

Abstract
Carriers of FMR1 alleles with 55-200 repeats in the 5' UTR are at risk for Fragile X associated tremor and ataxia syndrome. The cause of the neuropathology is unknown but is thought to be RNA-mediated. Maternally transmitted premutation alleles are also at risk of expansion of the repeat tract into the “full mutation” range (>200 repeats). The mechanism responsible for expansion is unknown. Full mutation alleles produce reduced amounts of the FMR1 gene product, FMRP, which leads to Fragile X mental retardation syndrome. We have developed a murine model for Fragile X premutation carriers that recapitulates key features seen in humans including a direct relationship between repeat number and Fmr1 mRNA levels, an inverse relationship with FMRP levels and Purkinje cell dropout that have not been seen in a previously described knock-in mouse model. In addition, these mice also show a differential deficit of FMRP in different parts of the brain that might account for symptoms of the full mutation that are seen in premutation carriers. As in humans, repeat instability is high with expansions predominating and, for the first time in a mouse model, large expansions into the full mutation range are seen that occur within a single generation. Thus, contrary to what was previously thought, mice may be good models not only for the symptoms seen in human carriers of FMR1 premutation alleles but also for understanding the mechanism responsible for repeat expansion, a phenomenon that is responsible for a number of neurological and neurodevelopmental disorders.
Keywords: repeat expansion, DNA instability, full mutation
1. Introduction
The 5′ UTR of the human Fragile X mental retardation 1 (FMR1) gene contains a CGG•CCG-repeat tract whose size varies from 5 to >2000 repeats. Individuals with 55-200 repeats are said to be carriers of Fragile X premutation alleles. They are at risk for Fragile X associated tremor and ataxia syndrome (FXTAS) (Hagerman and Hagerman, 2004) and premature ovarian failure (Sherman, 2000). Premutation alleles produce elevated levels of FMR1 mRNA and this RNA is thought to have toxic effects (Jin et al., 2003; Handa et al., 2005). Premutation alleles are also prone to expand, with female carriers being at risk of having children with alleles with >200 repeats. These carriers of so-called full mutation alleles are likely to have Fragile X mental retardation syndrome (FXS) (Verkerk et al., 1991; Yu et al., 1991). The symptoms of FXS result from a deficiency of the FMR1 gene product, Fragile X mental retardation protein (FMRP), which arises due to a combination of repeat-induced promoter silencing and difficulties in translating mRNA with large numbers of repeats (Feng et al., 1995). There is some overlap of symptoms in premutation and full mutation carriers probably because the negative effect of the repeat on translation is apparent even in the premutation range and results in a significant decrease in the level of FMRP (Kenneson et al., 2001; Primerano et al., 2002; Tassone et al., 2004).
The mechanism of expansion is not known and large expansions from a premutation sized allele to an allele in the full mutation size range have not been previously described in mice. This has led to the idea that expansions do not occur in these animals and thus that they are not good models for studying the expansion mechanism. We describe here Knock-in (KI) mice we have generated that share key features of human premutation carriers not seen in a previous KI mouse model (Bontekoe et al., 2001; Willemsen et al., 2003). These animals also provide the first examples in mice of large repeat expansions that transform a premutation sized allele into a full mutation sized allele in a single generation.
2. Materials and Methods
2.1 Generation of the Fragile X premutation KI mice
Mice were maintained in accordance with the guidelines of the NIDDK Animal Care and Use Committee and with the Guide for the Care and Use of Laboratory Animals (NIH publication no. 85-23, revised 1996). The outline of the strategy used to generate the targeting vector is shown in Fig. 1 and described in more detail in the figure legend. Briefly, the mouse Fmr1 gene was identified in a BAC clone (BAC 16411) derived from 129/SvJ mouse embryonic stem (ES) cells (Genome Systems, Inc, St. Louis, MO, USA). A targeting construct was generated as shown in Fig. 1 using 38LoxP (Genome Systems, Inc) as the vector backbone. This vector has a neomycin (Nm) resistance gene flanked by loxP sites situated between 2 multiple cloning sites. The repeats present in exon 1 of Fmr1 were replaced with 2 different Sfi I sites. The repeat tract was generated in vitro using a modification of a serial ligation strategy we had previously developed (Grabczyk and Usdin, 1999), which enabled us to generate long repeat tracts of defined length from shorter, more stable, repeats. The repeats were flanked by user-defined restriction sites, in this case Sfi I sites compatible with those inserted into exon 1. The repeat tract was ligated into the Sfi I digested targeting construct and electroporated into 129S6/SvEv derived TC1 ES cells (Deng et al., 1996). Mouse blastocysts were injected with ES cells containing a properly targeted insertion using standard procedures. These mice and all subsequent generations of these mice were bred to C57/BL6 animals. Crossing these mice to mice expressing the Cre recombinase in the germline allowed removal of the Nm gene (Wagner et al., 2001). No discernable difference was found between Nm+ and Nm− mice in any of the measures we tested.
