

Abstract
The lipid A from nitrogen-fixing bacterial species R. sin-1 is structurally unusual due to lack of phosphates and the presence of a 2-aminogluconolactone and a very long chain fatty acid, 27-hydroxyoctacosanoic acid (27OHC28:0), moiety. This structurally unusual lipid A can antagonize TNF-α production by human monocytes induced by E. coli LPS. To establish the relevance of the unusual long chain 27-hydroxyoctacosanoic acid for antagonistic properties, a highly convergent strategy for the synthesis of several derivatives of the lipid A of Rhizobium sin-1 has been developed. Compound 1 is a natural R. sin-1 lipid A having a 27-hydroxyoctacosanoic acid at C-2′, compound 2 contains an octacosanoic acid moiety at this position, and compound 3 is modified by a short chain tetradecanoic acid. Cellular activation studies with a human monocytic cell line have shown that the octacosanoic acid is important for optimal antagonistic properties. The hydroxyl of the natural 27-hydroxyoctacosanoic moiety does, however, not account for inhibitory activity. The resulting structure activity relationships are important for the design of compounds for the treatment of septic shock.
Keywords: lipopolysaccharides, lipid A, antagonist, tumor necrosis factor, septic shock
1. Introduction
Septicemia is a serious worldwide health problem associated with mortality rates of 40%–60%. Currently, no effective treatment exists for this life-threatening syndrome other than supportive therapy in an intensive care setting.1, 2 The development of septicemia is often linked to a systemic inflammatory response to lipopolysaccharide (LPS) in the blood of affected patients.
LPS induces cellular responses after binding to the cluster differentiation antigen CD14 on mononuclear phagocytes, or to soluble CD14 in plasma and then to cells lacking CD14.3–5 As CD14 is a glycosylphosphatidylinositol-anchored protein, it lacks transmembrane and cytoplasmic domains, and therefore is unable to directly transmit signals to the interior of the cell. The latter function is performed by the Toll-like receptor 4 (TLR4),6, 7 which contains extracellular transmembrane and intracellular domains, and an accessory protein MD2.8 While the precise mechanisms involved in the interactions among LPS, CD14, TLR4, and MD2 remain to be discovered,9, 10 it is clear that cellular activation leads to the induction of cytokine gene expression, primarily through the activation of NF-κB, and the MAP kinases, which results in the biosynthesis of a diverse set of inflammatory mediators such as TNF-〈, IL-6 and IL1-β to eradicate the immediate infection. Unfortunately, the presence of a large amount of LPS in the blood can cause the overproduction of the mediators, which can lead to septic inflammatory response syndrome (SIRS) and include life-threatening symptoms such as vascular fluid leakage, tissue damage, hypotension, shock, and organ failure.
LPS consists of an O-chain polysaccharide, a core oligosaccharide and an amphiphilic moiety referred to as lipid A. The latter moiety has been shown to be the toxic principle of LPS. Lipid A of most enteric bacteria consists of a β(1-6)-linked glucosamine disaccharide backbone with phosphate monoesters at C-1 and C-4′ and β-hydroxyl fatty acyl groups and acyloxyacyl residues at C-2, and C-3 and C-2′ and C-3′, respectively (Figure 1).11 Small modifications in the acylation pattern of lipid A are thought to contribute to the virulence of enteric pathogens. For example, fatty acyl components can be present that have shorter chain length, sites of unsaturation or keto functional groups.12, 13 Other modifications include the addition of a palmitoyl residue, the hydroxylation of a myristoyl substituent and the addition of aminoarabinosyl and phosphoethanolamine moieties.14
Figure 1.
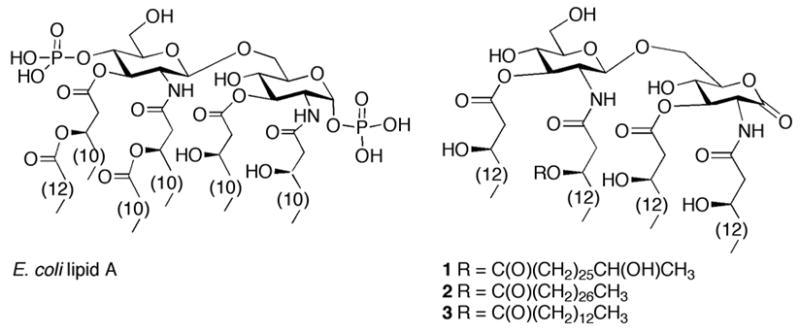
Structures of E. coli lipid A and target R. sin-1 lipid A derivatives 1–3.
