af. Am. | +7019 | 119 | 96/23/0 | 0.98 | 0.5 | 0.33 |
| Af. Am. | +7803 | 118 | 73/38/7 | 0.45 | 0.48 | 0.17 |
| Af. Am. | +9476 | 119 | 46/60/13 | 0.11 | 0.41 | 0.093 |
| Af. Am. | +11881 | 119 | 41/60/18 | 0.83 | 0.88 | 0.91 |
| UT Cauc | -804 | 122 | 81/41/4* | 0.75 | 0.0098 | 0.022 |
| UT Cauc | +390 | 122 | 106/20/0 | 0.2 | 0.86 | 0.88 |
| UT Cauc | +759 | 122 | 36/70/20 | 0.98 | 0.93 | 0.66 |
| UT Cauc | +9476 | 122 | 77/41/4* | 0.45 | 0.0011 | 0.067 |
| UT Cauc | +10350 | 122 | 101/21/0 | 0.44 | 0.13 | 0.1 |
| AR Cauc | -804 | 208 | 149/57/2* | 0.63 | 0.1 | 0.46 |
| AR Cauc | +390 | 208 | 183/25/0 | 0.0005 | 0.5 | 0.005 |
| AR Cauc | +759 | 208 | 86/95/27 | 0.58 | 0.51 | 0.32 |
| AR Cauc | +9476 | 208 | 148/55/5* | 0.64 | 0.039 | 0.29 |
| AR Cauc | +10350 | 207 | 181/26/0 | 0.91 | 0.11 | 0.21 |
Table 4 gives the p values for the difference between marginal means by genotype. P values are presented without correction for multiple testing of traits and SNPs, which are strongly correlated. Marginal means for genotypes showing nominally significant differences among genotypes are shown in Table 5. Af. Am., Arkansas African American metabolic study; UT Cauc, Utah Caucasian metabolic study; AR Cauc, Arkansas Caucasian Metabolic study; N, number of successfully typed samples; genotype counts, counts of common homozygotes/heterozygotes/rare homozygotes; SI, insulin sensitivity index from MinMod; AIRG, acute 2-10 min insulin response to intravenous glucose bolus; DI, disposition index, SI*AIRG.
, genotypes rescored to include rare homozygotes with heterozygotes for analysis.
Table 5.
Marginal Means for Nominally Significant Quantitative Traits
| Population | Trait | SNP | Mean 1 | Mean 2 | P value |
|---|---|---|---|---|---|
| UT Cauc | AIRG | −804 | 195 (161, 236) | 133 (103, 172) | 0.01 |
| UT Cauc | DI | −804 | 75.6 (58.0, 98.5) | 120.9 (84.4, 173.2) | 0.022 |
| AR Cauc | SI | +390 | 3.32 (2.90, 3.80) | 1.69 (1.18, 2.42) | 0.0005 |
| AR Cauc | DI | +390 | 1230 (1063, 1423) | 695 (474, 1019) | 0.005 |
| Af. Am. | AIRG | +3681 | 535 (455, 629) | 768 (582, 1013) | 0.026 |
| UT Cauc | AIRG | +9476 | 210 (172, 256) | 131 (104, 167) | 0.001 |
| AR Cauc | AIRG | +9476 | 402 (358, 452) | 321 (267, 385) | 0.039 |
Marginal means are shown after adjustment for age, gender, and BMI; significance is based on a general linear model, and p values are shown without correction for multiple testing. AIRG in the Utah Caucasian study is from the mean 2 min-10 min post-challenge insulin excursion in pmol/l; for the African American and Arkansas Caucasian studies, the AIRG is taken from the MinMod Millenium output and is the area under curve from 0 – 10 min. All means are transformed back to the linear scale from ln-transformed values; 95% confidence intervals are provided in parentheses. As noted in Table 4, SNPs -804 and +9476 were rescored to combine heterozygotes and rare homozygotes, and no rare homozygotes were observed for SNPs +390 and +3681; hence, we show only 2 means for these 4 SNPs.