Fig. 1. Outline of targeting vector construction strategy.

The Kpn I-Pst I fragment that includes exon 1 of the WT murine Fmr1 gene was modified so as to replace the normal mouse CGG•CCG-repeats with 2 different Sfi I sites. This modified fragment was substituted for the WT Kpn I-Pst I fragment in a Bam HI-Sst I subclone of the 5' end of the Fmr1 gene. After the insertion of the resultant modified 5' arm (grey rectangle) and the 3' arm (black rectangle) into the 38LoxP cloning vector (white rectangle), the construct was digested with Sfi I and ligated to the CGG•CCG-repeat fragment that was generated as previously described (Lavedan et. al., 1998) and was flanked by Sfi I sites compatible with those introduced into exon 1. Restriction enzyme sites that are abolished on cloning are shown in parentheses. Sites introduced into the targeting construct from vector derived E. coli sequences are shown in grey font.
2.2. Determination of repeat size
The size of the CGG•CCG-repeat tract was monitored by Polymerase chain reaction (PCR) as previously described (Lavedan et al., 1998) using the primers frax-m4 (5'-CTTGAGGCCCAGCCGCCGTCGGCC-3') and frax-m5 (5'-CGGGGGGCGTGCGGTAACGGCCCAA-3'). The binding sites for these primers are located immediately adjacent to the repeat tract and their 3' ends are unique to the KI allele. The primer pair, frax-c and frax-f (Fu et al., 1991), was used to detect both wildtype (WT) and KI alleles. The resultant PCR products were analyzed on a 5% polyacrylamide sequencing gels and confirmed where necessary by Southern blotting. PCR across long repeats typically produces multiple bands and the size of each allele was calculated based on the mobility of the central band in the cluster. Statistical analysis of instability was carried out using the Chi square and Student's T tests.
2.3. Methylation status of the expanded repeats
Tail DNA from 3 week old male and female mice with repeat numbers was digested with Sau96 I (New England Biolabs, Ipswich, MA, USA), a methylation-sensitive restriction enzyme with a recognition site within the upstream primer. The digested material was then amplified by PCR as described above. An aliquot of pGL3-Basic (Promega, Madison, WI, USA), which has 8 Sau96 I sites, was added prior to Sau96 I digestion as a control for digestion and for normalizing for material lost during processing.
2.4. Fmr1 mRNA measurements
Brains from 2 month old male mice with different repeat numbers were placed in RNALater (Ambion, Austin, TX, USA). Total RNA from these brains was prepared using Trizol as per the supplier's instructions (Invitrogen, Carlsbad, CA, USA). RNA quality was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Carlsbad, CA, USA). Reverse transcription was performed using SuperScript III (Invitrogen) as per the supplier's instructions. Quantitative PCR reactions were performed using the 7900 HT Fast real Time PCR System and TaqMan Universal PCR Master Mix (Applied Biosystems, Foster City, CA, USA) and the following mouse Fmr1 primer/probe set: Mouse Fmr1-F: 5'-GACAGATTCCATTCCATGATGTGA-3'; Mouse Fmr1-R; 5'-ACCACCAACAGCAAGGCTCT-3'; and the TaqMan probe, 5'-(FAM)–TGATGAAGTTGAGGTGTATTCCAGAGCAAATGA-(TAMRA)-3'. Fmr1 mRNA levels were assessed by comparison with 18S rRNA using a probe set supplied by Applied Biosystems. Reactions were performed in triplicate as previously described (Tassone et al., 2000b).
2.5. Western Blotting
Proteins were made from the brains of 2 month old male mice with different repeat numbers, resolved by SDS-PAGE and Western Blotted using standard procedures. FMRP was detected using a mouse monoclonal antibody to FMRP, 7G1-1 ((Brown et al., 2001) and an ECL™ kit as per the manufacturer's instructions (Amersham Biosciences, Piscataway, NJ, USA). Protein from the brains of Fmr1 knock-out animals (Dutch-Belgian Fragile X Consortium, 1994) was used as negative control. Quantitation was carried out by densitometric analysis of the Western blot using Quantity One® 1-D analysis software (Bio-Rad, Hercules, CA, USA) using Actin as a loading control.
2.6. Immunocytochemistry
Four WT males and 5 KI males with 120 repeats were anesthetized at ∼100 weeks of age and perfused as previously described (Hoffman et al., 1992). Immunocytochemistry was performed using standard protocols and rabbit anti-Ubiquitin (Dako, Carpenteria, CA, USA), anti-FMRP (Abcam, Cambridge, UK), anti-Calbindin and anti-Lamin C (Chemicon), and mouse anti-FMRP (Chemicon, Temecula, CA, US). After staining, slides were coded and analyzed by an individual blind to the genotype of the animal. The integrated optical density of staining was measured and multivariant analysis of variance (MANOVA) for, genotype, region, and their interaction was performed using JMP software. Post hoc analyses determined statistical differences for each region, with p<0.05 considered significant.
