

Abstract
Activation induced cell death (AICD) via Fas/FasL is the primary homeostatic molecular mechanism employed by the immune system to control activated T-cell responses and promote tolerance to self-antigens. We herein investigated the ability of a novel multimeric form of FasL chimeric with streptavidin (SA-FasL) having potent apoptotic activity to induce apoptosis in diabetogenic T cells and modulate insulin-dependent type 1 diabetes (IDDM) in an adoptive transfer model. Diabetogenic splenocytes from NOD/Lt females were co-cultured in vitro with SA-FasL, SA control protein, or alone without protein, and adoptively transferred into NOD/Lt-Rag1null recipients for diabetes development. All animals receiving control (Alone: n=16 or SA: n=17) cells developed diabetes on average by 6 weeks, whereas animals receiving SA-FasL-treated (n = 25) cells exhibited significantly delayed progression (p<.001) and decreased incidence (70%). This effect was associated with an increase in CD4+CD25+ T cells and correlated with FoxP3 expression in pancreatic lymph nodes. Extracorporeal treatment of peripheral blood lymphocytes using SA-FasL during disease onset represents a novel approach that may alter the ability of pathogenic T cells to mediate diabetes and have therapeutic utility in clinical management of IDDM.
Keywords: FasL, diabetes, immunomodulation, treatment, apoptosis
1. Introduction
Activation-induced cell death (AICD) via Fas/FasL is an important homeostatic molecular mechanism by which the immune system controls activated T-cell responses, promotes tolerance to self antigens, and prevents autoimmunity. T cells upregulate both Fas and FasL upon activation and become sensitive to autocrine and paracrine apoptosis following repeated reengagement with the challenge antigen (Krammer, 2000; Van Parijs and Abbas, 1998; Ju et al., 1995). The importance of Fas/FasL-mediated apoptosis to tolerance to self antigens is demonstrated in the lpr and gld mice wherein the lack of or low expression of Fas and FasL leads to lymphoproliferative disorders and autoimmunity (Nagata and Suda, 1995). Transgenic replacement of Fas expression in mice restores both AICD and a disease-free state (Wu et al., 1994).
The NOD mouse is a widely used spontaneous model for the study of insulin-dependent type 1 diabetes (IDDM). Although molecular and cellular mechanisms responsible for the development of IDDM in NOD are complex and not fully characterized, activation of autoreactive CD4+ and CD8+ T cells and their recruitment into pancreatic islets play an important role in the initiation of the disease (Bendelac et al., 1987; Yagi et al., 1992). A series of studies reported resistance to AICD in NOD mice which may contribute to the disease process (Decallonne et al., 2003; Leijon et al., 1994; Arreaza et al., 2003). Although the nature of these defects are not fully understood, over 20 susceptibility loci, termed insulin-dependent diabetes (idd), have been identified (Aoki et al., 2005). Some of these defects translate to dysregulated anti-apoptotic gene expression including Bcl-2 (Garchon et al., 1994) and Bcl-x (Lamhamedi-Cherradi, 1998), as well as expression of a less apoptotic allele of FasL (Kayagaki et al., 1997).
Despite these claims, a number of studies have reported that selective induction of apoptosis via AICD in diabetogenic lymphocytes can prevent autoimmune diabetes in NOD mice. In vivo administration of Jo-2, an agonistic antibody to Fas, resulted in diabetes remission. Immunohistochemical analysis of pancreata from treated animals demonstrated apoptosis of infiltrating cells, but not insulin and/or glucagon-producing cells, suggesting that remission was due to the induction of AICD in autoreactive lymphocytes (Dharnidharka et al., 2002). In a second study, lipopolysaccharide (LPS)-activated B cells have been shown to express FasL and induce diabetogenic T cells to undergo apoptosis in vitro (Tian et al., 2001). Transfusion of these LPS-activated B cells into prediabetic mice inhibited spontaneous development of diabetes in greater than 80% of the B-cell treated NOD mice at 52 weeks of age. Furthermore, co-transfer of LPS-activated B cells and diabetogenic splenic T cells prevented the disease in an adoptive transfer model. In addition, both membranous as well as soluble forms of human FasL were shown to engage Fas on naïve and diabetogenic T cells, induce apoptosis, and modulate development of diabetes by eliminating autoreactive diabetogenic lymphocytes and pre-activated CD4+ CD45RBlow memory T cells (Kim et al., 2000). Of further interest, human soluble FasL was shown to function in vivo to delete potentially autoreactive memory lymphocytes in peripheral blood lymphocytes from mice as well as IDDM patients, complementing membrane FasL in promoting AICD (Kim et al., 2002). Altogether, these studies demonstrate that diabetogenic T cells are sensitive to FasL-mediated apoptosis and as such FasL-based immune intervention may have therapeutic utility for the prevention/treatment of IDDM.
FasL, a type II membranous protein, is expressed on antigen-activated T cells in response to TCR signaling and costimulation (Askenasy et al., 2005). This protein is cleaved off from the cell surface in response to various physiologic stimuli by matrix metalloproteinases (Ju et al., 1995; Tanaka et al., 1998). Traditionally, the membranous form is noted for its ability to induce apoptosis in autoreactive and alloreactive T cells and promote tolerance; in contrast, the soluble form may inhibit apoptosis, initiate inflammatory responses, and promote the active recruitment of neutrophils, thereby accelerating disease or allograft rejection (Ottonello et al., 1999; Seino et al., 1998; Suda et al., 1997). However, this functional difference between membranous and soluble forms of FasL applies to murine FasL more than the human molecule, which appears to have apoptotic function in both forms (Kim et al., 2002; Tanaka et al., 1995).
These diverse functions of soluble and membraneous forms of FasL may be a manifestation of their differential ability to crosslink the Fas receptor upon engagement and the quality/quantity of the ensuing signals (Siegel et al., 2000). We have recently generated a chimeric SA-FasL protein that forms tetramers and higher structures and has potent apoptotic activity on Fas+ cells in soluble form. Immunomodulation with biotinylated donor cells decorated with the chimeric molecule resulted in effective elimination of alloreactive T cells, leading to the survival of transplanted islet and cardiac allografts without any sign of general toxicity or chemotactic function on neutrophils (Askenasy et al., 2005; Yolcu et al., 2002; Askenasy et al., 2003). In this study, we tested whether chimeric SA-FasL can be utilized to modulate autoreactive responses by altering the ability of transferred cells to mediate IDDM. Ex vivo treatment of diabetogenic splenocytes with SA-FasL resulted in significant apoptosis of T cells as well as slower progression and lower incidence of disease in an adoptive transfer model. The observed effect was associated with increased percentage and absolute number of CD4+CD25+ T regulatory cells. The implication of these findings for the use of chimeric FasL to alter autoreactive T cell populations from IDDM patients under extracorporeal conditions as a novel therapeutic intervention is discussed.
2. Materials and Methods
2.1. Mice and Glucose Monitoring
Eight week old female NOD/Lt mice and NOD/Lt-Rag1null breeder pairs were purchased from The Jackson Laboratory (Bar Harbor, ME) and maintained under specific pathogen-free conditions according to standards and guidelines stipulated by the University of Louisville Institutional Animal Care and Use Committee. NOD/Lt-Rag1null mice for adoptive transfer experiments were used from the breeding colony at 4-6 weeks. NOD/Lt-Rag1null mice lack functional T and B cells and do not spontaneously develop diabetes because they are deficient in RAG1, the gene responsible for V(D)J recombination in immunoglobulin and T-cell receptor genes. This NOD/Lt-Rag1null adoptive transfer model for type 1 diabetes has been previously established and described (Shultz et al., 2000). For NOD donors and adoptively transferred recipients urine glucose levels were monitored using Chemstrip uGK urine test sticks (Roche, Indianapolis, IN) twice per week. Positive urine glucose tests were confirmed by blood glucose measurement using Prestige Smart System (Home Diagnostics, Inc., Ft. Lauderdale, FL). Animals with blood glucose values >250 mg/dl on two consecutive occasions were considered positive for diabetes.
2.2. Protein construction: SA and chimeric SA-FasL
Soluble SA-FasL was produced as previously described (Yolcu et al., 2002). Briefly, DNA encoding core streptavidin (SA) from S. avidinii and the extracellular domains of rat FasL without the metalloproteinase cleavage site were cloned using specific primers by PCR. Both genes, SA alone as well as SA with FasL (SA-FasL), were subcloned into the PMT/BiP/V5-HisACuSO4-inducible vector for expression in an S2-Drosophila system (Invitrogen, Carlsbad, CA). A 6x-His tag was engineered into the SA control and SA-FasL test proteins for purification using ion metal affinity chromatography.