Discussion
RBP4 has been proposed as a circulating, adipocyte-derived signal that may contribute to secondary insulin resistance in muscle and liver, both in mice and humans [6;7]. A recent study [7] demonstrated an association of circulating RBP4 protein levels in serum with insulin resistance measured by euglycemic clamp in three populations, and an earlier study demonstrated increased serum RBP4 levels with obesity and T2DM. We are not aware of any published work that has examined the role of genetic variation in RBP4 with T2DM or insulin resistance. Such studies were called for in a recent editorial [8]. We have examined two populations: Caucasians and African Americans. We found no variants that altered the amino acid sequence of RBP4 in either population. Many of the variants identified were rare, and the variants typed were in strong linkage disequilibrium, particularly in Caucasians. Hence, although we did not fully resequence the large introns 4 and 5, we have likely captured the genetic variation in and around this gene.
Although no single SNP was associated with T2DM, we did identify a haplotype comprising all 8 common SNPs in Caucasians that was significantly increased in T2DM relative to controls after permutation testing. By standards required to show and confirm the generally modest effect sizes observed in other T2DM genes, our population is relatively small; hence, this finding requires replication. Over the range of minor allele frequencies observed in this study, we have over 70% power at p<0.05 (uncorrected for multiple tests) to detect a difference in allele frequency of 7%-10% between Caucasian cases and controls, corresponding to an odds ratio (OR) of 1.5 to 1.8, or approximately the range observed for the well replicated TCF7L2 diabetes gene [24]. Among African Americans our power was slightly higher, with at least 70% power to detect a frequency difference of 6% to 8%, or an OR range of 1.4 to 1.7. Using the single SNPs with the largest differences in minor allele frequency between cases and controls, we would need over 1600 cases and 1600 controls for 70% power to find a difference in African Americans (rs34571439 or rs10882273), or 505 Caucasian cases and 505 Caucasian controls to find a significant difference given the observed frequencies at rs10882273. Further evaluation of the latter SNP is clearly justified.
Given the lack of association of any individual SNP with T2DM, a variant not detected, perhaps in the flanking regions or unsequenced introns, may be responsible for the observed haplotype association. In contrast, among the African American population where linkage disequilibrium is weaker and more SNPs were required to cover the full gene, we found no evidence for an association of individual SNPs or haplotypes covering each of the blocks. The lack of replication in the African American case-control sample may have resulted from spurious association in the Caucasian sample, a lower effect size or frequency of the risk allele in the African American population, or different gene-environment interactions in the two population groups. Alternatively, because of lower levels of linkage disequilibrium in African Americans, the haplotypes examined may not have captured the full genetic diversity in the two large introns.
We did see an association of individual SNPs with quantitative traits related to glucose homeostasis in nondiabetic subjects, including SI (SNP +390 in Arkansas Caucasians), AIRG (SNP +9476 in both Caucasian studies; SNPs -804 in Utah Caucasians and +3681 in African Americans), and DI (SNP +390 in Arkansas Caucasians). Some of the associations in Table 4 may be spurious given the testing of 3 traits and 5-7 SNPs in each population. Indeed, the associations are not entirely consistent across populations. Thus, the -804 SNP association with AIRG was observed only in the Utah sample, SNP +390 was associated with SI only in Arkansas Caucasians, and SNP +3681 was associated with AIRG and DI only observed in African Americans. SNP +9476 showed the best consistency with an association with AIRG in both Caucasian populations, but the association with insulin secretion was unexpected. Other than spurious associations (type 1 error), the lack of consistency has several possible explanations: 1) relatively small samples combined with possibly small effect size, and hence low power for replication; 2) differences in environment and ascertainment, particular with respect to diet, BMI, and family history of diabetes; 3) different analytical methods, with Utah samples requiring correction for family membership and glucose tolerance status in addition to age, gender, and BMI.
The mechanisms by which noncoding RBP4 variants could increase diabetes risk, decrease insulin secretion, or decrease insulin sensitivity are unclear. Previous data suggested that insulin sensitivity and risk of T2DM might be related to circulating RBP4 protein levels [6;7]; these in turn were proposed to reflect adipocyte gene expression [6]. We have not measured serum RBP4 levels, nor do we have measures of adipocyte gene expression to address this hypothesis. RBP4 is expressed widely, with the highest level in the liver, but with significant expression also in pancreatic islets based on expressed sequence tags (ESTs). Hence, the gene may have non-endocrine effects. Furthermore, the exact structure of the gene is unclear, with various annotations assigning from 6 to 9 exons. Available ESTs predict at least 5 splice forms; furthermore, conservation and EST clones suggest that the large 4th intron (Figure 1) may harbor several exons. Hence, both tested SNPs and SNPs not tested but in strong linkage disequilibrium with typed variants may have a functional role in alternative splicing. Such SNPs may even alter the protein sequence of some splice forms. Such effects could be tissue specific, and might explain the role of RBP4 in insulin secretion, as suggested by our data. Such potential roles of RBP4 are clearly different from those suggested in the published data that led us to conduct the present study.