3. Results and Discussion
3.1. Generation of Fragile X premutation mice
Unlike the previous fragile X premutation mouse model which was generated by replacement of a region including the endogenous murine repeat tract with one derived from a yeast artificial chromosome (YAC) containing a human premutation allele (Bontekoe et al., 2001), the repeat tract in the premutation mice described here were generated by serial ligation of short, stable CGG•CCG-repeat tracts as previously described (Grabczyk and Usdin, 1998). The premutation-sized repeat tract was added as the last step in the construction of the targeting vector as described in the Materials and Methods (Fig. 1). The targeting vector was then electroporated directly into ES cells without propagation in bacteria or yeast. This allowed us to avoid problems related to the propensity of long repeat tracts to undergo deletion and rearrangement in these organisms. The resultant premutation mice thus contain minimal differences from the WT murine Fmr1 gene in the region flanking the repeat. Southern blot analysis of Eco RI digested genomic DNA from the tails of the resultant mice using a 3.1 kilobases (kb) WT Eco RI fragment (shown at the bottom of Fig. 1) as a probe produced bands of 1.6 and 2.0 kb in males consistent with the predicted sizes shown in Fig. 1. Heterozygous females have, in addition to the WT allele and the 1.6 and 2.0 kb bands, an additional band of 4.5 kb. This extra band results from the fact that the upstream Eco RI site, indicated by the asterisk in Fig. 1, overlaps a CpG site and is thus not cut when the Fmr1 gene is on the inactive X chromosome. PCR using a primer pair that amplifies both the normal and KI allele shows a single band of ∼700 base pairs (bp) in males with the KI allele consistent with a repeat number of ∼120 (Fig. 2B). A more accurate estimate of the number of repeats was obtained by PCR using primers that have their 3' ends immediately adjacent to the repeat and electrophoresis on a high resolution denaturing gel. This analysis demonstrated that the repeat number in the founder mice was ∼118 (Fig. 2C).
Fig. 2. Characterization of premutation mice generated with the targeting construct shown in Fig. 1.
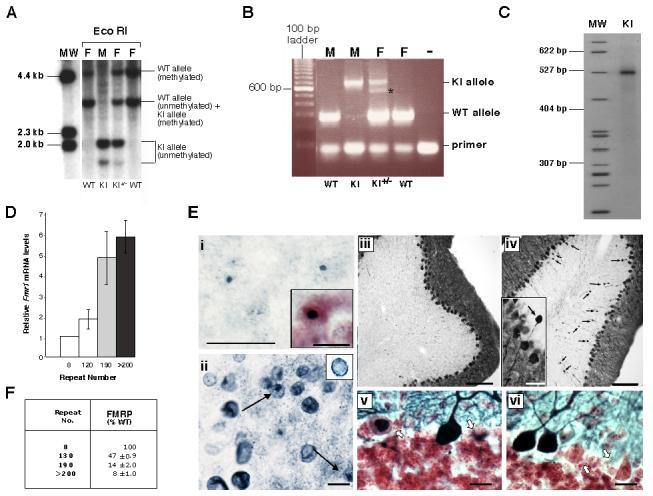
A) Southern blot analysis of Eco RI digested genomic DNA from the tails of WT and KI animals using a probe containing the Eco RI fragment that spans the CGG•CCG-repeat in WT mice (shown at the bottom of Fig. 1). MW, molecular weight marker; M, male; F, female. B) PCR genotyping of mice using frax-c and frax-f primers. Note that in reactions with DNA from females heterozygous for the KI allele, 2 bands are seen in the region expected for the KI allele. Only one band is seen on denaturing gels. The smaller of the 2 bands (indicated by the asterisk) results from a heteroduplex formed between the KI and WT alleles in the last round of the PCR. C) Determination of the repeat size in KI mice using primers frax-m4 and frax-m5. MW, molecular weight marker. D) Relative Fmr1 mRNA levels in 2 month old WT and KI male mice. The difference in RNA levels between WT and KI animals were significant at p<0.0001 by Students t-test. E) CNS pathology in male premutation mice with 120 repeats at ∼100 weeks of age. i) Ubiquitin immunoreactivity in the parafascicular nucleus of the thalamus. (Inset: a section from the reticular formation counterstained with Neutral Red showing a larger inclusion). ii) Lamin immunoreactivity in the nodulus of the cerebellum showing lamin-positive inclusions (arrows) and the irregular distribution of lamin (Inset: cell from WT mice showing the normal lamin distribution on the nuclear periphery) iii-vi) Calbindin immunoreactivity in the cerebellum. iii-iv) Calbindin staining in the cerebellum of WT (iii) and KI (iv) mice. Note that the KI mouse has increased numbers of torpedo axon swellings (arrows in iv). Some are almost as large as the Purkinje soma (iv, inset). v-vi) Cerebellum from KI mice stained for calbindin in blue/black and counterstained with Neutral Red showing Purkinje cells with abnormal calbindin distribution (arrows). In panels i, ii , v and vi the bars indicate 10 μm. The bars in panel iii and iv indicate 100 μm, and the bar in iv (inset) indicates 25 μm. F) Relative FMRP levels in 2 month old WT and KI male mice based on Western blots with FMRP 7G1-1 antibody and normalized using Actin antibodies as described in the text. The data presented represent the results of 3 independent experiments. The difference in protein levels in WT and KI animals were significant at p<0.0001 by Students t-test.
