

Abstract
β protein from bacteriophage λ promotes a single-strand annealing reaction that is central to Red-mediated recombination at double-strand DNA breaks and chromosomal ends. β protein binds most tightly to an intermediate of annealing formed by the sequential addition of two complementary oligonucleotides. Here we have characterized the domain structure of β protein in the presence and absence of DNA using limited proteolysis. Residues 1–130 form an N-terminal “core” domain that is resistant to proteases in the absence of DNA, residues 131–177 form a central region with enhanced resistance to proteases upon DNA complex formation, and the C-terminal residues 178–261 of β protein are sensitive to proteases in both the presence and absence of DNA. We probed the DNA binding regions of β protein further using biotinylation of lysine residues and mass spectrometry. Several lysine residues within the first 177 residues of β protein are protected from biotinylation in the DNA complex, whereas none of the lysine residues in the C-terminal portion are protected. The results lead to a model for the domain structure and DNA binding of β protein in which a stable N-terminal core and a more flexible central domain come together to bind DNA, whereas a C-terminal tail remains disordered. A fragment consisting of residues 1–177 of β protein maintains normal binding to sequentially added complementary oligonucleotides and has significantly enhanced binding to single-strand DNA.
The Red system of bacteriophage λ consists of three proteins that promote DNA recombination initiated at dsDNA4 breaks or at the overlapping ends of the linear λ chromosome (1–2). The exo gene (redα) encodes λ exonuclease, a 24-kDa protein with 5′-3′ exonuclease activity (3–4). The bet gene (redβ) encodes the 29-kDa β protein, which binds ssDNA and promotes annealing of complementary strands (5–6). The gam gene encodes the 16-kDa γ protein, which binds and inhibits host nuclease enzymes (7–8). Together, the three proteins promote recombination events that enhance the replication and repair of phage λ DNA within the Escherichia coli host. Certain strains of E. coli contain an analogous recombination system, RecET, encoded on a cryptic rac prophage, that is turned on to promote recombination in certain genetic backgrounds (9). RecE and RecT are functionally analogous and related in sequence to λ exonuclease and β protein, respectively (10–11). Both the RecET and Redαβ systems have in recent years been employed in novel and powerful methods for genetic engineering (12–13) and, thus, are of interest technologically in addition to their long-standing as model systems for understanding general principles of genetic recombination.
Purified β protein promotes renaturation of complementary strands of DNA (5–6) and annealing of shorter oligonucleotides (14). The protein binds weakly to ssDNA of minimal length of about 36 nucleotides with a site size of approximately one monomer per five nucleotides (15). β protein does not bind directly to dsDNA but remains tightly associated with the duplex product of annealing formed by the sequential addition of two complementary oligonucleotides to β protein (14). Although β protein is not an ATPase, it will promote some RecA-like strand exchange and invasion reactions in vitro but only when certain restrictive criteria are met, such as high A/T composition (16–17).
The structure of β protein has been studied by electron microscopy (18). In the presence of Mg2+, the protein alone forms rings of ~12 subunits, slightly larger rings (15–18 subunits) in the presence of ssDNA, and left-handed helical filaments in the presence of dsDNA or two complementary strands of ssDNA. A model was proposed in which ssDNA binds to the outer surface of the rings formed by β protein in such a way that the bases are exposed and available for interactions with a second strand of ssDNA, which may be bound by a separate ring of β protein. Initiation of annealing likely nucleates formation of a β protein filament, which coats the duplex product of annealing. Presumably the strongly favorable interaction of β protein with the newly formed dsDNA drives the reaction in favor of annealing.
β belongs to a family of proteins (10) including RecT, for which there is no representative crystal structure. β protein and RecT, the best-studied members of this family, share only about 15% sequence identity with one another. Secondary structure prediction based on a multiple sequence alignment suggests four α-helices and five β-strands for a conserved region corresponding to residues 19–205 of β protein (10). Notable features in the sequences include several conserved aromatic and apolar residues and two consecutive highly conserved acidic residues near the end of the conserved region. Beyond the low resolution electron microscopy studies and what can be gleaned from sequence analyses, very little is known about the structure of β protein.
Here we have used limited proteolysis, N-terminal sequencing, and mass spectrometry to probe the domain structure of β protein in the presence and absence of DNA. The results reveal a protease-resistant N-terminal core domain, a central region that is stabilized by DNA complex formation, and a flexible C-terminal tail. In addition, application of a recently developed technique for mapping out protein-DNA interactions, which involves biotinylation of lysine residues and mass spectrometry (19–20), suggests that both the N-terminal and central regions of β protein, but not the C-terminal tail, are involved in binding to DNA.
EXPERIMENTAL PROCEDURES
Expression and Purification of β Protein
The bet gene encoding β protein was PCR-amplified from phage λ genomic DNA (New England Biolabs) and cloned into pET-14b (Novagen) between the NdeI and BamH1 sites. The primers for PCR amplification were 5′-GGAA TTC CAT ATG AGT ACT GCA CTC GC-3′(forward) and 5′-CGC GGA TCC TCA TGC TGC CAC CTT CTGC-3′ (reverse). The resulting plasmid expresses β protein with an N-terminal His6 tag followed by a thrombin cleavage site. The integrity of the insert was verified by DNA sequencing, and the plasmid was transformed into BL21 (DE3) pLysS E. coli cells. Cell cultures (6 × 1 liter) were grown in Luria broth at 37 °C to an absorbance at 600 nm of 0.5, induced with 1 mM isopropyl 1-thio-β-D-galactopyranoside, and shaken at 18 °C for an additional 16 h. Cells were harvested by centrifugation at 10,000 × g, and the pellets were stored at −80 °C. Cell pellets were thawed and re-suspended in 50 ml of buffer A (300 mM NaCl, 30 mM NaH2PO4, 10 mM imidazole, pH 8.0) supplemented with 1 mg/ml lysozyme, 100 μg/ml α-phenylmethylsulfonyl fluoride, 1 μg/ml pepstatin A, and 1 μg/ml leupeptin. After sonication the lysate was centrifuged at 39,000 × g, and the supernatant was loaded onto a 10-ml column of Ni2+-loaded chelating Sepharose fast flow (Amersham Biosciences). The column was washed with 300 ml of buffer A containing 30 mM imidazole, and the protein was eluted with a 200-ml linear gradient from 30–500 mM imidazole in buffer A. Pooled fractions containing β protein were dialyzed into thrombin cleavage buffer (20 mM NaH2PO4, 150 mM NaCl, pH 7.4) and incubated with 50 units of thrombin (Amersham Biosciences) at 22 °C for ~24 h. The protein was then loaded onto the Ni2+ column and the flow-through was dialyzed into 20mM Tris, pH 8.0, loaded onto a 25-ml HiTrap Q HP anion exchange column (Amersham Biosciences), and eluted with a 180-ml linear gradient from 0 to 1 M NaCl. Pooled fractions were dialyzed into 20 mM Tris, pH 8.0, 1 mM DTT, concentrated to 47 mg/ml, and stored at −80 °C in small aliquots. The concentration of β protein was determined by absorbance at 280 nm in 6 M guanidine hydrochloride, 0.02 M NaH2PO4, pH 6.5, using an extinction coefficient of 34,850 M−1 cm−1, which was calculated from the amino acid sequence (21). The final protein contains the extra N-terminal sequence Gly-Ser-His. The K253A mutant of β protein was generating using the QuikChange procedure (Stratagene), and the protein was purified as described above for wild type.
Expression and Purification of β Protein Fragments
Regions of DNA encoding amino acids 1–177 and 1–188 of β protein were PCR-amplified from phage λ genomic DNA and cloned into pET-9a between the NdeI and BamH1 sites. PCR used the forward primer listed above and the reverse primers 5′-CGC GGA TCC TCA ACG TTC TGC AGT GTA TGC-3 ′(β1–188) and CGC GGA TCC TCA GCG CTC GGC TTC ATC CTT GTC (β1–177). The K11A, K36A, K69A, and K172A mutants of the β1–188 fragment were generated by the QuikChange procedure. Plasmids were transformed into BL21-AI E. coli cells (Invitrogen), grown in 1-liter cultures of Luria broth at 37 °C to an absorbance at 600 nm of 0.5, induced with 0.2% arabinose, and shaken at 15 °C for an additional 16 h. Cells were harvested by centrifugation, and the pellets were stored at −80 °C.