A number of lipid A derivatives have been shown to lack agonistic properties and instead can inhibit the production of TNF-α induced by enteric LPS.15–19 Not surprisingly, these compounds have attracted attention for the treatment of septic shock. For example, the lipid As from Rhodobacter sphaeroides, R. capsulatus, and Helicobacter pylori, deacylated LPS, and lipid IVa lack toxic properties, but can antagonize cytokine production induced by enteric LPS. Furthermore, chemical synthesis of lipid A analogs patterned after R. sphaeroides/R. capsulatus lipid A have been reported. A number of these compounds could prevent the pyrogenic effects of enteric LPS in rabbits,20 protects against LPS-induced lethality in mice,19 and blocks TLR4-mediated NF-kB activation by LPS.21
We have identified naturally occurring LPS from Rhizobium sin-1, a nitrogen-fixing bacterial species, that does not stimulate TNF-α production by human monocytes,22–24 and that prevents the induction of TNF-α by E. coli LPS. The lipid A of R. sin-1 is perhaps the most structurally unusual lipid A reported to date, its structure (Figure 1; e.a. compound 1) differing in almost every aspect from those known to contribute to the toxicity of enteric lipid A.25 In particular, the disaccharide moiety of Rhizobial lipid A is devoid of phosphate and the glucosamine phosphate is replaced by 2-aminogluconolactone. Furthermore, it contains a very long chain fatty acid, 27-hydroxyoctacosanoic acid (27OHC28:0).
Previously reported phosphate-containing lipid A antagonists are metabolically labile. Hence, it is attractive to study the structure-activity relationship of R. sin-1 lipid A to identify specific structural features that render R. sin-1 lipid A an antagonist. We have already investigated the importance of the lactone moiety of R. sin-1 lipid A for antagonistic properties. Here, we report the chemical synthesis of compounds 1-3 (Figure 1) to establish the relevance of the unusual long chain 27-hydroxyoctacosanoic acid for antagonistic properties. Thus, compound 1 is a natural R. sin-1 lipid A having a 27-hydroxyoctacosanoic acid at C-2′, compound 2 contains an octacosanoic acid moiety at this position, and compound 3 is modified by a short chain tetradecanoic acid.
2. Results and discussion
2.1. Chemical synthesis
In first instance, attention was focused on the preparation of compound 3 which is acylated with a simple tetradecanoic acid at the β-hydroxyl of the C-2 acyloxyacyl residue of the distal sugar moiety. It was envisaged that 3 could be obtained by a regio-, chemo-, and stereoselective glycosylation of glycosyl donor 426 with glycosyl acceptor 527, 28 to give disaccharide 11, which after a number of selective deprotections can be acylated at the C-2, C-2′, C-3 and C-3′, oxidized to a lactone and deprotected. Thus, an N-iodosuccinimide (NIS)/trifluoromethanesulfonic acid (TfOH)29 mediated coupling of 4 with 5 in dichloromethane (DCM) at −75 °C gave, after purification by silica gel column chromatography, disaccharide 11 in a yield of 84% (Figure 2 and Scheme 1). The coupling exploited the higher reactivity of the C-6 hydroxyl of glycosyl acceptor 5 compared to the C-3 hydroxyl of glycosyl donor 4. Furthermore, the reaction took advantage of the ability to selectively active a selenoglycoside in the presence of a thioglycoside.30 Interestingly, it was found that NIS/TfOH gave a higher yield of 11 than when AgOTf/K2CO331 was used as the promoter system.
Figure 2.
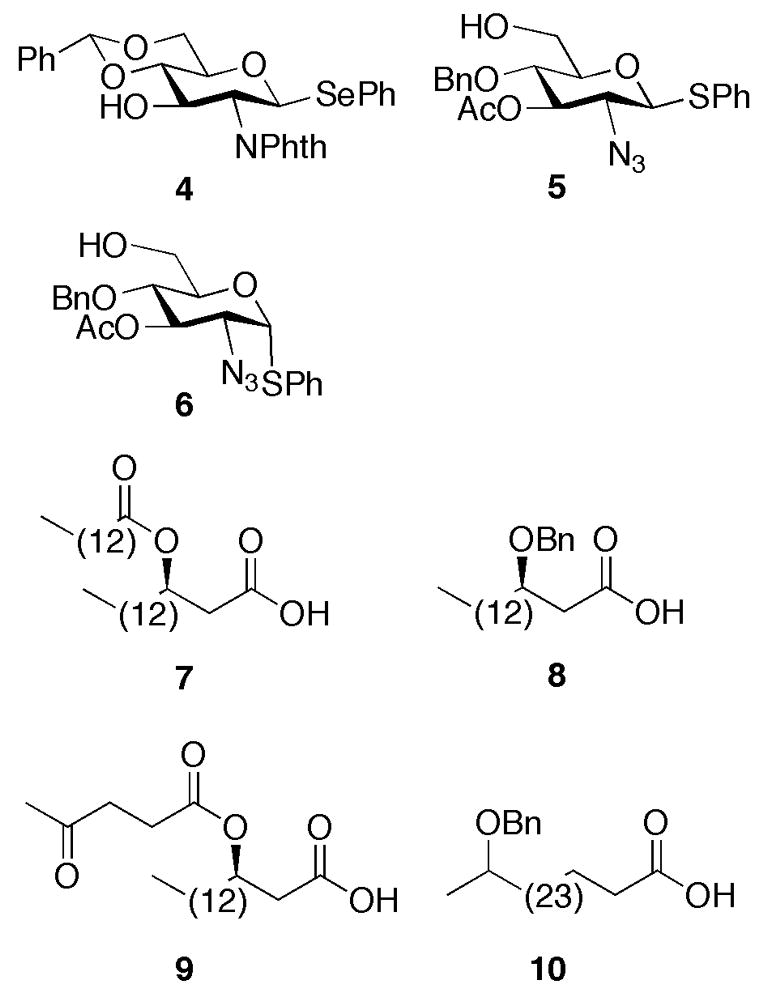
Structures of building blocks 4–10.