As will be demonstrated below, these mice share key features with human premutation carriers that were not seen in a previously described KI premutation mouse (Bontekoe et al., 2001; Willemsen et al., 2003; Brouwer et al., 2006). Why this should be is unclear since other than having an initial repeat tract 20 repeats shorter than ours, in principal the previously described mouse model differs from ours only in small details. Minor differences exist in regions of intron 1 resulting from the different targeting vectors that were used. In addition, the previous mouse model contains a human version of the region flanking the repeats that differs from the murine sequence by a just few bases (Bontekoe et al., 2001).
3.2. The KI mice have pathophysiological changes seen in human premutation carriers
Our KI premutation mice show a direct relationship between repeat number and Fmr1 mRNA levels (Fig. 2D) as reported for human premutation carriers (Tassone et al., 2000b). This is different for what has been reported in a previously described mouse KI model (Brouwer et al., 2006). Pathological changes are seen in our mice that are reminiscent of that seen in human carriers of FMR1 premutation alleles (Tassone et al., 2000b; Greco et al., 2002; Greco et al., 2006) including the presence of ubiquitin and lamin-positive neuronal intranuclear inclusions (Fig. 2E, i and ii). While ubiquitin-positive inclusions were reported in the earlier mouse model, no evidence of Purkinje cell degeneration like that seen in humans was seen in these animals (Willemsen et al., 2003). In contrast, the mice we have generated show evidence of Purkinje pathology including abnormal calbindin staining, swollen axons and torpedoes and Purkinje cell dropout (Fig. 2E, iv-vi).
In spite of the elevated level of Fmr1 mRNA in these mice, there is a negative relationship between the repeat number and the amount of FMRP in brain (Fig. 2F). A similar relationship over a similar repeat size range has been reported for cell lines obtained from human premutation carriers (Kenneson et al., 2001; Primerano et al., 2002; Tassone et al., 2004). Once again this differs from the previously described mouse model (Bontekoe et al., 2001; Willemsen et al., 2003; Brouwer et al., 2006). The FMRP deficiency in mice is not uniform in all regions in the central nervous system (CNS) (Fig. 3). For example, FMRP expression was only modestly reduced in the hippocampus of older male mice with 120 repeats (see Fig. 3I and compare Fig3A with 3B). In contrast, in the thalamus and inferior olivary complex, FMRP expression was only about 10% that seen in WT animals (see Fig. 3I and compare Fig. 3C with Fig. 3D and Fig. 3E with Fig. 3F). This raises the possibility that in human carriers of long premutation alleles, symptoms resulting from an FMRP deficiency in places like the thalamus and inferior olive may be more prominent than symptoms resulting from an FMRP deficiency elsewhere in the CNS. This might be germane since the thalamus is thought to act as a “gate” or filter of information from a variety of sensory sources and has been implicated in social anxiety, obsessive-compulsive disorder and autism. Inferior olive abnormalities have been reported in autism and disorders of speech and language. These sorts of symptoms can be seen in male carriers of premutation alleles with normal IQs (Aziz et al., 2003; Farzin et al., 2006).
Fig 3. Regional FMRP deficits in male premutation mice with 120 repeats.

A-H). Micrographs showing the FMRP localization in the hippocampal complex (A and B), the ventral posteriolateral thalamus (C and D), the inferior olive (E and F) and the cerebellum (G and H) from 2 year old WT (A, C, E, and G) and KI male mice with 120 repeats (B, D, F, and H) mice. Bars = 500 μm for A, B, E, F, G and H and 25 μm for C and D. GC, granule cells; MOL: molecular layer, PC: Purkinje cells. I). Bar graph showing the degree of change in FMRP expression in KI relative to WT. BL, basolateral; CB, cerebellum; DCN, deep cerebellar nuclei; CTX, cortex; DG, dentate gyrus; nLOT, nucleus of lateral olfactory tract. * Significantly different from WT p<0.05. The data represent the average obtained from 4 WT males and 5 KI males all ∼100 weeks in age.