For purification of β1–188 protein, cells were thawed and resuspended in 35 ml of buffer B (500mM NaCl, 50mM NaH2PO4, 10% glycerol, 10 mM imidazole, pH 8.0) supplemented with lysozyme and protease inhibitors as described above. After sonication, the lysate was centrifuged at 39,000 × g, and the supernatant was dialyzed into 20 mM Tris, pH 8.0, loaded onto a 25-ml HiTrap Q HP anion exchange column, and eluted with a 180-ml linear gradient from 0 to 1 M NaCl. Pooled fractions were dialyzed into 20 mM NaH2PO4, pH 7.0, loaded onto a 25-ml HiTrap heparin column (Amersham Biosciences), and eluted with a 180-ml linear gradient from 0 to 1.5 M NaCl. Pooled fractions were dialyzed into 20 mM Tris pH 8.0, 1 mM DTT, concentrated to 66 mg/ml, and stored at −80 °C in small aliquots.
For purification of β1–177, thawed cells were re-suspended in buffer B. After sonication and centrifugation of the cell lysate, polyethyleneimine was added to the supernatant to a final concentration of 0.5%, and the resulting suspension was stirred at 4 °C for 30 min and centrifuged at 12,000 × g. The supernatant was collected and dialyzed into 20 mM Tris, pH 8.0, 100 mM NaCl. The resulting suspension was centrifuged at 10,000 × g, and the protein from the supernatant was further purified by anion exchange and heparin chromatography as described above for β1–188 except that 100 mM NaCl was included in the buffer before the heparin column. Purification of β1–188 proteins with the K11A, K36A, K69A, and K172A mutations followed this same procedure, except the heparin chromatography step was omitted, and the final buffer included 100 mM NaCl.
Oligonucleotides
Two complementary 33-mer oligonucleotides, 33+ (ACA GCA CCA GAT TCA GCA ATT AAG CTC TAA GCC) and 33− (GGC TTA GAG CTT AAT TGC TGA ATC TGG TGC TGT), which are derived from a sequence present in M13 phage DNA (14), were purchased from Integrated DNA Technologies and purified by ion exchange HPLC. Oligonucleotide concentrations were determined from absorbance at 260 nm using extinction coefficients calculated from their sequences and are expressed in nucleotides. 33+ oligonucleotide was 32P-5′-end-labeled with T4 polynucleotide kinase (New England Biolabs), and free nucleotides were removed with a MicroSpin G-25 column (Amersham sham Biosciences).
DNA Binding Assays
The gel shift assay to measure binding of β protein to sequentially added oligonucleotides is based on a published protocol (14). Each 20-μl reaction contained 50mM Tris, pH 7.5, 1mM DTT, and 0.1 mg/ml bovine serum albumin. 32P-end-labeled 33+ and β protein of the indicated concentrations were incubated at 37 °C, and after 10 min the complementary oligonucleotide (33−) of the indicated concentration was added, and the reaction was incubated an additional 30 min at 37 °C. After the addition of 5 μl of 5× loading buffer (20% glycerol, 0.12% bromphenol blue, 0.12% xylene cyanol), each sample was loaded onto a 15% native polyacrylamide gel (5% stacking gel) and electrophoresed in TAE (40 mM Tris acetate, pH 8.3, 1 mM EDTA) buffer at room temperature. Dried gels were autoradiographed.
Binding of full-length β protein and the β1–177 fragment to a 33-mer oligonucleotide (33+) was measured using a double-filter method (22). Binding reactions (50 μl) were in buffer C (20 mM Tris-HCl, pH 7.5, 1 mM MgCl2, 1 mM DTT) and contained ~100 nM 32P-end-labeled 33+ oligonucleotide and 0–1 mM protein. The reactions were equilibrated at 37 °C for 30 min and then loaded onto a Minifold I 48-well slot-blot apparatus (Schleicher & Schuell) containing nitrocellulose (Bio-Rad) and DEAE (Whatman) filters. Samples were pulled through the filters under vacuum and washed with 1 ml of buffer C. The radioactivity on the filters was measured using a Storm 860 PhosphorImager (Amersham Biosciences) and ImageQuant 5.2 software (Molecular Dynamics). The percentage of protein-bound DNA was calculated from the intensities on the nitro-cellulose (protein-bound) and DEAE (unbound) filters.
Limited Proteolysis
Limited trypsin digestions were performed in 25mM Hepes, pH 7.9, 10% glycerol, 0.2 mM EDTA, 5 mM MgCl2, 20 mM CaCl2, and 60 mM KCl at 25 °C. The reactions contained 2.5 mg/ml (86 μM) β protein in the absence or presence of 250 μM each of sequentially added 33+ and 33− oligonucleotides, as described above. Trypsin was added to a final concentration of 5 ng/ μl, and 10 μl of the reaction was removed at the indicated times. Reactions were quenched with 10 μl of 2× SDS-PAGE loading buffer, heated at 95 °C for 5 min, electrophoresed on a 13.5% SDS-PAGE gel, and stained with Coomassie Blue. Digestions using the same procedure were also done with 50 ng/ μl chymotrypsin, 10 ng/ μl subtilisin, and 25 ng/μl thermolysin. For mass spectrometric analysis of limited proteolysis products, reactions were incubated for 30 min, quenched by adding 6 M guanidine hydrochloride, and injected directly onto the LC-MS instrument.
For N-terminal sequencing, unstained SDS-PAGE gels were blotted onto polyvinylidene fluoride membranes and subsequently stained with Coomassie Blue. Individual bands were cut out of the gel and sequenced by automated Edman degradation (Protein and Nucleic Acid Chemistry Laboratory, Washington University, St. Louis, MO) using an Applied Bio-systems model 494 Procise HT sequencer. Data analysis was performed with Applied Biosystems model 610A software. Standard reagents and conditions were employed.
Biotinylation Reactions
12 μl of 2.5 μg/ μl β protein or β protein-DNA complex was prepared as described above. 38 μl of phosphate-buffered saline (Sigma) and 2.5 μl of 40 mM NHS-biotin (Pierce) dissolved in Me2SO were mixed and incubated at room temperature for 40 min, and the reactions were quenched by adding 5 μl of 100 mM lysine (8.7 mM final concentration). The biotinylated protein was subjected to complete trypsin digestion as has been described (23). Briefly, the protein was precipitated from the reaction mixture by adding acetone, and the pellet was washed with acetone and dried. The pellet was dissolved in 20 μl of 8 M urea, 0.4 M NH4HCO3, reduced with 5 μl 45 mM DTT, and alkylated with 5 μl of 100 mM iodoacetic acid. 50 μl of double-distilled H2O was then added to dilute the digestion buffer to 2 M urea, 0.1 M NH4HCO3. 5 μl of 0.2 μg/ μl trypsin in 1% acetic acid was added, and the mixture was incubated at 37 °C for 24 h. The trypsin digestion was stopped by adding 10 μl of 2% trifluoro-acetic acid. A 10-μl aliquot of the mixture was then subjected to LC-MS analysis.
Mass Spectrometry
All mass spectrometric analyses used a ThermoFinnigan LCQ DECA ion trap mass spectrometer coupled with a Surveyor HPLC system (ThermoQuest, San Jose, CA). For mass spectrometry of limited proteolysis reactions, HPLC separations were performed with a PLRP-S column (8 μm, 1000 Å, 1.0 × 150 mm, Michrom Bioresources, Inc.) and a Vydas C4 guard column using H2O/acetonitrile gradients at a flow rate of 50 μl/min. 0.1% formic acid and 0.02% trifluoroacetic tic acid were included in both water and acetonitrile. The data were collected in positive ion mode with full scan ranging from 400–2000.