2.3. Ex vivo Treatment with SA-FasL
SA-FasL was titrated on splenocytes from diabetic NOD mice to determine concentration and time required to achieve 50-60% total apoptosis. Spleens from NOD mice positive for diabetes were harvested, processed, erythrocytes hemolyzed, washed three times, and resuspended at 4×106/mL in complete mixed lymphocyte reaction (MLR) medium (base medium 95% DMEM and 5% fetal bovine serum supplemented with 1mM sodium pyruvate, 10mM HEPES, 100U/mL Penicillin/Streptomycin, 2mM L-glutamine, 548 μM L-arginine-HCL, 13.6 μM folic acid, 270 μM L-asparagine, 50 μM 2-mercaptoethanol). Cells were bulk cultured with PBS (Alone) as a volume control, control protein (SA), or test protein (SA-FasL) at approximately 400 ng protein/mL at 37°C and 5% CO2. Cultures were checked for apoptosis at various times and harvested at 7.5 hours (mean time) to reach targeted apoptotic cell percentage. Cells were then washed 4 times with ice cold PBS, phenotyped for apoptosis, and used for adoptive transfer experiments. In total, eight ex vivo treatment sets were performed.
2.4. Adoptive Transfer
Twenty million diabetogenic splenocytes ex vivo treated with PBS (Alone), control protein (SA), and test protein (SA-FasL), were adoptively transferred by intravenous tail vein infusion into 4-6 week old male and female NOD/Lt-Rag1null mice. A total of 58 adoptive transfers were performed (Alone: n=16, SA: n=17, SA-FasL: n=25). Animals were monitored twice weekly for diabetes as described above.
2.5. Flow Cytometry
During treatment and at transfer, ex vivo treated cultures were stained with fluoroisothiocyanate (FITC)-AnnexinV (BD Pharmingen, San Jose, CA) and 7-amino-actinomycin D (7-AAD; Molecular Probes, Eugene, OR) for determination of apoptotic and dead cells. Post-transfer recipient spleen, mesenteric, peripheral, and pancreatic lymph nodes were harvested and phenotyped. Antibodies used to characterize lymphocyte subpopulations sensitive to ex vivo treatment as well as to determine resident and repopulation of cells within recipients after transfer included FITC-CD3, phycoerythrin (PE)-CD25, peridinin chlorphyll protein (PerCp)-CD8, and allophycocyanin (APC)-CD4 and CD19 (BD Pharmingen). Expressed percentages were based on gating using forward and side scatter parameters and analyzed on a FACSCalibur using Cellquest software (BD Pharmingen, Immunocytometry Systems, San Jose, CA).
2.6. In vitro Suppression Assay
CD4+CD25+ and CD4+CD25- T cells from healthy, naïve NOD peripheral lymph nodes and individual recipient pooled peripheral and pancreatic lymph nodes were sorted using a FACSCalibur high speed cell sorter (>97% purity). The function of CD4+CD25+ T cells was tested using CD3 stimulation based suppression assay. Briefly, CD4+CD25- T cells (25,000 - 12,500) were cocultured with CD4+CD25+ T cells at various ratios in the presence of irradiated antigen presenting cells (APC) (105) and 0.5 μg/mL anti-CD3 antibody. Cells were cultured in complete MLR medium in 5% CO2 at 37°C for 3 days and pulsed with 0.5 μCi 3[H]thymidine for the last 16-24 hours. Cells were harvested and 3[H]thymidine incorporation measured by scintillation counter.
2.7. Real-time PCR
Total RNA was extracted from whole pancreatic lymph nodes of adoptively transferred recipients, sorted CD4+CD25+ and CD4+CD25- T cells as well as AnnexinV negative CD4+CD25+ sorted T cells from ex vivo cultures (>97% purity) using TRIZOL (Molecular Research Center, Cincinnati, OH). RNA was then reverse transcribed using AMV reverse transcriptase and Oligo DT12 primers (Invitrogen). cDNA was amplified in duplicate by real-time PCR using SYBR Green PCR kit (Applied Biosystems, Foster City, CA) with primers for GAPDH (FWD: 5’-GGA/GCG/AGA/CCC/CAC/TAA/CA-3’ and RVS: 5’- ACA/TAC/TCA/GCA/CCG/GCC/TC-3’) and FoxP3 (FWD: 5’-CCC/ACC/TAC/AGG/CCC/TTC/TC-3’ and RVS: 5’-GC/ATG/GGC/ATC/CAC/AGT-3’) (Integrated DNA Technologies, Coralville, IA). FoxP3 mRNA levels were normalized relative to GAPDH mRNA expression. Data are presented as the fold-change relative to CD4+CD25- T cells.
2.8. Statistical Analysis
The incidence of diabetes was assessed by Kaplan-Meier life table analysis using the Log-Rank test (Lee and Wang, 2003). Mann-Whitney U test was used for cell population comparisons between recipients administered alone, SA, SA-FasL ex vivo treated NOD splenocytes and Kruskal-Wallace test was used for FoxP3 real-time PCR comparisons (Zar, 1999). Differences were considered significant at values of p<0.05. Animals that received ex vivo treated SA-FasL splenocytes and surpassed 73 days, the point at which all controls were positive for disease, were considered long-term SA-FasL survivors. All statistical tests were performed using SPSS 12.0 for Windows (SPSS, Inc., Chicago, IL).
3. Results
3.1. Ex vivo SA-FasL treatment induces apoptosis in NOD T cells
T cells upregulate Fas and become sensitive to Fas/FasL-mediated apoptosis upon repeated antigenic stimulation (Krammer, 2000; Van Parijs and Abbas, 1998; Ju et al., 1995). Inasmuch as T cells are critical to the induction of diabetes, we reasoned that NOD mice with active disease will have a pool of autoantigen activated T cells that may be sensitive to Fas-mediated apoptosis. This notion was tested by treating splenocytes from diabetic NOD mice with equimolar amounts of SA (control protein) and SA-FasL for 7.5 hours. Cultures were then analyzed for total and T cell subset-specific apoptosis using AnnexinV and T cell specific markers by flow cytometry. Treatment with SA-FasL significantly increased the total number of apoptotic cells (11.71×106±1.01) compared to control (SA: 6.37×106±0.84, p=0.001 and Alone: 6.21×106±0.86, p=0.002) cultures (Fig. 1A). As such, FasL treated cultures contained significantly less live cells at time of transfer compared to controls (SA-FasL: 8.29×106±1.01 vs. SA: 13.63×106±0.84 and Alone: 13.79×106±0.86, p=0.001). For CD4+ T cells treated with SA-FasL, there was greater than two-fold increase in apoptotic death, as compared with cells left untreated (Alone) or treated with SA (Fig. 1B). Although the apoptosis level for CD4+ T cells in SA-FasL treated cultures was significant (2.72×106±1.3) compared to those for CD4+ T cells in control cultures (SA: 0.92×106±0.4, p=0.008 and Alone: 0.94×106±0.32, p=0.013), this difference was not significant for CD8+ T cells (SA-FasL: 1.61×106±1.36 vs. SA: 0.69×106±0.54 or Alone: 0.68×106±0.46, p>0.05). These results demonstrate that SA-FasL treatment induced apoptosis in T cells harvested from diabetic NOD mice and that CD4+ T cells appeared to be more sensitive to apoptosis than CD8+ T cells.
Fig. 1. Effect of SA-FasL on ex vivo treated cells at transfer.