3.3. Repeat instability is high in the KI mice
To test whether the stability of the KI allele undergoes large intergenerational changes in repeat length as it does in humans, we monitored the transmission of the allele through the male and female germline. Instability is high in mice with 120 repeats, with expansions predominating and more expansions being seen on paternal than on maternal transmission (Table 1). This instability is more than twice that seen in the previously reported mouse model (Brouwer et al., 2006). Most expansions were small involving the gain of <5 repeats. The increased frequency of paternal transmissions of small expansions is consistent with what is seen in a number of human repeat expansion diseases. The elevated level of small paternal expansions is usually attributed to the greater number of cell divisions involved in the generation of mature sperm.
Table 1.
Repeat Instability
| Expansions | Deletions | No change | |||||
|---|---|---|---|---|---|---|---|
| Cross (Male × Female) |
No. of alleles |
total | <5 repeats |
>5 repeats |
% | % | % |
| 120 repeats |
|||||||
| KI × WT* | 43 | 26 | 19 | 7 | 61 | 12 | 27 |
| WT × KI+/−* | 85 | 31 | 31 | 0 | 37 | 27 | 36 |
| KI × KI+/+ | 118 | 54 | 43 | 11 | 46 | 3 | 51 |
| 190 repeats |
|||||||
| KI × WT | 33 | 27 | 1 | 26# | 82 | 6 | 12 |
Chi square differences between KI×WT vs WT×Ki are significant at p ≤ 0.001
Of these 20 were >20 repeats
While large expansions are rare, we have observed 4 expansions of alleles containing ∼120 repeats to ones containing 200 repeats or more, i.e., close to or above the threshold for full mutation alleles in humans (∼0.5%; see Fig 4 for example). The largest expansion observed to date results in an allele with ∼265 repeats. No such “large jumps” have been reported in any previous mouse model for CGG•CCG-repeat expansion. No evidence of any rearrangements or the introduction of point mutations was found (data not shown). Both paternal and maternal expansions into the full mutation range were seen.
Fig. 4. Detection of a large expansion that generates an allele in the full mutation range on germline transmission.

The repeat size in a breeding pair involving a male with a premutation allele of ∼120 repeats and a female homozygous for the same sized repeat and one of their litters showing one expansion into the full mutation range indicated by the arrow. MW, molecular weight marker, M, male, F, female, KI, PCR positive control (DNA from a mouse previously shown to carry the premutation), B, PCR negative control (DNA from a WT animal),
Unlike human full mutation alleles that show significant methylation at birth that persists throughout life, no evidence of DNA methylation was seen in full mutation mice at 3 weeks of age (Fig. 5). While it is possible that limited methylation occurs that is below the detection limit of our assay, significant methylation is unlikely since Fmr1 mRNA levels are higher in these mice than in mice with premutation alleles (Fig. 2). Not all full mutations are completely methylated in humans (Tassone et al., 2000a) and different mice strains show different amounts of de novo methylation of other genes (Schumacher et al., 2000). It is thus reasonable to assume that trans-acting factors affect the methylation threshold in both humans and mice.
Fig. 5. Analysis of the methylation status of mice with different numbers of repeats.

Tail DNA from mice taken at weaning was digested with Sau96 I and then amplified by PCR using primers frax-m4 and frax-m5 that overlap the Sau96 I sites. The PCR products were then compared to the PCR products generated from equivalent amounts of tail DNA that had not been digested with Sau96 I. MW, molecular weight marker
3.4. Large premutation alleles do not show a paternal contraction bias
Large FMR1 alleles contract on paternal transfer in humans. However, this is not true of mice (Fig. 6A). In this respect the murine Fmr1 locus behaves like the human FMR2 locus (Hamel et al., 1994), and this may account for the detection of paternally transmitted alleles with repeat numbers in the full mutation range. In fact not only do large CGG-repeat tracts not show a predisposition to contract on paternal transmission, the expansion frequency actually increases from 60% in males with 120 repeats to 82% in males with 190 repeats (Table 1). The average expansion size also increases from 3 repeats in males with 120 repeats to 40 in males with 190 repeats (Fig. 6B). This increase in expansion size and frequency with increasing repeat numbers is also different from the previously described mouse KI model (Brouwer et al., 2006) and would be consistent with there being a higher threshold for large expansions in mice.
Fig. 6. Paternal transmission of an allele with 190 repeats.

A) PCR analysis of a male mouse with 190 repeats (M) and the progeny produced when this mouse was crossed to a WT female. MW, molecular weight marker, B) Comparison of allele sizes in the progeny of male mice with 120 and 190 repeats. The gray bars represent the number of alleles of the indicated size seen in the progeny of a cross between a male with 120 repeats and a WT female. The black bars indicate the same data obtained from a cross between a male with 190 repeats and a WT female.