For mass spectrometry of trypsin digests of biotinylation reactions, collection of all MS and MS/MS data used a C18 column (Vydas, 1.0-mm inner diameter × 250 mm long, 5-μm particles, and 300 Å pore size; Hesperia, CA) with Vydas C18 guard column. In the survey run the mass spectrometer was operated in data-dependent mode and cycled through a single MS and five MS/MS experiments. MS/MS experiments (collision energy 35%) were performed on the five most abundant ions (3-Da window; precursor m/z ± 1.5 Da) in each full scan mass spectrum. MS/MS spectra were matched to amino acid sequences from β protein using SEQUEST with differential modification of 226.3 Da to lysine and a static modification of 57 Da to cysteine. Once the unbiotinylated and biotinylated peaks were assigned to individual peptides, the retention time and best response charge states were recorded. The mass spectrometer was then switched to full scan ion mode with the scan range of 400–1500. When signal responses of analytes were compared, chromatographic peak area was used. The doubly charged peak of “VGMDSVDPQELITTLR” (16–31)2+ was selected as internal loading control, since this peptide fragment is not affected by biotinylation and is always formed by tryptic digestion. All other peptide responses were normalized to the response of this peptide.
RESULTS
Limited Proteolysis of β Protein Alone and in Complex with DNA
We first verified that our preparation of β protein, which has the extra N-terminal residues GSH, has the expected DNA binding properties. In the assay (14), β protein is first incubated with a 32P-end-labeled 33-mer (33+) oligonucleotide, and as the complementary oligonucleotide (33−) is titrated in, the resulting β protein-DNA complex is shifted to the top of a 15% polyacrylamide gel (supplemental Fig. S1). Binding of β protein to only one oligonucleotide (lane 3) or to pre-annealed duplex 33-mer (not shown) is not observed. Thus, as has been concluded previously (14), a tight complex is formed only when the two complementary oligonucleotides are added sequentially to β protein. As seen by electron microscopy, this preparation of β protein also forms rings and filaments in the presence of heat-denatured dsDNA (data not shown) that appear very similar to those reported for native β protein (18).
The domain structure of β protein both alone and in complex with DNA was investigated using limited proteolysis with trypsin. In the absence of DNA, β protein is rapidly digested into a distinct fragment that appears as a ~15-kDa band on the SDS-PAGE gel (Fig. 1). N-terminal sequencing and mass spectrometric analysis of this cleavage product unambiguously identified it to be residues 1–134 of β protein. In complex with annealed duplex DNA (as prepared in Fig. S1, panel A, lane 8), ), parts of β protein are significantly protected from trypsin digestion. In particular, three new distinct cleavage products are observed as ~19-, 21-, and 27-kDa bands, which correspond to residues 16–177, 1–177, and 1–230 of β protein, respectively. The fragment of residues 1–177 is the most stable and persists for greater than 2 h.
FIGURE 1. Limited protease digestion of β protein in the presence and absence of DNA.
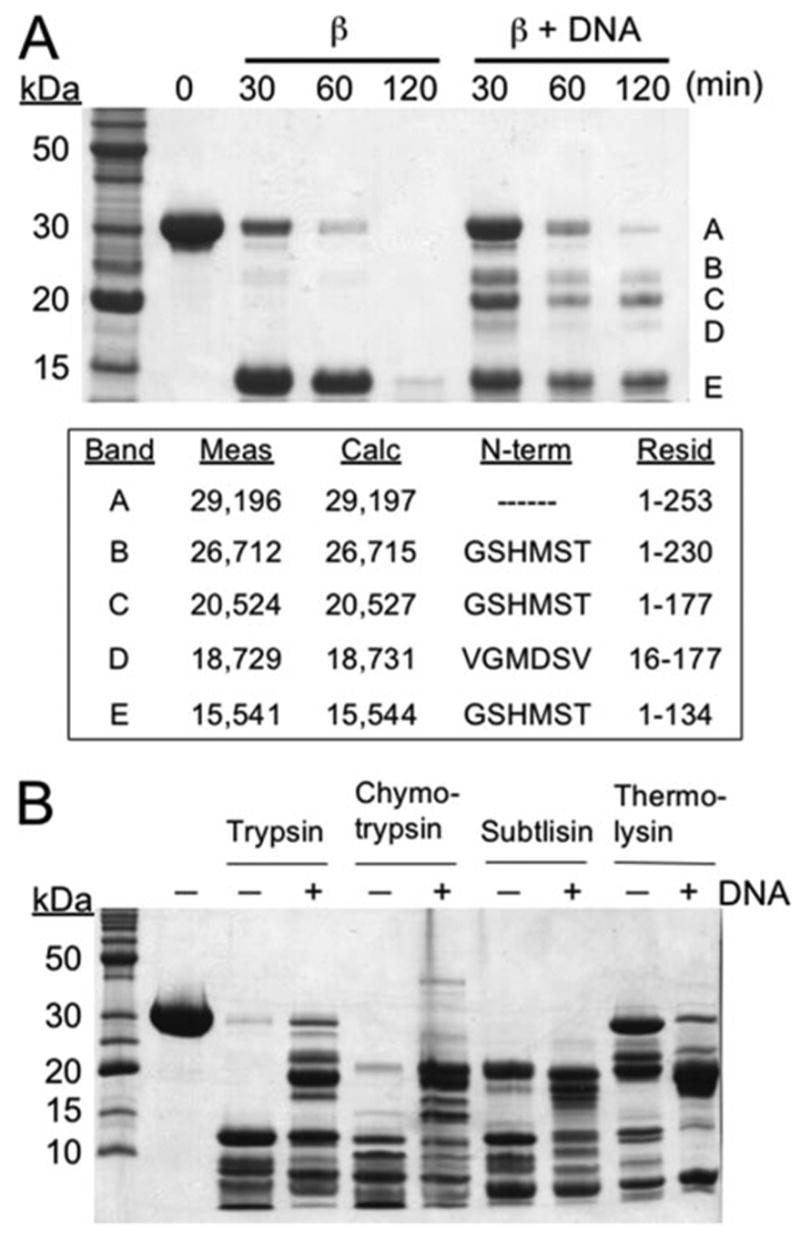
The digestion was performed on β protein alone or in complex with DNA as formed by the sequential addition of two complementary 33-mer oligonucleotides. A, time course of limited trypsin digestion. The text box shows for each of the major fragments the mass measured by LC-MS, the mass calculated from the amino acid sequence, the results from N-terminal sequencing, and the corresponding residues of β protein. Notice that formation of the DNA complex confers protease resistance to residues 135–230 of β protein, as seen by the appearance of bands B, C, and D. B, limited digestion with trypsin, chymotrypsin, subtilisin, and thermolysin for 30 min in the presence and absence of DNA. The identities of each of the fragments produced were determined by mass spectrometry and are presented in supplemental Fig. S2.
In addition to stabilizing larger fragments of β protein, DNA complex formation also stabilizes the N-terminal domain (residues 1–134) since it persists for 120 min in the presence of DNA but is completely digested within 120 min in the absence of DNA. DNA does not stabilize full-length β protein, since the upper band disappears at the same rate in both the presence and absence of DNA. Mass spectrometric analysis reveals that the upper band from the samples digested in the presence of DNA does not actually correspond to full-length β protein but rather to residues 1–253 that result from removal of residues 254–261 by trypsin cleavage at Lys-253.
The single-strand annealing activity and oligomeric properties of β protein have previously been shown to be sensitive to Mg2+ ions (5, 15, 18). However, the limited trypsin digestion patterns in both the presence and absence of DNA are not affected at all by the omission of Mg2+ from the reaction (data not shown). In addition, essentially the same results seen in Fig. 1 are observed when the buffer is the same as that used for the gel-shift assay of Fig. S1, which does not contain Mg2+ or other divalent cations.