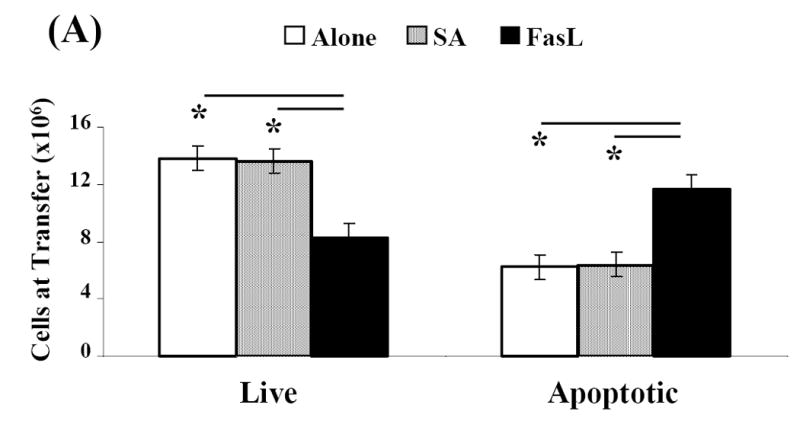
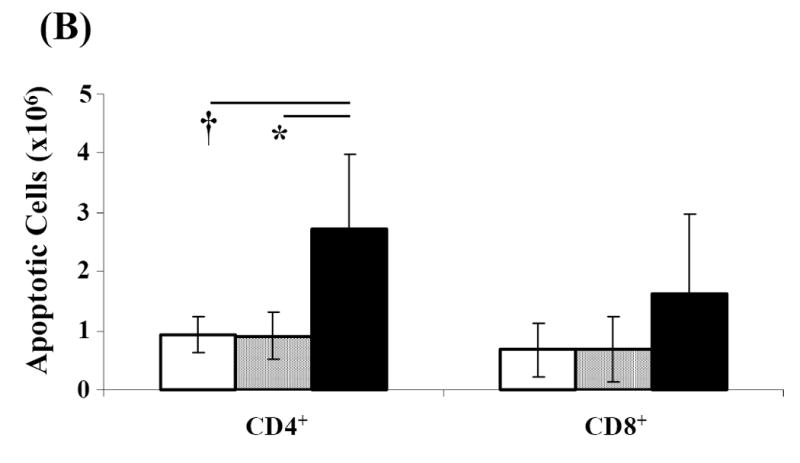
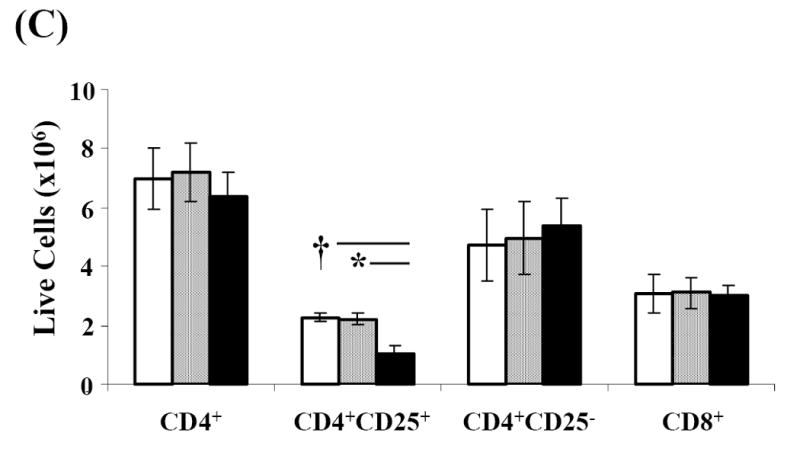
Spleens from NOD mice positive for diabetes were harvested, processed, and resuspended at 4×106/mL in complete MLR medium. Splenocytes were bulk cultured and treated with PBS, SA, or SA-FasL for 7.5 hours. Ex vivo treated cultures were analyzed for apoptosis (AnnexinV-FITC positive and 7-AAD negative) by flow cytometry. CD4+, CD4+CD25+, and CD8+ T-cell populations were analyzed for apoptosis using AnnexinV-FITC and T-cell antibodies: CD25-PE, CD8-PerCp, and CD4-APC. Effect of SA-FasL on ex vivo treated cells was statistically analyzed by Mann-Whitney U test. (A) Absolute number of live and apoptotic cells transferred. (B) Absolute number of CD4+ and CD8+ apoptotic cells transferred. (C) Absolute number of CD4+, CD4+CD25+, CD4+CD25-, and CD8+ live cells transferred. * P<0.01. †P<0.05.
Antigen-experienced T cells have been demonstrated to be the primary target of Fas/FasL-mediated apoptosis due to their upregulation of the Fas receptor (Krammer, 2000; Van Parijs and Abbas, 1998; Ju et al., 1995). We, therefore, tested whether FasL-treatment preferentially induced apoptosis in recently activated CD4+ T cells expressing CD25 as an activation marker. There was a significant reduction in the absolute number of live CD4+CD25+ T cells (1.02×106±0.29) in SA-FasL treated cultures as compared with those in control (SA: 2.22×106±0.21, p=0.009 and Alone: 2.26×106±0.14, p=0.014) cultures. In contrast, the absolute numbers of live CD4+CD25- T cells were similar among all groups (Alone: 4.73×106 ±1.20, SA: 4.97×106 ±1.23, SA-FasL: 5.36×106 ±0.97; Fig. 1C). Additionally, there was no difference in the absolute numbers of live CD4+ or CD8+ T cells in untreated (Alone: 6.98×106±1.02, 3.08×106±0.64), control treated (SA: 7.19×106±0.98, 3.11×106±0.51), or FasL treated (SA-FasL: 6.37×106±0.824, 3.00×106±0.33) cultures (p>0.05, Fig. 1C). Taken together, these results suggest that SA-FasL preferentially induced apoptosis in CD4+CD25+ T cells. In addition, transfer of live CD4+ and CD8+ T cells, which play an important role in disease initiation, were equal among the experimental groups.
3.2. SA-FasL ex vivo treatment appears to preferentially spare FoxP3 expressing CD4+CD25+ T cells
CD4+CD25+ T cell population includes activated T cells as well as naturally occurring CD4+CD25+FoxP3+ T regulatory (Treg) cells that are critical for tolerance to self antigens (Sakaguchi et al., 1995). The sensitivity of Treg cells to Fas/FasL-mediated apoptosis has been the subject of few recent studies with conflicting observations (Taams et al., 2001; Banz et al., 2002; Papiernik et al., 1997; Fritzsching et al., 2005). To test whether the CD4+CD25+ T cells physically eliminated by SA-FasL represents newly activated CD4+ T cells, rather than Treg cells, AnnexinV negative CD4+CD25+ T cells were sorted from ex vivo treated cultures and used for total RNA isolation and quantitative real-time PCR for FoxP3 mRNA expression. Relative expression of FoxP3 showed a perceptive, but insignificant decrease in CD4+CD25+ T cells sorted from untreated (Alone: 74.95±35.21), control treated (SA: 48.57±39.60), and FasL treated (SA-FasL: 34.59±33.77) cultures (p=0.109, Fig. 2). These results show that FoxP3+CD4+CD25+ Treg cells survive SA-FasL treatment and suggest that these cells may be less sensitive to SA-FasL-mediated apoptosis compared with newly activated CD4+CD25+ T effector cells.
Fig. 2. FoxP3 expression in live CD4+CD25+ T-cells.
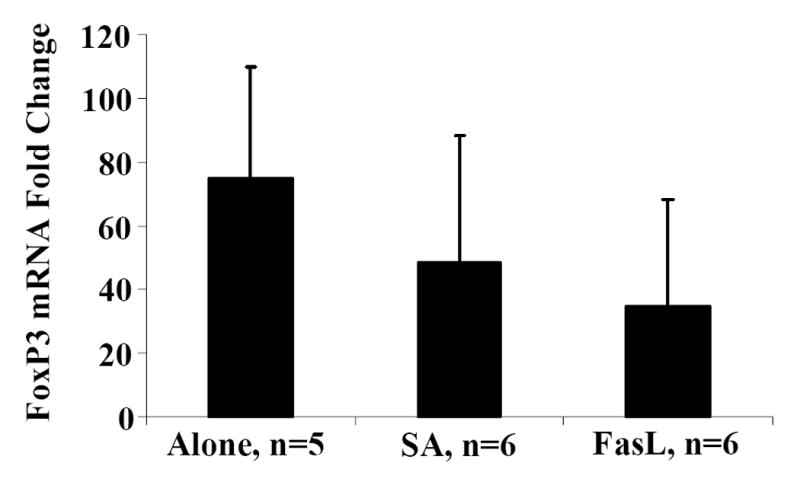
Spleens from NOD mice positive for diabetes were harvested, processed, and resuspended at 4×106/mL in complete MLR medium. Splenocytes were bulk cultured and treated with PBS, SA, or SA-FasL for 7.5 hours. Ex vivo treated cultures were washed and stained with a saturating amount of AnnexinV-FITC and T-cell antibodies: CD25-PE and CD4-APC. Using a high speed cell sorter, live (AnnexinV negative) CD4+CD25+ from SA-FasL and control ex vivo treated cultures were sorted and total RNA prepared. CD4+CD25+ and CD4+CD25- T cells from healthy NOD peripheral lymph node were sorted for FoxP3 mRNA controls. cDNA from sorted cells was prepared and then amplified in duplicate by real-time PCR for GAPDH and FoxP3. FoxP3 mRNA levels were normalized relative to GAPDH mRNA expression and statistically analyzed by Kruskal-Wallace test. Data are presented as the fold-change relative to CD4+CD25- T cells. P>0.05.