3.5. Little, if any, somatic instability is seen
No significant heterogeneity in the PCR product from tail DNA was seen in more than 700 premutation alleles tested even when small pool PCR was used (data not shown). We have also tested organs of ectodermal, endodermal and mesodermal origin from animals with 120 repeats. No Organs tested included the brain, liver, gonads and kidneys and the repeat size in these organs was indistinguishable from the alleles seen in the tail samples taken 3 weeks after birth (Fig. 7A). Similar results were seen using small pool PCR and no change in this profile was seen even in mice 18 months of age or older. Only very limited somatic instability was seen in mice with ∼190 repeats (Fig. 7B and C). A number of mice were mosaic for 2 different sized versions of an inherited allele. Mosaic animals were sometimes seen in the first litter of a breeding pair (Fig. 8), and no evidence was seen for a parental age effect (data not shown). In all instances both derivative alleles differed from the parental allele from which they originated. Both alleles were present in roughly equal amounts, were present in the same quantities in all tissue examined and were transmitted to their offspring at about the same frequency (Fig. 8). This mosaicism is reminiscent of the high frequency of post-meiotic segregation of CGG•CCG-repeats seen in yeast and suggests that, as in yeast, heteroduplexes containing these repeats often escape repair (Moore et al., 1999). The somatic stability of premutation alleles together with the characteristics of the mosaic alleles would be consistent with expansion occurring in the very early embryo or in the perigametic interval between the initiation of premeiotic DNA synthesis and the initiation of pronuclear DNA synthesis in the fertilized egg. Differences in the length of the perigametic interval which lasts decades in human females, but is of the order of weeks or months in mice, together with the higher threshold for expansion in mice, may account for some of the differences seen in the frequency of large expansions into the full mutation range.
Fig. 7. Somatic stability of premutation alleles.

PCR products containing the repeat were generated from tail DNA of mice taken 3 weeks after birth, and the DNA from the indicated organs of adult animals 6-18 months old. The sex of the mice, the number of repeats in their tail DNA and the organ from which the DNA was derived, are indicated above the gel. The samples shown in lanes 14-18 are from a female who inherited 2 premutation alleles and is mosaic for one of them.
Fig. 8. Germline transmission of alleles from a female mosaic for the premutation.
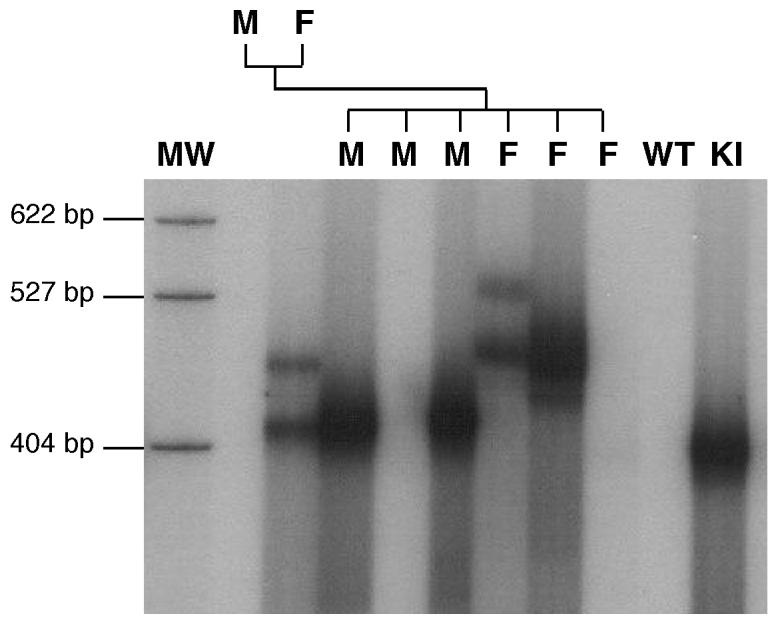
PCR analysis of the repeat size in a mosaic but heterozygous female, the WT male to which she was bred and 1 litter from this cross. MW, molecular weight marker, M, male, F, female, -, PCR negative control (DNA from a WT animal), +, PCR positive control (DNA from a mouse previously shown to carry the premutation). Note that one of the offspring of this cross, the female shown in lane 7, is also mosaic. The mice in lanes 5 and 9 inherited the normal maternal allele as assessed by PCR using primers that amplify both WT and KI alleles (data not shown).
In summary, we have demonstrated that the Fragile X premutation mice we have generated have many features similar to those seen in human premutation carriers, including a direct relationship between repeat number and Fmr1 mRNA levels and an inverse relationship between repeat number and FMRP in brain. These mice also show CNS pathology reminiscent of what is seen in humans. Moreover, they demonstrate for the first time in a mouse model, a large expansion that transforms a premutation allele to full mutation in a single generation. While the frequency of these large expansions is lower than it is in humans, understanding the basis of this difference may help us understand the expansion mechanism.