Limited proteolysis of β protein was also investigated using chymotrypsin, subtilisin, and thermolysin (Figs. 1B and supplemental Fig. S2). Although in general chymotrypsin cleaves at sites very different from trypsin (after aromatic or aliphatic residues instead of basic residues), the two give very similar cleavage patterns for β protein; that is, a stable N-terminal domain (residues 1–131) in the absence of DNA and a larger series of fragments (residues 1–184, 9–184, 13–184, and 36–184) in the presence of DNA. With subtilisin, a non-sequence-specific protease, β protein alone is digested into two similar fragments, an N-terminal domain (residues 1–134) and a longer fragment (residues 1–184), which in this case persists even in the absence of DNA. DNA does, however, change the cleavage pattern such that several new fragments (residues 1–177, 13–182, 20–182) are observed. Similarly with thermolysin, which cleaves before apolar residues, N-terminal (residues 1–138) and longer (1–177) fragments are observed for the protein alone, whereas DNA shifts the pattern to a series of intermediate fragments (residues 1–177, 8–177, and 12–177). Interestingly, with thermolysin a fragment consisting of nearly full-length β -protein (residues 1–258) is significantly more stable in the absence of DNA than in its presence, indicating that some sites within β protein are more accessible in the DNA complex than in the free protein.
We have also investigated the trypsin digestion of β protein preparations that give distinct oligomeric forms (rings and filaments) as seen by electron microscopy (18). For the small ring preparation with Mg2+ in the absence of DNA, only the N-terminal core domain (residues 1–134) is observed. For the large ring preparation in the presence of a 33-mer oligonucleotide, a small amount of the fragment corresponding to residues 1–177 is also observed. For the filament preparation formed in the presence of a heat-denatured fragment of ΦX174 dsDNA, increased amounts of full-length and fragments corresponding to residues 1–177 and 1–230 are observed, and the pattern is similar to that of the DNA complex with sequentially added 33-mer oligonucleotides described above. This analysis is consistent with the notion that in the DNA complex formed with sequentially added complementary oligonucleotides, β protein likely exists as short filaments. Using size-exclusion chromatography on Superdex S-200, our preparation of β protein in the absence of DNA elutes as a distinct peak corresponding to a large oligomer (566 kDa or ~ 19 subunits), consistent with a ring (data not shown). The complex with sequentially added oligonucleotides elutes as a distinct peak with a larger apparent size. Similarly, with native acrylamide gel electrophoresis the complex of β protein with sequentially added oligonucleotides forms a distinct band that is of lower mobility (presumably larger size) than several bands formed by β protein alone (data not shown).
Probing the DNA Binding Regions of β Protein by Biotin Modification and Mass Spectrometry
To further probe the structure of β protein and its interactions with DNA, we used a method involving mass spectrometric analysis of protein modified by biotinylation at lysine residues. NHS-biotin reacts efficiently and specifically with the primary ε-amino group of lysine side chains to become covalently attached via a stable amide linkage. The modification alters the pattern of peptides that are detected in a mass spectrometric analysis of protein fragments produced by complete digestion with trypsin. Lysine residues often participate directly in binding to the sugar phosphate backbone of DNA and may thereby be protected in the DNA complex from modification by NHS-biotin. The protected lysine residues can be determined from the mass spectrometry experiment. This approach has been successfully employed to identify DNA binding regions of human replication protein A (19) and human immunodeficiency virus-1 reverse transcriptase (20). Here we have employed this approach to determine regions of β protein that interact with DNA.
First, the effect of the NHS-biotin reagent on the binding of β protein to DNA was determined. The addition of NHS-biotin to β protein completely prevents its subsequent binding to sequentially added complementary oligonucleotides, as determined by gel-shift assay (supplemental Fig. S1, panel B). ). The effect of NHS-biotin is concentration-dependent, with only partial inhibition of DNA binding at 60 μM NHS-biotin and complete inhibition at 300–600 μM NHS-biotin. By contrast, modification of the preformed β protein-DNA complex with NHS-biotin does not result in its dissociation (supplemental Fig. S1, panel B, lane 3). ). These data suggest that one or more of the 14 lysine residues of β protein is positioned at the protein-DNA interface such that its modification with biotin completely disrupts DNA binding. These lysine residue(s) of β protein are, however, protected from modification in the complex that is formed with DNA.
To determine which lysine residues of β protein are modified by biotin, the biotin-modified protein was subjected to complete trypsin digestion, and the peptide mixture was analyzed by mass spectrometry as outlined in supplemental Fig. S3. Briefly, the entire digest was injected onto a reversed phase column (C18), and the resulting fractions were analyzed by mass spectrometry. Peptide peaks in the mass spectra were sequenced by MS/MS analysis. A chromatograph of each extract ion was used to quantify the intensity of each peptide. Based on the MS/MS experiments, 86% of the amino acid sequence of β protein was covered, including 10 of the 14 lysine residues (Fig. 2). Incomplete coverage arises primarily because some tryptic peptides are too small to be detected. Biotin modification alters the observed pattern of peptides in several ways. Whereas trypsin normally cleaves peptide bonds after lysine or arginine residues, it does not cleave after lysine residues that are biotinylated. This results in a reduction in the total number of cleavage sites, a dramatic reduction in intensity of peaks for peptides that begin after or end in a modified lysine, and the appearance of peaks for larger peptides with an attached biotin group. Thus, biotinylation of a particular lysine residue can be observed by decreased detection of a peptide ending in or immediately after that lysine residue and/or appearance of a new peak corresponding to a larger peptide that is biotinylated.
FIGURE 2. Sequence of β protein summarizing the limited proteolysis and biotin modification data.
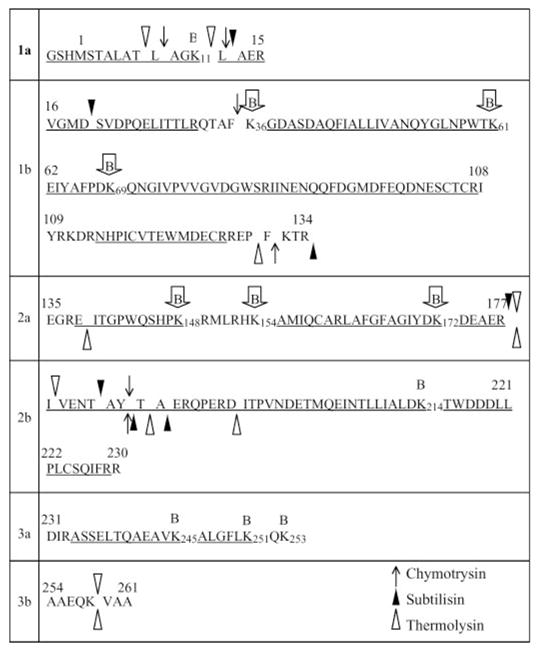
The sequence is divided into segments based on limited trypsin digestion. Region 1 is a stable N-terminal core domain that is resistant to trypsin in the presence and absence of DNA and is divided into 1a and 1b based on cleavage at Lys-15 to generate band D in Fig. 1A. Domain 2 becomes resistant to trypsin in the DNA complex and is divided into 2a and 2b based on cleavage at Arg-177 to generate band C. Domain 3 is the C-terminal “tail,” which is highly susceptible to trypsin both in the free protein and in the complex with DNA, and is further divided into 3a and 3b based on cleavage at Lys-253 to generate band A. For the other three proteases, the symbols above and below the sequence indicate the observed proteolytic cleavage sites in the presence and absence of DNA, respectively. Lysine residues that are observed to be biotinylated are indicated above the sequence with the B symbols, where those enclosed in arrows are protected from biotinylation in the DNA complex. Underlined portions of the sequence indicate the regions covered by the MS/MS analysis. The MS analysis of biotinylated protein did not provide information for Lys-111, Lys-132, Lys-253, and Lys-258.
Fig. 3 shows how this experiment reveals which lysine residues of the protein are protected from biotinylation in the DNA complex. A portion of the mass spectrum for a selected region of the LC chromatograph is shown for β protein alone, β protein with NHS-biotin, and β protein-DNA complex with NHS-biotin. For β protein alone (Fig. 3A), ), a strong peak is observed at m/z 601, which corresponds to a peptide of residues 162–172, generated by trypsin cleavage at Arg-161 and Lys-172. For biotinylated β protein (Fig. 3B), ), this peak essentially disappears due to modification of Lys-172, and a new peak appears at m/z 677 for a peptide corresponding to residues 162–177 with biotin attached to Lys-172. For β protein biotinylated in the presence of DNA, the m/z 601 peak reappears, and the m/z 677 peak is reduced, indicating that the DNA protects Lys-172 from biotinylation. This region of the spectrum also shows peaks for two peptides that contain lysine residues that are not protected in the DNA complex. These are m/z 648 for residues 246–251, generated from cleavage at Lys-245 and Lys-251, and m/z 673 for residues 1–11, generated from cleavage at Lys-11. These peaks are essentially absent in biotinylated samples of both β protein and β protein-DNA complex, indicating that Lys-11, Lys-245, and Lys-251 are not protected by the DNA. Also seen in this region of the spectrum are peaks corresponding to two control peptides, C1 (residues 16–31) and C2 (residues 114–127). Because these peptides are each generated from trypsin cleavage at two arginine residues, their intensities are not affected by biotinylation and, therefore, remain constant in the three samples.