3.3. SA-FasL ex vivo treatment delays transfer of IDDM
To further provide evidence that ex vivo treatment of diabetogenic splenocytes with SA-FasL preferentially targets activated autoreactive T effector cells for apoptosis, we performed adoptive transfer experiments in NOD/Lt-Rag1null mice. One hundred percent of the animals receiving untreated (Alone: n=16) or control treated (SA: n=17) splenocytes developed diabetes on average by six weeks and all animals in these two groups developed overt diabetes by 11 weeks. In contrast, mice receiving FasL treated cells (SA-FasL: n=25) exhibited significantly delayed onset (p=0.0001) and decreased incidence of diabetes (70%) (Fig. 3). These results demonstrate that SA-FasL preferentially eliminates diabetogenic T effector cells and modulates the ability of SA-FasL treated cells to mediate diabetes. Furthermore, these results are consistent with our previous observations that immunomodulation with donor cells decorated with SA-FasL or direct display of this molecule on graft endothelium was effective in blocking alloreactive responses with therapeutic consequences (Yolcu et al., 2002; Askenasy et al., 2003).
Fig. 3. Incidence of diabetes after adoptive transfer of ex vivo treated NOD splenocytes.
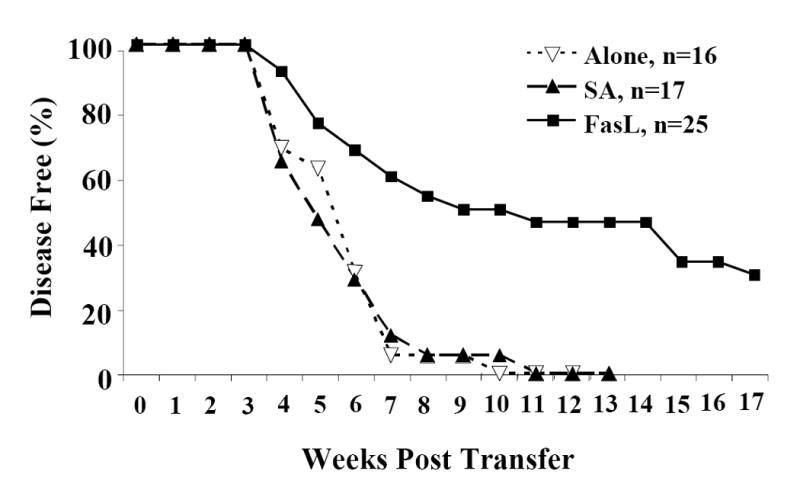
Diabetogenic splenocytes from NOD/Lt females were bulk cultured in vitro with PBS (Alone), control (SA), and test protein (SA-FasL) for 7.5 hours. After culture, cells were washed and 20×106ex vivo treated cells were adoptively transferred by intravenous tail vein infusion into 4-6 week old male and female NOD/Lt-Rag1null mice. For 17 weeks all recipients were monitored for diabetes development twice weekly. Diabetes incidence was assessed by Kaplan-Meier life table analysis using the Log-Rank test. All animals receiving untreated (Alone: ∇ white triangle, n=16) or control treated (SA: ▲ black triangle, n=17) cells developed overt diabetes by 73 days (11 weeks). Animals receiving FasL treated (SA-FasL: ■ black square, n=25) cells exhibited significantly delayed onset and decreased incidence. P<0.001.
3.4. Long-term survivors repopulate
To address whether the protective effect of SA-FasL is due to the low number of total live cells transferred (approximately <10×106 treated cells; Fig. 1A) and as a result fewer number of repopulating T cells, spleen, mesenteric, pancreatic, and peripheral lymph nodes were harvested from various groups of animals and total cell recovery was calculated. Recall, live CD4+, CD8+, and CD4+CD25- T cell transfer were equal among the experimental groups (Fig. 1C). Compared to controls (Alone: 11.88×106±5.13, p=0.012 and SA: 9.67×106±8.55, p=0.004), total cell recovery from long-term SA-FasL recipients, those who surpassed 73 days, the point at which all controls were positive for disease, was significantly increased (45.10×106±30.18) (Fig. 4A). There was no significant difference in splenic B-cell recovery between untreated, SA, and long-term SA-FasL survivors, demonstrating that ex vivo SA-FasL treatment had no differential effect on B-cell populations in NOD/Lt-Rag1null recipients (data not shown). However, long-term SA-FasL survivors had a significant increase in the absolute number of T cells (18.06×106±10.30) recovered from spleen as compared to controls (Alone: 3.49×106±1.92, p=0.006 and SA: 3.42×106±1.30, p=0.003) (Fig. 4B). These data demonstrate that ex vivo treatment with SA-FasL and subsequent disease modulation was not due to a general defect in T cell repopulation and expansion in vivo.
Fig. 4. Repopulation analyses.
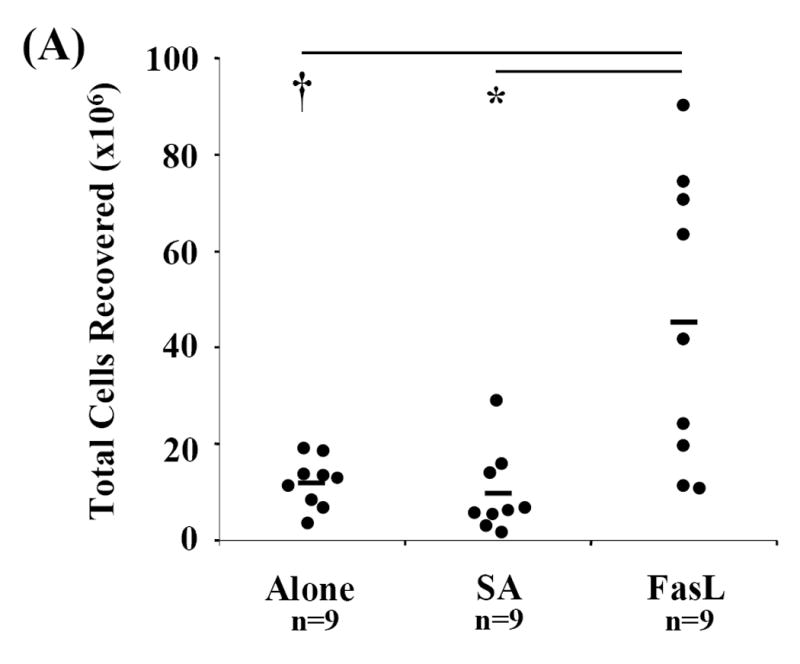
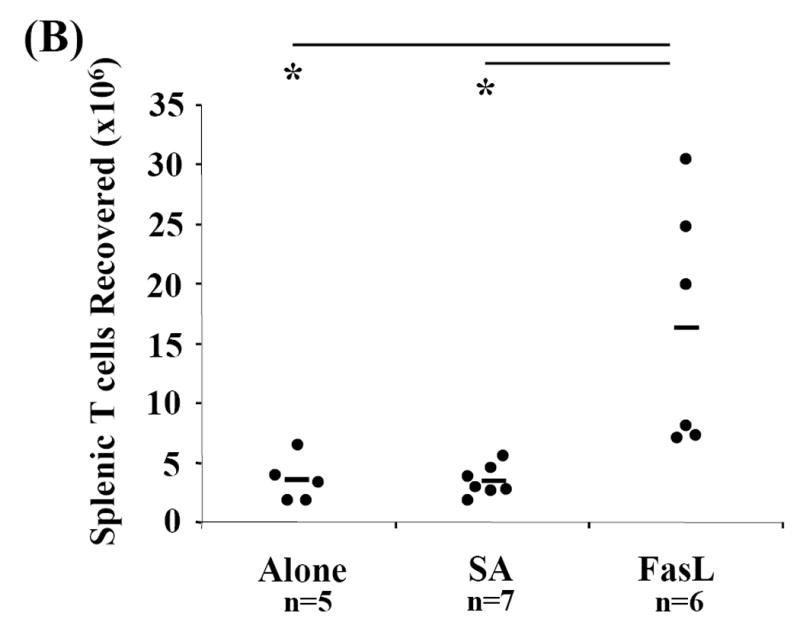
Spleen, mesenteric, pancreatic, and peripheral lymph nodes from recipients were harvested, processed, counted, and phenotyped by flow cytometric analysis to determine effective T cell expansion and repopulation. Each cellular compartment was phenotyped using the T cell specific surface marker CD3-FITC and statistically analyzed by Mann-Whitney U test. (A) Cell counts for each compartment and individual animals were obtained and totaled for determination of total cells recovered. (B) Absolute numbers of splenic T cells in individual recipients were calculated based on percent of CD3+ cells recovered in spleen. *P<0.01. †P<0.05.