Acknowledgements
We would like to thank Lisa Garrett in the NHGRI Embryonic Stem Cell and Transgenic Core Facility, and Sonia Farmer and Maria Jorge in the NIDDK mouse facility. This research was supported in part by the Intramural Research Program of the NIDDK and NHGRI (NIH).
Abbreviations
- bp
base pairs
- CNS
central nervous system
- ES
embryonic stem
- FXTAS
Fragile X associated tremor and ataxia syndrome
- FMR1
Fragile X mental retardation 1
- FMRP
Fragile X mental retardation protein
- FXS
Fragile X mental retardation syndrome
- kb
kilobases
- KI
knock-in
- Nm
neomycin
- PCR
polymerase chain reaction
- WT
wildtype
- YAC
yeast artificial chromosome
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aziz M, Stathopulu E, Callias M, Taylor C, Turk J, Oostra B, Willemsen R, Patton M. Clinical features of boys with fragile X premutations and intermediate alleles. Am J Med Genet B Neuropsychiatr Genet. 2003;121:119–27. doi: 10.1002/ajmg.b.20030. [DOI] [PubMed] [Google Scholar]
- Bontekoe CJ, Bakker CE, Nieuwenhuizen IM, van der Linde H, Lans H, de Lange D, Hirst MC, Oostra BA. Instability of a (CGG)98 repeat in the Fmr1 promoter. Hum Mol Genet. 2001;10:1693–9. doi: 10.1093/hmg/10.16.1693. [DOI] [PubMed] [Google Scholar]
- Brouwer JR, Mientjes EJ, Bakker CE, Nieuwenhuizen IM, Severijnen LA, Van der Linde H, Nelson DL, Oostra BA, Willemsen R. Elevated Fmr1 mRNA levels and reduced protein expression in a mouse model with an unmethylated Fragile X full mutation. Experimental Cell Research. 2006 doi: 10.1016/j.yexcr.2006.10.002. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown V, Jin P, Ceman S, Darnell JC, O'Donnell WT, Tenenbaum SA, Jin X, Feng Y, Wilkinson KD, Keene JD, Darnell RB, Warren ST. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107:477–87. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- Dutch-Belgian Fragile X Consortium Fmr1 knockout mice: a model to study fragile X mental retardation. Cell. 1994;78:23–33. [PubMed] [Google Scholar]
- Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911–21. doi: 10.1016/s0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- Farzin F, Perry H, Hessl D, Loesch D, Cohen J, Bacalman S, Gane L, Tassone F, Hagerman P, Hagerman R. Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J Dev Behav Pediatr. 2006;27:S137–44. doi: 10.1097/00004703-200604002-00012. [DOI] [PubMed] [Google Scholar]
- Feng Y, Zhang F, Lokey LK, Chastain JL, Lakkis L, Eberhart D, Warren ST. Translational suppression by trinucleotide repeat expansion at FMR1. Science. 1995;268:731–4. doi: 10.1126/science.7732383. [DOI] [PubMed] [Google Scholar]
- Fu YH, Kuhl DP, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S, Verkerk AJ, Holden JJ, Fenwick RG, Jr., Warren ST, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. 1991;67:1047–58. doi: 10.1016/0092-8674(91)90283-5. [DOI] [PubMed] [Google Scholar]
- Grabczyk E, Usdin K. Generation of microgram quantities of trinucleotide repeat tracts of defined length, interspersion pattern, and orientation. Anal Biochem. 1999;267:241–3. doi: 10.1006/abio.1998.2962. [DOI] [PubMed] [Google Scholar]
- Greco CM, Berman RF, Martin RM, Tassone F, Schwartz PH, Chang A, Trapp BD, Iwahashi C, Brunberg J, Grigsby J, Hessl D, Becker EJ, Papazian J, Leehey MA, Hagerman RJ, Hagerman PJ. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS) Brain. 2006;129:243–55. doi: 10.1093/brain/awh683. [DOI] [PubMed] [Google Scholar]
- Greco CM, Hagerman RJ, Tassone F, Chudley AE, Del Bigio MR, Jacquemont S, Leehey M, Hagerman PJ. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain. 2002;125:1760–71. doi: 10.1093/brain/awf184. [DOI] [PubMed] [Google Scholar]
- Hagerman PJ, Hagerman RJ. Fragile X-associated tremor/ataxia syndrome (FXTAS) Ment Retard Dev Disabil Res Rev. 2004;10:25–30. doi: 10.1002/mrdd.20005. [DOI] [PubMed] [Google Scholar]
- Hamel BC, Smits AP, de Graaff E, Smeets DF, Schoute F, Eussen BH, Knight SJ, Davies KE, Assman-Hulsmans CF, Oostra BA. Segregation of FRAXE in a large family: clinical, psychometric, cytogenetic, and molecular data. Am J Hum Genet. 1994;55:923–31. [PMC free article] [PubMed] [Google Scholar]