FIGURE 3. Use of mass spectrometry to detect biotinylation of lysine residues and the protection from biotinylation upon DNA complex formation.
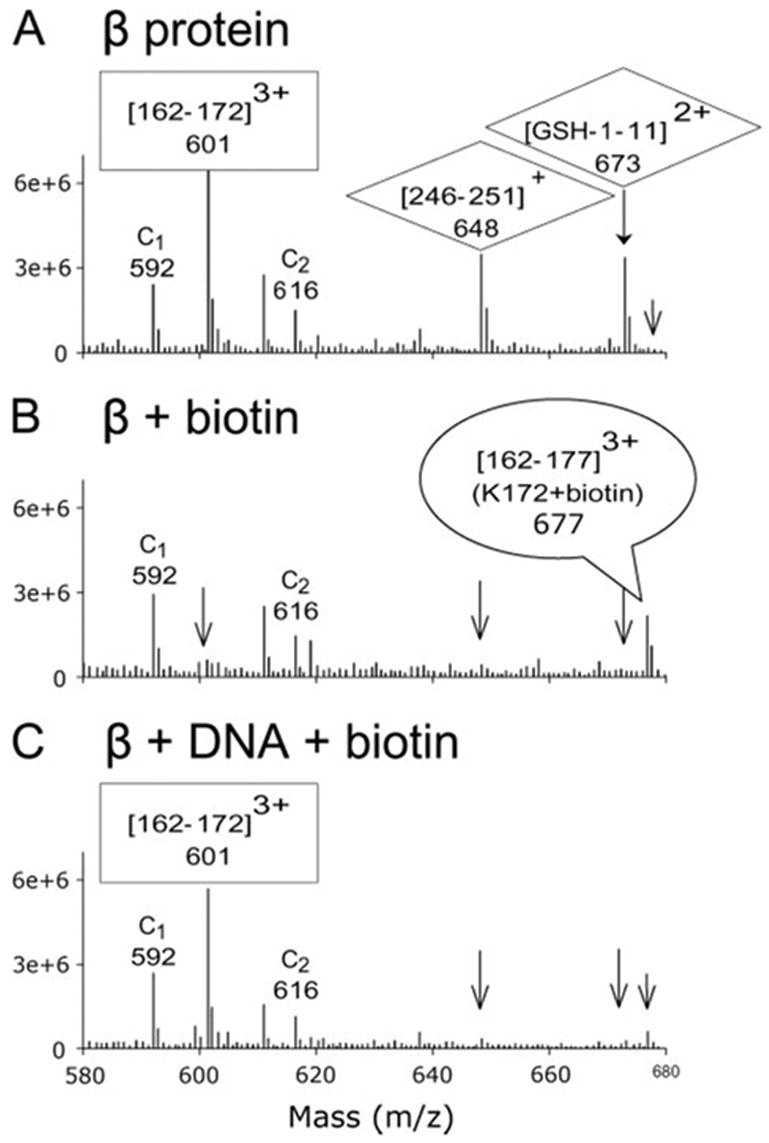
Each panel shows an average of a selected region of the spectra taken during the 30–40-min elution times during the LC-MS analysis. The spectra are for β protein alone (A), β protein modified by NHS-biotin (B), and β protein-DNA complex modified by NHS-biotin (C). The peak at m/z 601 in panel A, which corresponds to residues 162–172 of β protein, disappears after biotinylation in panel B since modification of Lys-172 prevents the trypsin digestion necessary to generate this peptide. In addition, a peak at m/z 677 for the triply charged peak of fragment 162–177 with biotin attached to Lys-172 appears upon biotin modification in panel B. For the DNA complex in panel C, the peak at m/z 601 returns, and the peak at m/z 677 is reduced, indicating that DNA binding protects Lys-172 from biotin modification. By the same type of reasoning, Lys-245, Lys-251, and Lys-11 are not protected from biotin modification by DNA binding, since the peaks at m/z 648 and 673 are reduced upon biotinylation of both β protein (panel B) and β protein-DNA complex (panel C). Also seen in the spectra are peaks for two control peptides, C1 at m/z 592 ((16–31)3+, and C2 at m/z 616 ((104–127)3+), which are generated by trypsin cleavage at two arginine residues. Because their MS responses are not affected by biotinylation of lysine residues, they can serve as internal controls for quantitative purposes.
In this way information on the extent of protection from biotinylation of 10 out of 14 lysine residues in the β protein-DNA complex was obtained (Table 1). For quantitatively comparing the extent of protection of the lysine residues, the intensities for the peaks corresponding to unmodified peptides (for example m/z 601, 648, and 673 of Fig. 3) were found to be the most useful. The intensity for each peptide was determined from the peak area in the extract ion chromatograph (supplemental Fig. S3, E–F), and intensities were normalized to that of m/z 888 for residues 16–31, which is generated from cleavage at Arg-15 and Arg-31 and, therefore, not affected by biotinylation. In Table 1, two different intensity ratios for nine peptides are reported. Each value in the table is based on the average of five independent measurements and is reported along with its S.D. β/(β + biotin) is the peak intensity ratio for peptides from samples of β protein alone and β protein with NHS-biotin. A high number indicates a high degree of biotinylation on the corresponding lysine residue of that peptide, whereas a value of one would indicate no biotinylation. The variation in the value for this ratio among the nine peptides presumably reflects the differences in accessibilities of each of the lysine residues to the NHS-biotin reagent in the free β protein. (β + DNA)/(β + DNA + biotin) is the peak intensity ratio for peptides from samples of β protein-DNA complex and β protein-DNA complex with NHS-biotin. A high value for this ratio indicates that the corresponding lysine residue(s) of that peptide is biotinylated and is, therefore, not protected by the DNA in the complex, whereas a low value indicates that the particular lysine residue is protected by DNA. Using a ratio of 1.3 as a cutoff, these data indicate that lysine residues within the central region of β protein (Lys-36, -61, -69, -148, -154, -172) are protected by DNA, whereas lysine residues at the N- and C-terminal regions of β protein (Lys-11, -214, -245, and -251) are not. The data do not provide any information for Lys-111, -132, -253, and -258.
TABLE 1. Quantitative analysis of the protection of lysine residues of β protein from biotinylation upon DNA complex formation.
The values in the table indicate the ratio of intensities for each lysine-containing peptide before and after biotinylation of the free β protein (β/(β + biotin)) or the β protein-DNA complex ((β + DNA)/(β + DNA + biotin)). Each value in the table is derived from integration of peaks in the extraction chromatograph and is the average of five separate experiments. A high value indicates that the corresponding lysine residue is biotinylated.
| Fragments | Modified K | β/(β+biotin) | (β+DNA)/(β+ DNA + biotin) | Protection |
|---|---|---|---|---|
| 1–11 | 11 | 21.4 ± 4.2 | 17.4 ± 3.9 | − |
| 37–61 | 36, 61 | 11.4 ± 3.1 | 1.1 ± 0.1 | + |
| 62–69 | 69 | 5.0 ± 2.5 | 1.2 ± 0.3 | + |
| 138–148 | 148 | 2.0 ± 0.8 | 1.3 ± 0.3 | + |
| 155–161 | 154 | 12.4 ± 2.9 | 1.2 ± 0.2 | + |
| 162–172 | 172 | 21.4 ± 3.8 | 1.3 ± 0.3 | + |
| 215–229 | 214 | 5.4 ± 1.7 | 5.3 ± 2.8 | − |
| 234–245 | 245 | 96.0 ± 20.5 | 68.3 ± 9.9 | − |
| 246–251 | 251 | 67.6 ± 15.3 | 103.3 ± 30.4 | − |
To summarize how this experiment provides information on the environment of lysine residues in the complex, Fig. 4 shows the typical MS response changes of three selected peptides that end in lysine. When the lysine is not protected by DNA, as for the peptide shown in Fig. 4A, the corresponding intensity decreases dramatically upon biotinylation both in the absence and presence of DNA. When the lysine residue is protected by DNA, as for the peptide shown in Fig. 4B, the corresponding intensity decreases dramatically upon biotinylation in the absence of DNA but not in the presence of DNA. For the peptide shown in Fig. 4C, the decrease in intensity upon biotinylation is less dramatic, indicating that its lysine residue (Lys-148) is less accessible to NHS-biotin in the free protein. However, DNA still protects Lys-148 from biotinylation, since the intensity for this peptide increases in the presence of DNA. Examination of this peptide at higher NHS-biotin levels provides more convincing evidence that Lys-148 is indeed protected by DNA, since higher levels of biotin results in its increased modification in the free protein but not in the protein-DNA complex.