3.5. Long-term survivors have significantly increased absolute numbers of CD4+CD25+ cells
CD4+CD25+ Treg cells appeared to be less sensitive to apoptosis induced by SA-FasL treatment ex vivo (Fig. 2). Furthermore, the anti-inflammatory and immunomodulatory properties of apoptotic bodies may induce/expand this cell population via effects on APC or other undefined mechanisms (Ferguson et al., 2003; Voll et al., 1997). We, therefore, tested whether the lower incidence of diabetes in animals adoptively transferred with SA-FasL-treated splenocytes correlated with increased numbers of Treg cells. Spleen, mesenteric, pancreatic, and peripheral lymph node cells were harvested and phenotyped for various cell surface markers using flow cytometry from long-term SA-FasL survivors and at the onset of diabetes from controls (Alone and SA groups). Long-term SA-FasL survivors had a significant increase in total CD4+CD25+ T cells (4.01×106±2.63) as compared to controls (Alone: 1.11×106±0.53, p=0.011 and SA: 1.13×106±0.52, p=0.039) (Fig. 5A). This increase in CD4+CD25+ cell number applied to spleen (SA-FasL: 2.72×106±2.01 vs. SA: 0.76×106±0.25, p=0.007 and Alone: 0.63×106±0.47, p=0.006) and pancreatic lymph nodes (SA-FasL: 117.17×103±103.56 vs. SA: 12.95×103±12.01, p=0.014 and Alone: 38×103±8.37, p=0.590) (Fig. 5B and C). Of note, in addition to the increase in absolute cell numbers, an increase in the percentage of CD4+CD25+ cells in pancreatic lymph node was observed (SA-FasL: 11.90±4.51% vs. SA: 5.13±3.30%, p=0.007 and Alone: 7.66±4.15%, p=0.101) (data not shown).
Fig. 5. CD4+CD25+ T cells are increased in SA-FasL treated long-term survivors.
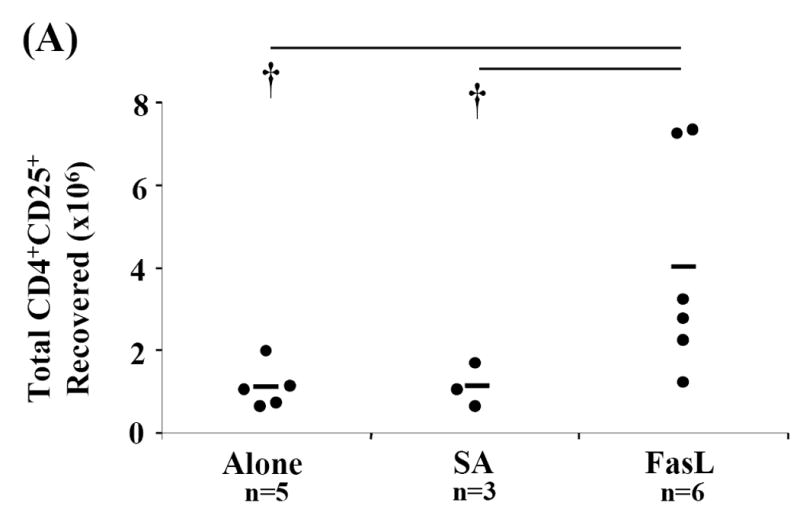
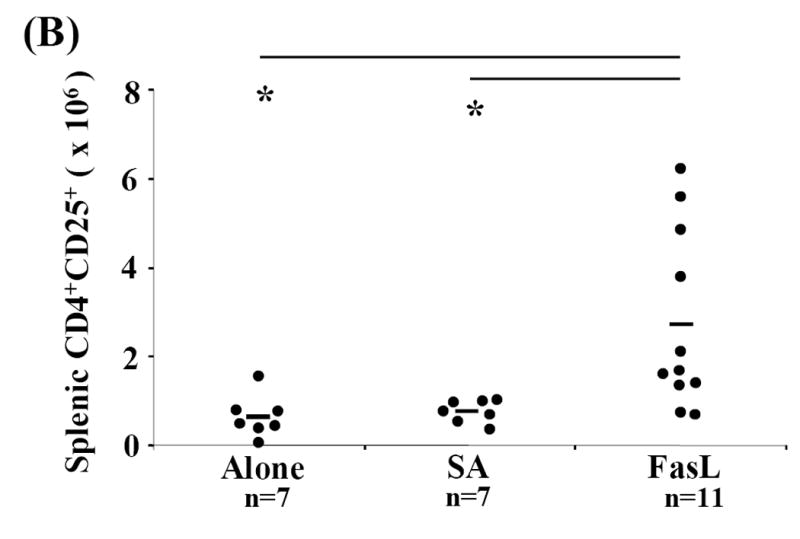
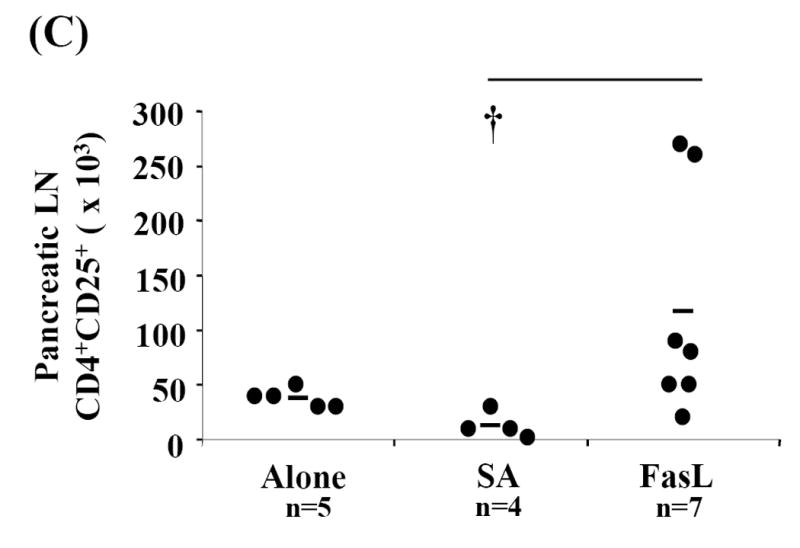
Spleens, mesenteric, pancreatic, and peripheral lymph nodes from individual recipients were harvested, processed, counted, and phenotyped by flow cytometric analysis to determine effective CD4+CD25+ T cell expansion and repopulation. Each cellular compartment was phenotyped using the surface markers specific for CD4+CD25+ T cells, CD3-FITC, CD25-PE, and CD4-APC and statistically analyzed by Mann-Whitney U test. (A) Absolute numbers of CD4+CD25+ T cells in individual recipients were calculated for each cellular compartment based on percent of CD3+CD4+CD25+ cells recovered. Absolute numbers of CD4+CD25+ cells calculated for each compartment were added to determine total cell recovery. Absolute numbers of (B) splenic and (C) pancreatic lymph node CD4+CD25+ T cells in individual recipients were calculated based on percent of CD3+ CD4+CD25+ cells recovered in spleen and pancreatic lymph nodes, respectively. * P<0.01. †P<0.05.
Ten weeks post-transfer, the point at which all control animals were overtly diabetic, four of the remaining 11 long-term survivors eventually developed diabetes (Fig. 3). Splenic CD4+CD25+ T cell numbers between non-diabetic and diabetic long-term survivors were compared. Consistent with the number of CD4+CD25+ cells found in Alone and SA controls that developed diabetes (less than 2×106), three of the four long-term survivors that were positive for diabetes demonstrated low absolute numbers of splenic CD4+CD25+ T cells (Fig. 5B). Furthermore, despite the fact that four long-term survivors eventually developed diabetes after 10 weeks, it is important to note that increased absolute numbers and percentages of CD4+CD25+ cells were present, particularly in pancreatic lymph nodes (Figs. 5A and 5C). These data suggest that for the animals that remained disease free beyond 10 weeks, CD4+CD25+ cells may play an important role in modulating diabetes.
3.6. CD4+CD25+ from long-term SA-FasL survivors are suppressive and express FoxP3
To test whether CD4+CD25+ T cells in long-term SA-FasL survivors are Treg cells and not newly activated T effector cells, FoxP3 mRNA expression and in vitro suppressive function were analyzed. Given that long-term SA-FasL survivors demonstrated an increase in percentage (data not shown) and absolute number of CD4+CD25+ T cells in pancreatic lymph nodes (Fig. 5C), cells were not sorted and FoxP3 mRNA expression in whole pancreatic lymph node was analyzed by real-time PCR. There was a relative increase in FoxP3 mRNA expression in whole pancreatic lymph node cells from long-term SA-FasL survivors (2.05±0.99) compared to Alone (1.03±0.96) and SA (1.30±0.20) controls (Fig. 6A).
Fig. 6. Analysis of FoxP3 mRNA expression and suppressive activity of CD4+CD25+ T cells in lymph node of long-term SA-FasL survivors.