In summary, recent data have added RBP4 to the circulating factors that influence insulin sensitivity and diabetes risk. The present study is to our knowledge the first to search for genetic variants in that gene. We find evidence in Caucasians that noncoding variants in or near RBP4 contribute to the risk of T2DM, are associated with reduced insulin sensitivity (SI), and appear to reduce insulin secretion and compensation for insulin resistance. In contrast, we find little role for RBP4 in either diabetes or intermediate traits among African Americans. Replication of our results in additional studies, particularly for the diabetes-associated haplotype, will be important in determining the role of genetic variation in RBP4 in diabetes susceptibility.
Supplementary Material
Acknowledgments
This work was supported by grants from NIH/NIDDK (DK39311 and DK54366), by the Research Service of the Department of Veterans Affairs, and by the American Diabetes Association. Subject ascertainment and metabolic studies were supported by the General Clinical Research Center, grant M01RR14288 from National Center for Research Resources (NIH) to the University of Arkansas for Medical Sciences. We thank S.K. Das for helpful discussions, and we thank the GCRC nursing staff for invaluable assistance with the testing of subjects for insulin sensitivity and insulin secretion.
Footnotes
Funding: NIH/NIDDK grants DK39311 and DK54366; the Research Service of the Department of Veterans Affairs, the American Diabetes Association, and grant M01RR14288 from National Center for Research Resources (NIH)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet. 2005;365:1333–1346. doi: 10.1016/S0140-6736(05)61032-X. [DOI] [PubMed] [Google Scholar]
- 2.Das SK, Elbein SC. The Genetic Basis of Type 2 Diabetes. Cellscience Reviews. 2006;2:1–32. doi: 10.1901/jaba.2006.2-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeFronzo RA. Pathogenesis of type 2 diabetes mellitus. Med Clin North Am. 2004;88:787–835. ix. doi: 10.1016/j.mcna.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 4.Hutley L, Prins JB. Fat as an endocrine organ: relationship to the metabolic syndrome. Am J Med Sci. 2005;330:280–289. doi: 10.1097/00000441-200512000-00005. [DOI] [PubMed] [Google Scholar]
- 5.Abel ED, Peroni O, Kim JK, Kim YB, Boss O, Hadro E, Minnemann T, Shulman GI, Kahn BB. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature. 2001;409:729–733. doi: 10.1038/35055575. [DOI] [PubMed] [Google Scholar]
- 6.Yang Q, Graham TE, Mody N, Preitner F, Peroni OD, Zabolotny JM, Kotani K, Quadro L, Kahn BB. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature. 2005;436:356–362. doi: 10.1038/nature03711. [DOI] [PubMed] [Google Scholar]
- 7.Graham TE, Yang Q, Bluher M, Hammarstedt A, Ciaraldi TP, Henry RR, Wason CJ, Oberbach A, Jansson PA, Smith U, Kahn BB. Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. N Engl J Med. 2006;354:2552–2563. doi: 10.1056/NEJMoa054862. [DOI] [PubMed] [Google Scholar]
- 8.Polonsky KS. Retinol-binding protein 4, insulin resistance, and type 2 diabetes. N Engl J Med. 2006;354:2596–2598. doi: 10.1056/NEJMe068091. [DOI] [PubMed] [Google Scholar]
- 9.Meigs JB, Panhuysen CI, Myers RH, Wilson PW, Cupples LA. A genome-wide scan for loci linked to plasma levels of glucose and HbA(1c) in a community-based sample of Caucasian pedigrees: The Framingham Offspring Study. Diabetes. 2002;51:833–840. doi: 10.2337/diabetes.51.3.833. [DOI] [PubMed] [Google Scholar]