FIGURE 4. Comparison of peak areas of three peptide fragments after biotinylation in the presence and absence of DNA.
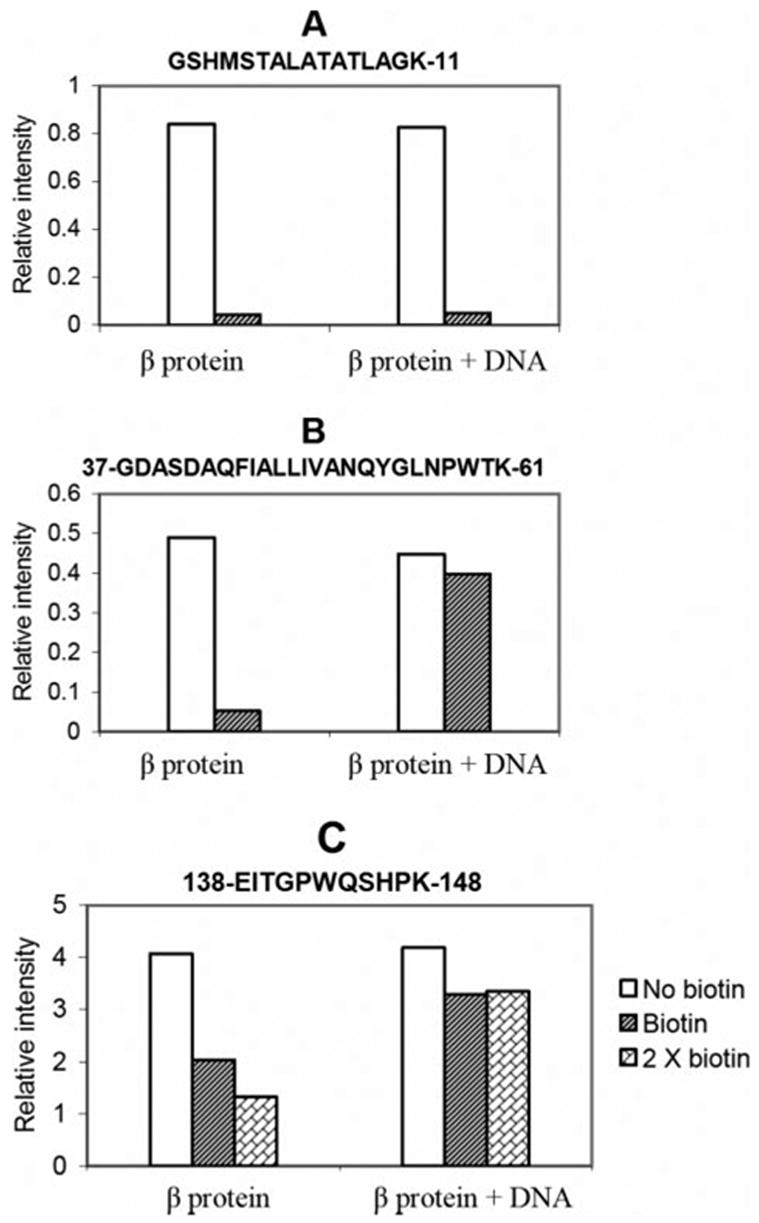
The value in the graph is the integrated peak intensity from the extract ion chromatograph of an ion corresponding to the indicated peptide. A, Lys-11 is not protected from biotinylation in the DNA complex, since the peak intensity for this peptide decreases upon biotinylation of free β protein and β protein DNA complex. B, Lys-61 and Lys-36 are protected from biotinylation in the DNA complex, since the peak intensity for this peptide decreases upon biotinylation of free β protein but much less so when the β protein-DNA complex is biotinylated. C, Lys-148 is protected from biotinylation in the DNA complex. The signal for peptide 138–148 is only partially reduced upon biotinylation, indicating low surface accessibility of Lys-148 in the free protein. The signal for this peptide is less reduced upon biotinylation of the DNA complex, indicating Lys-148 is partially protected by DNA. Because the intensity changes for this peptide are less dramatic than those in panels A and B, the analysis was repeated with a 2× dose of NHS-biotin, which results in increased modification of Lys-148 in the free protein but not in the protein-DNA complex.
DNA Binding of Fragments of β Protein Containing Lysine to Alanine Mutations
Based on the limited proteolysis and biotin protection studies, two N-terminal fragments of β protein corresponding to residues 1–177 and 1–188 were cloned and purified. Both fragments bind to sequentially added oligonucleotides at least as well as full-length β protein (Fig. 5A). Interestingly, based on a nitrocellulose filter binding assay, the β1–177 fragment binds to ssDNA (the 33+ oligonucleotide) considerably more tightly than full-length β-protein, which binds very weakly (Fig. 5B). ). The oligomeric state of the β1–177 fragment in the absence of DNA appears similar to that of full-length β protein, based on its elution as a distinct peak from a Superdex S-200 column at a volume corresponding to a mass of 333 kDa or ~17 subunits (data not shown).
FIGURE 5. DNA binding of N-terminal fragments of β protein containing lysine to alanine mutations.
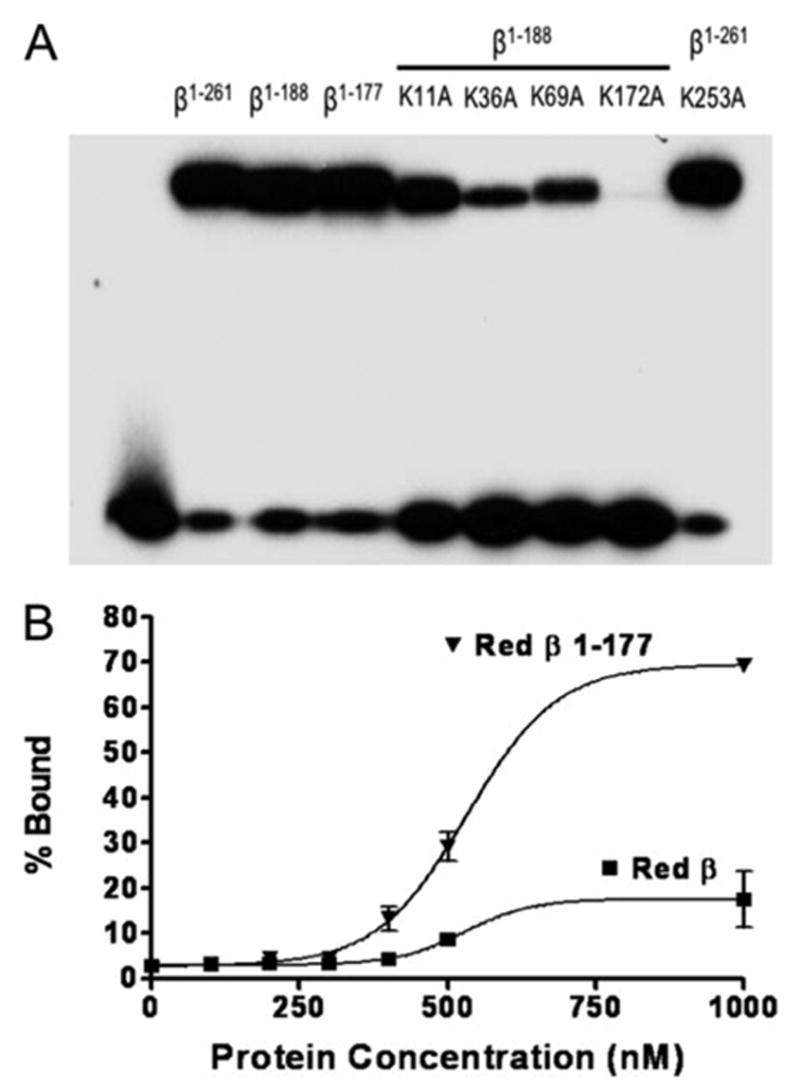
A, the binding of full-length or N-terminal fragments of β protein to sequentially added complementary 33-mer oligonucleotides was carried out as described in “Experimental Procedures.” The band at the top of the gel shows each protein in complex with the 33-mer duplex product of annealing, whereas the band at the bottom corresponds to unbound 33-mer duplex. Notice that the two fragments of β protein (β1–188 and β1–177) bind to DNA about as well as full-length β protein (residues 1–261) and that the lysine to alanine mutants of β1–188 bind less well, with the degree of impairment following the trend K172A > K36A > K69A > K11A. The K253A mutation within full-length β protein (at the far right) does not significantly impair DNA binding. B, binding of full-length β protein and β1–177 fragment to a single 33-mer ssDNA oligonucleotide (33+) was measured using a nitrocellulose filter binding assay (22), as described under “Experimental Procedures.” Notice that the β1–177 fragment binds to the 33+ oligonucleotide with considerably higher stability than full-length β protein.