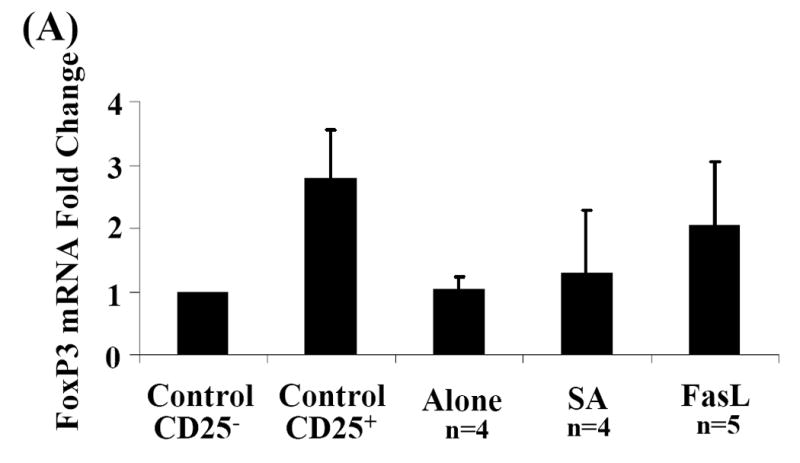
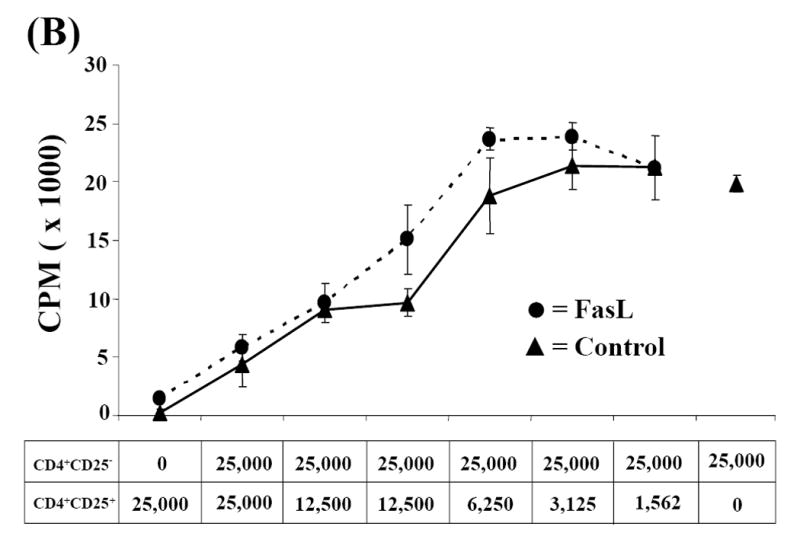
(A) Total RNA was extracted from whole pancreatic lymph node. cDNA was amplified in duplicate by real-time PCR for GAPDH and FoxP3. FoxP3 mRNA levels were normalized relative to GAPDH mRNA expression and statistically analyzed by Kruskal-Wallace test. Data are presented as the fold-change relative to control, sorted CD4+CD25- T cells. P>0.05. (B) CD4+CD25+ cells sorted from healthy naïve NOD peripheral lymph node (control) and long-term SA-FasL survivor pooled peripheral and pancreatic lymph nodes were cultured with control CD4+CD25- T cells at different ratios and evaluated for suppressive function. Data presented is representative of three suppression assays performed evaluating suppressive activity of CD4+CD25+ from long-term SA-FasL survivors.
We next tested whether CD4+CD25+ cells harvested from long-term SA-FasL survivors exhibited the functional properties of regulatory cells. CD4+CD25+ cells were sorted and tested for their ability to suppress the proliferation of CD4+CD25- T effector cells in co-culture experiments. Due to the small number of CD4+CD25+ T cells recovered, peripheral and pancreatic lymph nodes from individual ex vivo SA-FasL recipients were pooled and CD4+CD25+ sorted. Suppression assays from individual long-term SA-FasL survivors demonstrated that these CD4+CD25+ were anergic and could suppress the proliferative capacity of CD4+CD25- cells in a dose-dependent manner (Fig. 6B), providing evidence that these cells are Treg cells, rather than pathogenic T effectors. CD4+CD25+ from untreated and SA treated diabetic controls were also anergic and showed suppressive function on T effector cell proliferation (data not shown).
4. Discussion
We herein demonstrate that a novel form of chimeric FasL with potent apoptotic activity induced apoptosis in CD4+ T cells without a significant effect on CD8+ T and CD4+CD25+ Treg cell populations from diabetic NOD mice. Ex vivo SA-FasL-treated cells caused delayed onset and decreased incidence of diabetes in an adoptive transfer model. The observed immunomodulatory effect of SA-FasL may operate through inducing apoptosis in pathogenic T cells while sparing CD4+CD25+ Treg cells. A limited number of published reports have addressed this issue of Treg cell sensitivity to Fas-mediated apoptosis (Taams et al., 2001; Banz et al., 2002; Papiernik et al., 1997; Fritzsching et al., 2005). In humans, there exists an anergic and suppressive population of CD45RO+CD45RBlowCD4+CD25+ cells that are prone to AICD due to decreased expression of Bcl-2. IL-2 significantly inhibited cell death in this CD4+CD25+ population and TCR stimulation in the absence of costimulation did not significantly induce AICD (Taams et al., 2001). In addition, recent study of human CD4+CD25+FoxP3+ Tregs demonstrated relative resistance of these cells to AICD (Fritzsching et al., 2005). In the mouse, CD4+CD25+ Treg cells were shown to be resistant to apoptosis via Fas stimulation and do not undergo deletion by viral superantigens (Banz et al., 2002; Papiernik et al., 1997). Furthermore, DNA array and genetic analyses have also demonstrated that IL-2 signaling may trigger expression of anti-apoptotic genes that are both necessary and sufficient for the survival of CD4+CD25+ Treg cells (Gavin et al., 2002; Zheng et al., 2003). Taken together with these published studies, our data suggest that CD4+CD25+ T regulatory cells may be selectively spared from apoptosis, thus rendering them resistant to death-inducing signals delivered through Fas.
In conflict with our findings that CD4+ T cells of NOD are sensitive to apoptosis by SA-FasL are several published studies reporting resistance of NOD T cells to apoptosis (Decallonne et al., 2003; Leijon et al., 1994; Arreaza et al., 2003; Garchon et al., 1994). Our data, however, is consistent with several studies demonstrating that NOD T cells are sensitive to apoptosis by multiple forms of FasL, including FasL+ LPS-activated B cells, aberrant FasL expression on NOD lpr/lpr T cells, as well as treatment with Jo-2 agonistic Fas antibody and other membranous and soluble forms of FasL (Dharnidharka et al., 2002; Tian et al., 2001; Kim et al., 2000; Kim et al., 2002). Although the source of these discrepancies is unknown, the form of FasL and model systems used may serve as possible explanations. However, we observed a lack of significant apoptosis in CD8+ T cells, which is consistent with a recent study demonstrating the resistance of NOD CD8+ T cells to AICD due to their failure to downregulate c-FLIP after TCR-engagement (Arreaza et al., 2003).
Treg cells may preferentially expand following adoptive transfer into NOD/Lt-Rag1null animals as a result of homeostatic proliferation or response to autoantigens (Tang et al., 2004; Zelenay et al., 2005; Masteller et al., 2005). This notion is consistent with recent human studies in autologous stem cell transplantation as a treatment for severe refractory autoimmune disease and cancer demonstrating that Treg cells preferentially expand in lymphopenic hosts (de Kleer et al., 2006; Zhang et al., 2005). Alternatively, pathogenic T cells destined for apoptosis as a result of SA-FasL treatment ex vivo in our model may induce the conversion of CD4+CD25- T cells into CD4+CD25+ Treg cells and/or facilitate the expansion of Treg cells (Curotto et al., 2004). Consistent with this contention are observations that apoptotic lymphocytes secrete anti-inflammatory cytokines, such as TGF-β and IL-10 (Ferguson et al., 2003; Voll et al., 1997), that are implicated in the development, maintenance, expansion, and function of Treg cells (Peng et al., 2004; Green et al., 2003; Bacchetta et al., 2005).
Moreover, recognition and phagocytosis of apoptotic cells by macrophages and dendritic cells is shown to have immune regulatory consequences. Phagocytes engulf apoptotic cells before lysis and as such, curb potential inflammatory reactions by preventing release of immunogenic as well as toxic intracellular contents. Interestingly, it has been shown that apoptotic cells, as compared to necrotic cells, differentially affect macrophage production of cytokines as well as modulate dendritic cell maturation whereby anti-inflammatory cytokines and tolerance induction is promoted (Ferguson et al., 2003; Voll et al., 1997). However, known deficiencies in NOD macrophages in recognition, clearance, and response to apoptotic cells, argue against the notion that increased apoptotic cell death in our model contribute to the protective effect of SA-FasL treatment (O’Brien et al., 2002; Stoffels et al., 2004). Although CD4+CD25+ from long-term SA-FasL survivors were not more suppressive than CD4+CD25+ from diseased controls or recipients on a one to one cell basis, this study demonstrates that ex vivo treatment of diabetogenic splenocytes with SA-FasL modulates IDDM development by selectively targeting activated pathogenic CD4+ T cells for apoptosis. Likewise, SA-FasL ex vivo treatment may also preserve immunoregulatory networks and facilitate the active expansion/generation of Treg cells in vivo.