- Handa V, Goldwater D, Stiles D, Cam M, Poy G, Kumari D, Usdin K. Long CGG-repeat tracts are toxic to human cells: Implications for carriers of Fragile X premutation alleles. FEBS Lett. 2005;579:2702–8. doi: 10.1016/j.febslet.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Hoffman GE, Smith MS, Fitzsimmons MD. Detecting steroidal effects on immediate early gene expression in the hypothalamus. Neuroprotocols. 1992;1:52–66. [Google Scholar]
- Jin P, Zarnescu DC, Zhang F, Pearson CE, Lucchesi JC, Moses K, Warren ST. RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron. 2003;39:739–47. doi: 10.1016/s0896-6273(03)00533-6. [DOI] [PubMed] [Google Scholar]
- Kenneson A, Zhang F, Hagedorn CH, Warren ST. Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediatelength and premutation carriers. Hum Mol Genet. 2001;10:1449–54. doi: 10.1093/hmg/10.14.1449. [DOI] [PubMed] [Google Scholar]
- Lavedan C, Grabczyk E, Usdin K, Nussbaum RL. Long uninterrupted CGG repeats within the first exon of the human FMR1 gene are not intrinsically unstable in transgenic mice. Genomics. 1998;50:229–40. doi: 10.1006/geno.1998.5299. [DOI] [PubMed] [Google Scholar]
- Moore H, Greenwell PW, Liu CP, Arnheim N, Petes TD. Triplet repeats form secondary structures that escape DNA repair in yeast. Proc Natl Acad Sci U S A. 1999;96:1504–9. doi: 10.1073/pnas.96.4.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Primerano B, Tassone F, Hagerman RJ, Hagerman P, Amaldi F, Bagni C. Reduced FMR1 mRNA translation efficiency in fragile X patients with premutations. RNA. 2002;8:1482–8. [PMC free article] [PubMed] [Google Scholar]
- Schumacher A, Koetsier PA, Hertz J, Doerfler W. Epigenetic and genotype-specific effects on the stability of de novo imposed methylation patterns in transgenic mice. J Biol Chem. 2000;275:37915–21. doi: 10.1074/jbc.M004839200. [DOI] [PubMed] [Google Scholar]
- Sherman SL. Premature ovarian failure in the fragile X syndrome. Am J Med Genet. 2000;97:189–94. doi: 10.1002/1096-8628(200023)97:3<189::AID-AJMG1036>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Tassone F, Hagerman RJ, Garcia-Arocena D, Khandjian EW, Greco CM, Hagerman PJ. Intranuclear inclusions in neural cells with premutation alleles in fragile X associated tremor/ataxia syndrome. J Med Genet. 2004;41:e43. doi: 10.1136/jmg.2003.012518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassone F, Hagerman RJ, Loesch DZ, Lachiewicz A, Taylor AK, Hagerman PJ. Fragile X males with unmethylated, full mutation trinucleotide repeat expansions have elevated levels of FMR1 messenger RNA. Am J Med Genet. 2000a;94:232–6. doi: 10.1002/1096-8628(20000918)94:3<232::aid-ajmg9>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Tassone F, Hagerman RJ, Taylor AK, Gane LW, Godfrey TE, Hagerman PJ. Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am J Hum Genet. 2000b;66:6–15. doi: 10.1086/302720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–14. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- Wagner KU, McAllister K, Ward T, Davis B, Wiseman R, Hennighausen L. Spatial and temporal expression of the Cre gene under the control of the MMTV-LTR in different lines of transgenic mice. Transgenic Res. 2001;10:545–53. doi: 10.1023/a:1013063514007. [DOI] [PubMed] [Google Scholar]
- Willemsen R, Hoogeveen-Westerveld M, Reis S, Holstege J, Severijnen LA, Nieuwenhuizen IM, Schrier M, van Unen L, Tassone F, Hoogeveen AT, Hagerman PJ, Mientjes EJ, Oostra BA. The FMR1 CGG repeat mouse displays ubiquitin-positive intranuclear neuronal inclusions; implications for the cerebellar tremor/ataxia syndrome. Hum Mol Genet. 2003;12:949–59. doi: 10.1093/hmg/ddg114. [DOI] [PubMed] [Google Scholar]
- Yu S, Pritchard M, Kremer E, Lynch M, Nancarrow J, Baker E, Holman K, Mulley JC, Warren ST, Schlessinger D, et al. Fragile X genotype characterized by an unstable region of DNA. Science. 1991;252:1179–81. doi: 10.1126/science.252.5009.1179. [DOI] [PubMed] [Google Scholar]