To further investigate the importance of the lysine residues implicated by biotinylation protection, four lysine to alanine substitutions were introduced into the 1–188 fragment (K11A, K36A, K69A, and K172A), and the resulting proteins were purified and characterized for DNA binding (Fig. 5A, fifth through eighth lanes). In addition, the K253A substitution was introduced into full-length β protein as a control, since no protection of Lys-253 was observed upon DNA binding. As expected, the K253A mutation does not disrupt DNA binding (Fig. 5A, right lane). All of the lysine to alanine residues within the N-terminal 188 residues disrupt DNA binding to some extent, with the severity in the order of K172A > K36A > K69A > K11A. This is in general agreement with the biotinylation protection, since Lys-11 was not significantly protected from biotinylation in the DNA complex, and its mutation to alanine has a smaller effect on DNA binding than the other three. The observation that a particular lysine residue is protected from biotinylation in the DNA complex would indicate it is near the protein-DNA interface but not necessarily important energetically for the stability of the protein DNA interaction. This is reflected in the observation that the K172A mutation has a much more dramatic effect on DNA binding than the K69A mutation despite the fact that the two are protected from biotinylation upon DNA binding to similar degrees.
DISCUSSION
All together, the limited proteolysis and biotinylation protection studies suggest that β protein consists of essentially three regions; an N-terminal core domain (residues 1–130) that is protease-resistant in the absence of DNA, a “central” region (residues 131–177) that has enhanced protease resistance upon DNA binding, and a C-terminal region (residues 178–261) that is susceptible to proteases in the presence and absence of DNA. In the absence of DNA, the central region is apparently folded, since after subtilisin digestion a significant amount of the fragment consisting of residues 1–184 persists. It is, thus, possible that residues 1–184 form a single domain and that DNA stabilizes a surface loop formed by residues 131–138. The fact that a stable fragment corresponding to the first ~130 residues is observed for all of the proteases in the absence of DNA tends to indicate that it corresponds to a distinct folding domain, with the central region, residues 131–177, likely forming a separate module. Interestingly, the proteolysis studies suggest that the N-terminal ~30 residues of β-protein become more flexible upon DNA binding, as all four proteases cleave within this region in the DNA complex but not in the free protein (Figs. 1B and 2 and supplemental Fig. S2).
It is possible that there is a folded region within the C-terminal portion of β protein (residues 178–261) and that the absence of these residues in most of the observed proteolytic fragments is due to the high protease susceptibility of residues 178–193 in both the presence and absence of DNA. Consistent with this notion, a trypsin fragment corresponding to residues 1–230 of β protein is stabilized by DNA binding, and Lys-214 has low accessibility to biotinylation in the absence of DNA (Table 1). By contrast, residues at the very C-terminal end of β protein (Lys-245 and Lys-251) have dramatically higher accessibility to biotinylation.
The lysine residues of β protein that are protected from biotin modification in the DNA complex generally map to the regions protected from protease digestion in the presence of DNA. Thus, the results from limited proteolysis and biotin modification are very much consistent with one another and together point to the N-terminal residues 1–177 of β protein as forming the core DNA binding region. In agreement with this, the purified fragment of residues 1–177 binds to sequentially added oligonucleotides at least as well as full-length β protein, indicating that the C-terminal residues 178–261 are apparently dispensable for DNA binding. Moreover, the β1–177 fragment binds to the 33+ oligonucleotide considerably better than full-length β protein (Fig. 5B), suggesting a role for the C-terminal region of β protein in tempering its affinity for ssDNA. In future studies it will be interesting to compare this fragment with full-length β protein in a more complete functional analysis.
The enhanced protease resistance and biotinylation protection upon DNA complex formation implicate certain residues of β protein, including the protected lysines and residues 131–138 between the N-terminal core and central regions, as being directly involved in DNA binding. However, their enhanced protection in the presence of DNA could also be explained by a conformational change, such as oligomerization, that leads to a decrease in their exposure. Indeed, based on what we know from electron microscopy studies (18) as well as gel filtration and native gel analyses, it is likely that upon formation of the complex with sequentially added complementary oligonucleotides β protein undergoes a transition from an oligomeric ring to a helical filament. Thus, it is possible that for some of the sites, the enhanced protection observed in the presence of DNA results from new inter-subunit contacts that are formed as opposed to direct contacts with the DNA. We consider this possibility less likely for the sites cleaved by trypsin and protected from biotinylation, which involve positively charged residues that are more likely to be involved in binding to DNA than located at a protein-protein interface. The interaction of one or more of the protected lysine residues with DNA is consistent with the observation that binding of β protein to ssDNA is weakened at high salt (15).
It is interesting that nearly all of the lysine residues within the first 177 residues of β protein (except Lys-11 and Lys-132) exhibit protection from biotin modification in the DNA complex. One might expect the protein to have several lysine residues dues at its surface, with only a fraction located at the interface with DNA. Without knowledge of the three-dimensional structure of the protein, there is no information concerning how near the lysine residues are to one another in the protein structure. None of the lysine residues in β protein is highly conserved in the family of related proteins (10), although the sequences are highly diverged. The DNA binding properties of the five lysine to alanine mutants that were prepared demonstrate that, indeed, only a subset of these lysine residues is energetically important for DNA complex formation.
In the biotinylation experiments we focused on the complex of β protein with sequentially added oligonucleotides, since for full-length β protein that complex is far more stable than the complex with ssDNA. At present we cannot distinguish if a particular lysine residue might be involved in binding to the initial oligonucleotide or to the subsequently added complementary oligonucleotide. In future studies it will be interesting to examine the multiple oligomeric states of β protein (rings and filaments) to see if distinct roles of particular lysine residues can be dissected. Toward this end, the β1–177 fragment, which forms a tighter complex with a single oligonucleotide (Fig. 5B), ), may prove useful.
In a previous study, a stable ~20-kDa N-terminal fragment of β protein produced by limited digestion with chymotrypsin was identified (15). This fragment would correspond most closely with fragment A of supplemental Fig. 2B (residues 1–184; Mr 21,319). In this earlier study essentially the same digestion patterns were observed for β protein alone and in complex with a 36-mer oligonucleotide. The ~20-kDa N-terminal fragment was photochemically cross-linked to the oligonucleotide, nucleotide, providing direct evidence that it contains the DNA binding regions. The results of the present study are essentially in agreement with this earlier study. Here we have more precisely defined the domain boundaries by N-terminal sequencing and mass spectrometry and have used four different proteases. Moreover, using the complex with sequentially added oligonucleotides, we have identified regions of β protein for which the susceptibility to protease digestion is altered upon DNA binding.