- 10.Duggirala R, Blangero J, Almasy L, Dyer TD, Williams KL, Leach RJ, O′Connell P, Stern MP. Linkage of Type 2 Diabetes Mellitus and of Age at Onset to a Genetic Location on Chromosome 10q in Mexican Americans. Am J Hum Genet. 1999;64:1127–1140. doi: 10.1086/302316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elbein SC, Hoffman MD, Teng K, Leppert MF, Hasstedt SJ. A genome-wide search for type 2 diabetes susceptibility genes in Utah Caucasians. Diabetes. 1999;48:1175–1182. doi: 10.2337/diabetes.48.5.1175. [DOI] [PubMed] [Google Scholar]
- 12.Das SK, Chu W, Zhang Z, Hasstedt SJ, Elbein SC. Calsquestrin 1 (CASQ1) gene polymorphisms under chromosome 1q21 linkage peak are associated with type 2 diabetes in Northern European Caucasians. Diabetes. 2004;53:3300–3306. doi: 10.2337/diabetes.53.12.3300. [DOI] [PubMed] [Google Scholar]
- 13.Elbein SC, Hasstedt SJ, Wegner K, Kahn SE. Heritability of pancreatic beta-cell function among nondiabetic members of Caucasian familial type 2 diabetic kindreds. J Clin Endocrinol Metab. 1999;84:1398–1403. doi: 10.1210/jcem.84.4.5604. [DOI] [PubMed] [Google Scholar]
- 14.McVean G, Spencer CC, Chaix R. Perspectives on Human Genetic Variation from the HapMap Project. PLoS Genet. 2005;1:e54. doi: 10.1371/journal.pgen.0010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carlson CS, Eberle MA, Rieder MJ, Yi Q, Kruglyak L, Nickerson DA. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am J Hum Genet. 2004;74:106–120. doi: 10.1086/381000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 17.Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, LiuCordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 18.Pacini G, Bergman RN. MINMOD: a computer program to calculate insulin sensitivity and pancreatic responsivity from the frequently sampled intravenous glucose tolerance test. Comput Methods Programs Biomed. 1986;23:113–122. doi: 10.1016/0169-2607(86)90106-9. [DOI] [PubMed] [Google Scholar]
- 19.Boston RC, Stefanovski D, Moate PJ, Sumner AE, Watanabe RM, Bergman RN. MINMOD Millennium: a computer program to calculate glucose effectiveness and insulin sensitivity from the frequently sampled intravenous glucose tolerance test. Diabetes Technol Ther. 2003;5:1003–1015. doi: 10.1089/152091503322641060. [DOI] [PubMed] [Google Scholar]
- 20.Bergman RN. Toward physiological understanding of glucose tolerance: minimal model approach. Diabetes. 1989;39:1512–1527. doi: 10.2337/diab.38.12.1512. [DOI] [PubMed] [Google Scholar]
- 21.Kahn SE, Prigeon RL, McCulloch DK, Boyko EJ, Bergman RN, Schwartz MW, Neifing JL, Ward WK, Beard JC, Palmer JP, Porte D., Jr Quantification of the relationship between insulin sensitivity and beta-cell function in human subjects: evidence for a hyperbolic function. Diabetes. 1993;42:1663–1672. doi: 10.2337/diab.42.11.1663. [DOI] [PubMed] [Google Scholar]
- 22.Nyholt DR. A simple correction for multiple testing for single-nucleotide polymorphisms in linkage disequilibrium with each other. Am J Hum Genet. 2004;74:765–769. doi: 10.1086/383251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wheeler DL, Church DM, Lash AE, Leipe DD, Madden TL, Pontius JU, Schuler GD, Schriml LM, Tatusova TA, Wagner L, Rapp BA. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2001;29:11–16. doi: 10.1093/nar/29.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grant SF, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J, Helgason A, Stefansson H, Emilsson V, Helgadottir A, Styrkarsdottir U, Magnusson KP, Walters GB, Palsdottir E, Jonsdottir T, Gudmundsdottir T, Gylfason A, Saemundsdottir J, Wilensky RL, Reilly MP, Rader DJ, Bagger Y, Christiansen C, Gudnason V, Sigurdsson G, Thorsteinsdottir U, Gulcher JR, Kong A, Stefansson K. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet. 2006;38:320–323. doi: 10.1038/ng1732. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.