The major focus of this work was to test the direct effect of a novel form of FasL on diabetogenic T cells and its ability to modulate the transfer of diabetes. Despite demonstration of 30% remaining disease free after receiving SA-FasL treated cells, because animals were only monitored for 17 weeks, it is not known if all SA-FasL recipients would eventually develop diabetes. Further studies are required to definitively elucidate the molecular and cellular mechanisms responsible for the protective effect of SA-FasL in our model. To determine the therapeutic potential of ex vivo treatment with SA-FasL, experiments which include the secondary transfer of sorted CD4+CD25+ from long-term SA-FasL survivors into naïve NOD/Lt mice and/or co-transfer of these cells with diabetogenic NOD splenocytes into NOD/Lt-Rag1null may strengthen the evidence presented that SA-FasL treatment may alter the balance of pathogenic and regulatory T cells in diabetogenic population and alter progression of IDDM. Furthermore, these studies will help to determine if in fact T regulatory cells play a role in modulating diabetes progression through ex vivo SA-FasL treatment.
Functional and/or physical elimination of autoreactive diabetogenic T cells by ex vivo treatment with a novel form of FasL molecule with potent apoptotic activity may have implications in clinical management of IDDM. Treatment of peripheral blood lymphocytes with SA-FasL under extracorporeal conditions (ex vivo) from newly diagnosed patients in the honeymoon period with a considerable pancreatic islet mass and return of these cells into the patients may provide a novel means of immune intervention. A similar approach, where highly purified, protein-A bound to a silica matrix known as Prosorba columns is used to remove autoantibodies and immune complexes from the blood of rheumatoid arthritis patients, has already been tested in multiple clinical trials with reported efficacy (Felson et al., 1999; Levy and Degani, 2003). Inasmuch as SA-FasL can easily be attached to biotinylated beads, the application to IDDM or other autoimmune diseases with T cell etiology are possible. Importantly, unlike Prosorba, FasL-based immunomodulation may have long-lasting immunotherapeutic effects on patients due to the potential of this approach not only physically eliminating pathogenic T cells that sustain the disease, but also facilitating the development/expansion of Treg cells that may further control autoimmunity (Tang et al., 2004; Zelenay et al., 2005; Masteller et al., 2005; Wraith et al., 2004).
Acknowledgments
This research was supported by grants from NIH DK61333, AI47864, AI57903, Juvenile Diabetes Research Foundation (1-2001-328) and the Commonwealth of Kentucky Research Challenge Trust Fund. Additionally, we would like to thank Heather Stowers, Lisa Colbert, Janice Ditslear, and Victoria Kyle for their technical support and maintenance of our animal colony and Sarah Parnell for her time and technical expertise in executing the real-time PCR analyses.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aoki CA, Borchers AT, Ridgway WM, et al. NOD mice and autoimmunity. Autoimmun Rev. 2005;4:373–379. doi: 10.1016/j.autrev.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Arreaza G, Salojin K, Yang W, et al. Deficient activation and resistance to activationinduced apoptosis of CD8+ T cells is associated with defective peripheral tolerance in nonobese diabetic mice. Clin Immunol. 2003;107:103–115. doi: 10.1016/s1521-6616(03)00049-4. [DOI] [PubMed] [Google Scholar]
- Askenasy N, Yolcu ES, Wang Z, et al. Display of Fas ligand protein on cardiac vasculature as a novel means of regulating allograft rejection. Circulation. 2003;107:1525–1531. doi: 10.1161/01.cir.0000064893.96179.7e. [DOI] [PubMed] [Google Scholar]
- Askenasy N, Yolcu ES, Yaniv I, et al. Induction of tolerance using Fas ligand: a double-edged immunomodulator. Blood. 2005;105:1396–1404. doi: 10.1182/blood-2004-06-2364. [DOI] [PubMed] [Google Scholar]
- Bacchetta R, Gregori S, Roncarolo MG. CD4+ regulatory T cells: mechanisms of induction and effector function. Autoimmun Rev. 2005;4:491–496. doi: 10.1016/j.autrev.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Banz A, Pontoux C, Papiernik M. Modulation of fas-dependent apoptosis: a dynamic process controlling both the persistence and death of CD4 regulatory T cells and effector T cells. J Immunol. 2002;169:750–757. doi: 10.4049/jimmunol.169.2.750. [DOI] [PubMed] [Google Scholar]
- Bendelac A, Carnaud C, Boitard C, et al. Syngeneic transfer of autoimmune diabetes from diabetic NOD to healthy neonates. J Exp Med. 1987;166:823–832. doi: 10.1084/jem.166.4.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curotto de Lafaille MA, Lino AC, Kutchukhidze N, et al. CD25- T cells generate CD25+Foxp3+ regulatory T cells by peripheral expansion. J Immunol. 2004;173:7259–7268. doi: 10.4049/jimmunol.173.12.7259. [DOI] [PubMed] [Google Scholar]
- de Kleer I, Vastert B, Klein M, et al. Autologous stem cell transplantation for autoimmunity induces immunologic self-tolerance by reprogramming autoreactive T cells and restoring the CD4+CD25+ immune regulatory network. Blood. 2006;107:1696–1702. doi: 10.1182/blood-2005-07-2800. [DOI] [PubMed] [Google Scholar]
- Decallonne B, van Etten E, Giulietti A, et al. Defect in activation induced cell death in non-obese diabetic (NOD) T lymphocytes. J Autoimm. 2003;20:219–226. doi: 10.1016/s0896-8411(03)00025-8. [DOI] [PubMed] [Google Scholar]
- Dharnidharka VR, Patten YV, Bahjat FR, et al. Fas stimulation results in selective islet infiltrate apoptosis in situ and reversal of diabetes. Ann N Y Acad Sci. 2002;958:60–162. doi: 10.1111/j.1749-6632.2002.tb02960.x. [DOI] [PubMed] [Google Scholar]
- Felson DT, LaValley MP, Baldassare AR, et al. The Prosorba column for treatment of refractory rheumatoid arthritis: a randomized, double-blind, sham-controlled trial. Arthritis Rheum. 1999;42:2153–2159. doi: 10.1002/1529-0131(199910)42:10<2153::AID-ANR16>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Ferguson TA, Stuart PM, Herndon JM, et al. Apoptosis, tolerance, and regulatory T cells – old wine, new wineskins. Immunol Reviews. 2003;193:111–123. doi: 10.1034/j.1600-065x.2003.00042.x. [DOI] [PubMed] [Google Scholar]
- Fritzsching B, Oberle N, Eberhardt N, et al. In contrast to effector T cells, CD4+CD25+FoxP3+ regulatory T cells are highly susceptible to CD95 ligand- but not to TCR-mediated cell death. J Immunol. 2005;175:32–36. doi: 10.4049/jimmunol.175.1.32. [DOI] [PubMed] [Google Scholar]
- Garchon HJ, Luan JJ, Eloy L, et al. Genetic analysis of immune dysfunction in non-obese diabetic (NOD) mice: mapping of a susceptibility locus close to the Bcl-2 gene correlates with increased resistance of NOD T cells to apoptosis induction. Eur J Immunol. 1994;24:380–384. doi: 10.1002/eji.1830240217. [DOI] [PubMed] [Google Scholar]
- Gavin MA, Clarke SR, Negrou E, et al. Homeostasis and anergy of CD4+CD25+ suppressor T cells in vivo. Nat Immunol. 2002;3:33–41. doi: 10.1038/ni743. [DOI] [PubMed] [Google Scholar]
- Green EA, Gorelik L, McGregor CM, et al. CD4+CD25+ T regulatory cells control anti-islet CD8+ T cells through TGF-β- TGF-β receptor interactions in type 1 diabetes. Proc Natl Acad Sci USA. 2003;100:10878–10883. doi: 10.1073/pnas.1834400100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju ST, Panka DJ, Cui H, et al. Fas(CD95)/FasL interactions required for programmed cell death after T-cell activation. Nature. 1995;373:444–448. doi: 10.1038/373444a0. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Yamaguchi N, Nagao F, et al. Polymorphism of murine Fas ligand that affects the biological activity. Proc Natl Acad Sci USA. 1997;94:3914–3919. doi: 10.1073/pnas.94.8.3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Ki JY, Lee TH, et al. Soluble Fas ligand-susceptible “memory” cells in mice but not in human: Potential role of soluble Fas ligand in deletion of auto-reactive cells. Autoimm. 2002;35:15–20. doi: 10.1080/08916930290005882. [DOI] [PubMed] [Google Scholar]