Based on sequence alignment (10), residues 19 –205 of β protein form a region that is conserved in a family of related proteins including E. coli RecT, which has also been well characterized biochemically. This conserved region of β protein closely overlaps with the region that is protease-resistant. Some protection is seen for the additional residues 189 –230 in the trypsin digest, which extend beyond the conserved served region. A secondary structure prediction for this family results in an ααββαβββα pattern, with the first helix beginning at Gln-24 and the last helix ending in Arg-161. Two highly conserved acidic residues, Asp-173 and Glu-174 of β protein, are located near the C-terminal end of the protected region. It was hypothesized that these conserved acidic residues might be involved in binding Mg2+ ions (10), which modulate the oligomerization and DNA binding activity of β protein (5, 15, 18). Interestingly, however, the limited trypsin digestion of both free β protein and the β protein-DNA complex showed no differences in the presence and absence of Mg Mg2+, suggesting that Mg Mg2+ does not cause any major structural changes in the protein.
The possibility that β protein has a disordered C-terminal tail is interesting in light of the fact that several other single-stranded stranded DNA-binding proteins have similar disordered regions at their C termini. These include E. coli RecA (24) and single-stranded DNA-binding protein (25), T4 gene 32 protein (26), and T7 gene 2.5 protein (27). For many of these proteins, the C-terminal tail has a preponderance of negatively charged residues, and removal of the tail enhances the DNA binding activity of the protein (25–30). The disordered negatively charged tails of these proteins could electrostatically shield the DNA, preventing nonspecific binding modes. In the case of E. coli RecA protein, the C-terminal tail (residues 329 –352) is disordered in crystal structures and contains seven acidic, no basic, and six hydroxyl-containing residues. When the tail is removed, the protein has an increased rate of binding to dsDNA and displaces single-stranded DNA-binding protein from ssDNA more efficiently (29 –30). In the case of β protein, the C-terminal region (residues 178 –261) has a net charge of −5 (15 acidic and 10 basic residues), and removing it appears to significantly enhance the binding of the resulting N-terminal fragment to ssDNA.
Two previous studies have combined modification of lysine residues with NHS-biotin and mass spectrometry to map out the DNA binding regions of a protein. This study for β protein is unique in that there is no structure of β protein to provide a three-dimensional context for the lysine residues of interest. In the cases of human immunodeficiency virus-1 reverse transcriptase and replication protein A, the information from this technique was easily reconciled with the three-dimensional structures, providing strong validation for the method. In our case, the results from this technique are a prediction for which further validation will have to await structure determination.
Supplementary Material
The abbreviations used are
- dsDNA
double-stranded DNA
- ssDNA
single-stranded DNA
- DTT
dithiothreitol
- HPLC
high performance liquid chromatography
- LC
liquid chromatography
- MS
mass spectroscopy
Footnotes
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
References
- 1.Poteete AR. FEMS Microbiol Lett. 2001;201:9–14. doi: 10.1111/j.1574-6968.2001.tb10725.x. [DOI] [PubMed] [Google Scholar]
- 2.Kuzminov A. Annu Rev Genet. 1999;28:49 –70. [Google Scholar]
- 3.Little JW. J Biol Chem. 1967;242:679 –686. [PubMed] [Google Scholar]
- 4.Carter DM, Radding CM. J Biol Chem. 1971;246:2502–2512. [PubMed] [Google Scholar]
- 5.Kmiec E, Holloman WK. J Biol Chem. 1981;256:12636 –12639. [PubMed] [Google Scholar]
- 6.Muniyappa K, Radding CM. J Biol Chem. 1986;261:7472–7478. [PubMed] [Google Scholar]
- 7.Marsic N, Roje S, Stojiljkovic I, Salaj-Smic E, Trgovcevic Z. J Bacteriol. 1993;175:4738 –4743. doi: 10.1128/jb.175.15.4738-4743.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kulkarni SK, Stahl FW. Genetics. 1989;123:249 –253. doi: 10.1093/genetics/123.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kolodner R, Hall SD, Luisi-Deluca C. Mol Microbiol. 1994;11:23–30. doi: 10.1111/j.1365-2958.1994.tb00286.x. [DOI] [PubMed] [Google Scholar]
- 10.Iyer LM, Koonin EV, Aravind L. BMC Genomics. 2002;3:8. doi: 10.1186/1471-2164-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aravind L, Makarova KS, Koonin EV. Nucleic Acids Res. 2000;28:3417–3432. doi: 10.1093/nar/28.18.3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Copeland NG, Jenkins NA, Court DL. Nat Rev Genet. 2001;2:769 –779. doi: 10.1038/35093556. [DOI] [PubMed] [Google Scholar]
- 13.Muyrers JPP, Zhang Y, Stewart AF. Trends Biochem Sci. 2001;26:325–331. doi: 10.1016/s0968-0004(00)01757-6. [DOI] [PubMed] [Google Scholar]
- 14.Karakousis G, Ye N, Li Z, Chiu SK, Reddy G, Radding CM. J Mol Biol. 1998;276:721–731. doi: 10.1006/jmbi.1997.1573. [DOI] [PubMed] [Google Scholar]
- 15.Mythili E, Kumar KA, Muniyappa K. Gene. 1996;182:81–87. doi: 10.1016/s0378-1119(96)00518-5. [DOI] [PubMed] [Google Scholar]
- 16.Li Z, Karakousis G, Chiu SK, Reddy G, Radding CM. J Mol Biol. 1998;276:733–744. doi: 10.1006/jmbi.1997.1572. [DOI] [PubMed] [Google Scholar]
- 17.Rybalchenko N, Golub EI, Baoyuan B, Radding CM. Proc Natl Acad Sci U S A. 2004;101:17056–17060. doi: 10.1073/pnas.0408046101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Passy SI, Yu X, Li Z, Radding CM, Egelman EH. Proc Natl Acad Sci U S A. 1999;96:4279–4284. doi: 10.1073/pnas.96.8.4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shell SM, Hess S, Kvaratskhelia M, Zou Y. Biochemistry. 2005;44:971–978. doi: 10.1021/bi048208a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kvaratskhelia M, Miller JT, Budihas SR, Pannell LK, Le Grice SF. Proc Natl Acad Sci U S A. 2002;99:15988–15993. doi: 10.1073/pnas.252550199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gill SC, von Hippel PH. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 22.Wong I, Lohman TM. Proc Natl Acad Sci U S A. 1993;90:5428–5432. doi: 10.1073/pnas.90.12.5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stone KL, Williams KR. In: The Protein Protocols Handbook. Walker JM, editor. Humana Press Inc; Totowa, NJ: 2002. pp. 511–521. [Google Scholar]
- 24.Story RM, Weber IT, Steitz TA. Nature. 1992;355:318–325. doi: 10.1038/355318a0. [DOI] [PubMed] [Google Scholar]
- 25.Williams KR, Spicer EK, LoPresti MB, Guggenheimer RA, Chase JW. J Biol Chem. 1983;258:3346–3355. [PubMed] [Google Scholar]
- 26.Williams KR, Konigsberg W. J Biol Chem. 1978;253:2463–2470. [PubMed] [Google Scholar]
- 27.Hollis T, Stattel JM, Walther DS, Richardson CC, Ellenberger T. Proc Natl Acad Sci U S A. 2001;98:9557–9562. doi: 10.1073/pnas.171317698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lonberg N, Kowalczykowski S, Paul L, von Hippel P. J Mol Biol. 1981;145:123–138. doi: 10.1016/0022-2836(81)90337-5. [DOI] [PubMed] [Google Scholar]
- 29.Tateishi S, Horii T, Ogawa T, Ogawa H. J Mol Biol. 1992;223:115–129. doi: 10.1016/0022-2836(92)90720-5. [DOI] [PubMed] [Google Scholar]
- 30.Eggler AL, Lusetti SL, Cox MM. J Biol Chem. 2003;278:16389–16396. doi: 10.1074/jbc.M212920200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.