- Kim S, Kim KA, Hwang DY, et al. Inhibition of autoimmune diabetes by Fas ligand: The paradox is solved. J Immunol. 2000;164:2931–2936. doi: 10.4049/jimmunol.164.6.2931. [DOI] [PubMed] [Google Scholar]
- Krammer PH. CD95’s deadly mission in the immune system. Nature. 2000;407:789–795. doi: 10.1038/35037728. [DOI] [PubMed] [Google Scholar]
- Lamhamedi-Cherradi SE, Luan JJ, Eloy L, et al. Resistance of T-cells to apoptosis in autoimmune diabetic (NOD) mice is increased early in life and is associated with dysregulated expression of Bcl-x. Diabetologia. 1998;41:178–184. doi: 10.1007/s001250050887. [DOI] [PubMed] [Google Scholar]
- Lee ET, Wang JW. Statistical Methods for Survival Data Analysis. 3. John Wiley & Sons; Hoboken, New Jersey: 2003. [Google Scholar]
- Leijon K, Hammarstrom B, Holmberg D. Non-obese diabetic (NOD) mice display enhanced immune responses and prolonged survival of lymphoid cells. Int Immunol. 1994;6:339–345. doi: 10.1093/intimm/6.2.339. [DOI] [PubMed] [Google Scholar]
- Levy J, Degani N. Correcting immune imbalance: the use of Prosorba column treatment for immune disorders. Ther Apher Dial. 2003;7:197–205. doi: 10.1046/j.1526-0968.2003.00043.x. [DOI] [PubMed] [Google Scholar]
- Masteller EL, Warner MR, Tang Q, et al. Expansion of functional endogenous antigen-specific CD4+CD25+ regulatory T cells from nonobese diabetic mice. J Immunol. 2005;175:3053–3059. doi: 10.4049/jimmunol.175.5.3053. [DOI] [PubMed] [Google Scholar]
- Nagata S, Suda T. Fas and Fas ligand: lpr and gld mutations. Immunol Today. 1995;16:39–43. doi: 10.1016/0167-5699(95)80069-7. [DOI] [PubMed] [Google Scholar]
- O’Brien BA, Huang Y, Geng X, et al. Phagocytosis of apoptotic cells by macrophages from NOD mice is reduced. Diabetes. 2002;51:2481–2488. doi: 10.2337/diabetes.51.8.2481. [DOI] [PubMed] [Google Scholar]
- Ottonello L, Tortolina G, Amelotti M, et al. Soluble Fas ligand is chemotactic for human neutrophilic polymorphonuclear leukocytes. J Immunol. 1999;162:3601–3606. [PubMed] [Google Scholar]
- Papiernik M, do Carmo Leite-de-Moraes M, Pontoux C, et al. T cell deletion induced by chronic infection with mouse mammary tumor virus spares a CD25-positive, IL-10 producing T cell population with infectious capacity. J Immunol. 1997;158:4642–4653. [PubMed] [Google Scholar]
- Peng Y, Laouar Y, Li MO, et al. TGF-β regulates in vivo expansion of Foxp3-expressing CD4+CD25+ regulatory T cells responsible for protection against diabetes. Proc Natl Acad Sci USA. 2004;101:4572–4577. doi: 10.1073/pnas.0400810101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Sakaguchi N, Asano M, et al. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25): Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- Seino K, Iwabuchi K, Kayagaki N, et al. Chemotactic activity of soluble Fas ligand against phagocytes. J Immunol. 1998;161:4484–4488. [PubMed] [Google Scholar]
- Shultz LD, Lang PA, Christianson SW, et al. NOD/LtSz-Rag1null mice: an immunodeficient and radioresistant model for engraftment of human hematolymphoid cells, HIV infection, and adoptive transfer of NOD mouse diabetogenic T cells. J Immunol. 2000;164:2496–2507. doi: 10.4049/jimmunol.164.5.2496. [DOI] [PubMed] [Google Scholar]
- Siegel RM, Frederiksen JK, Zacharias DA, et al. Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science. 2000;288:2354–2357. doi: 10.1126/science.288.5475.2354. [DOI] [PubMed] [Google Scholar]
- Stoffels K, Overbergh L, Giulietti A, et al. NOD macrophages produce high levels of inflammatory cytokines upon encounter of apoptotic or necrotic cells. J Autoimm. 2004;23:9–15. doi: 10.1016/j.jaut.2004.03.012. [DOI] [PubMed] [Google Scholar]
- Suda T, Hashimoto H, Tanaka M, et al. Membrane Fas ligand kills human peripheral blood T lymphocytes and soluble Fas ligand blocks the killing. J Exp Med. 1997;186:2045–2050. doi: 10.1084/jem.186.12.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taams LS, Smith J, Rustin MH, et al. Human anergic/suppressive CD4+CD25+ T cells: a highly differentiated and apoptosis-prone population. J Immunol. 2001;31:1122–1131. doi: 10.1002/1521-4141(200104)31:4<1122::aid-immu1122>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Itai T, Adachi M, et al. Downregulation of Fas ligand by shedding. Nat Med. 1998;4:31–36. doi: 10.1038/nm0198-031. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Suda T, Takahashi T, et al. Expression of the functional soluble form of human Fas ligand in activated lymphocytes. EMBO J. 1995;14:1129–1135. doi: 10.1002/j.1460-2075.1995.tb07096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Henriksen KJ, Bi M, et al. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. 2004;199:1455–1465. doi: 10.1084/jem.20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J, Zekzer D, Hanssen L, et al. Lipopolysaccharide-activated B cells down-regulate Th1 immunity and prevent autoimmune diabetes in nonobese diabetic mice. J Immunol. 2001;167:1081–1089. doi: 10.4049/jimmunol.167.2.1081. [DOI] [PubMed] [Google Scholar]
- Van Parijs L, Abbas AK. Homeostasis and self-tolerance in the immune system: turning lymphocytes off. Science. 1998;280:243–248. doi: 10.1126/science.280.5361.243. [DOI] [PubMed] [Google Scholar]
- Voll RE, Herrmann M, Roth EA, et al. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–351. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- Wraith DC, Nicolson KS, Whitley NT. Regulatory CD4+ T cells and the control of autoimmune disease. Curr Opin Immunol. 2004;16:695–701. doi: 10.1016/j.coi.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Wu J, Zhou T, Zhang J, et al. Correction of accelerated autoimmune disease by early replacement of the mutated lpr gene with the normal Fas apoptosis gene in the T cells of transgenic MRL-lpr/lpr mice. Proc Natl Acad Sci USA. 1994;91:2344–2348. doi: 10.1073/pnas.91.6.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi H, Matsumoto M, Kunimoto K, et al. Analysis of the roles of CD4+ and CD8+ T cells in autoimmune diabetes of NOD mice using transfer to NOD athymic nude mice. Eur J Immunol. 1992;22:2387–2393. doi: 10.1002/eji.1830220931. [DOI] [PubMed] [Google Scholar]
- Yolcu ES, Askenasy N, Singh NP, et al. Cell membrane modification for rapid display of proteins as a novel means of immunomodulation: FasL-decorated cells prevent islet graft rejection. Immunity. 2002;17:795–808. doi: 10.1016/s1074-7613(02)00482-x. [DOI] [PubMed] [Google Scholar]
- Zar JH. Biostatistical Analysis. 4. Prentice Hall; Upper Saddle River, New Jersey: 1999. [Google Scholar]
- Zelenay S, Lopes-Carvalho T, Caramalho I, et al. Foxp3+ CD25- CD4 T cells constitute a reservoir of committed regulatory cells that regain CD25 expression upon homeostatic expansion. Proc Natl Acad Sci USA. 2005;102:4091–4096. doi: 10.1073/pnas.0408679102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Chua KS, Guimond M, et al. Lymphopenia and interleukin-2 therapy alter homeostasis of CD4+CD25+ regulatory T cells. Nat Med. 2005;11:1238–1243. doi: 10.1038/nm1312. [DOI] [PubMed] [Google Scholar]
- Zheng XX, Sánchez-Fueyo A, Sho M, et al. Favorably tipping the balance between cytopathic and regulatory T cells to create transplantation tolerance. Immunity. 2003;19:503–514. doi: 10.1016/s1074-7613(03)00259-0. [DOI] [PubMed] [Google Scholar]