Scheme 1.
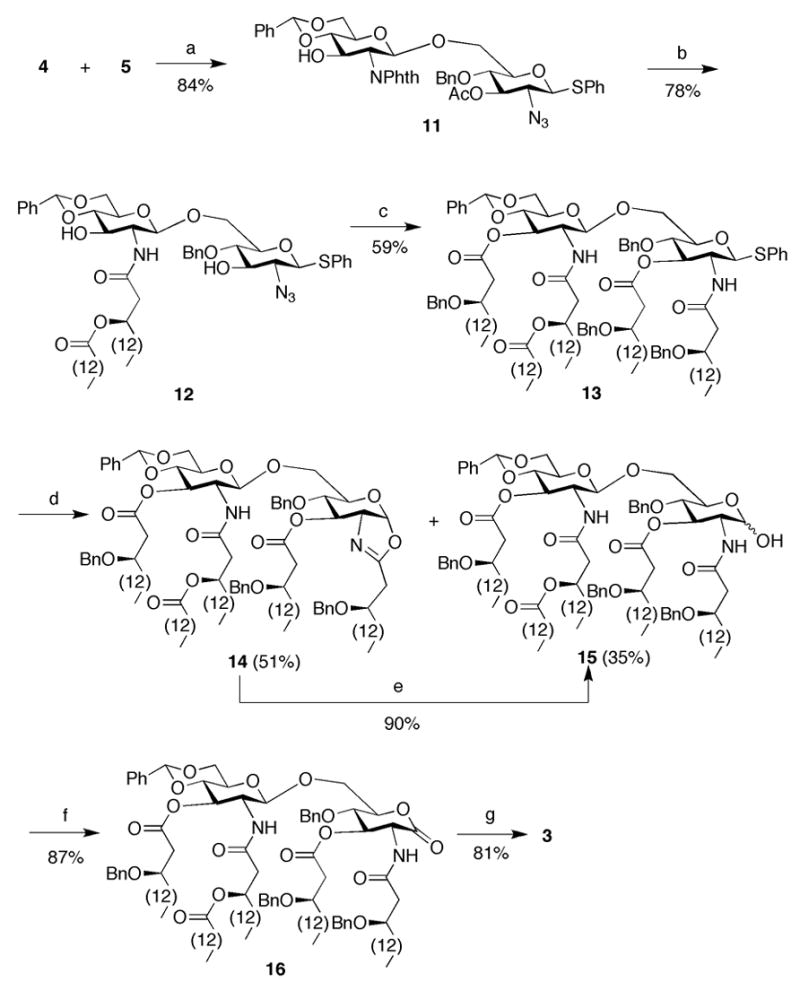
Reagents and conditions: (a) NIS, TfOH, MS 3 Å, DCM, −75 °C; (b) 1: H2N(CH2)2, n-BuOH, 90 °C; 2: 7, DCC, DCM; (c) 1: HS(CH2)3SH, pyridine, Et3N, H2O; 2: 8, DCC, DMAP, DCM; (d) NIS, TfOH, DCM, H2O; (e) dibenzylphosphate, H2O, DCM; (f) PCC, MS 3 Å, DCM; (g) Pd/C, H2, t-BuOH, THF.
Having advanced disaccharide 11 in hand, attention was focused on removal of the protecting groups and selective introduction of the fatty acids. Thus, the phthalimido group and acetyl ester of 11 were cleaved by treatment with ethylenediamine in refluxing n-butanol.32 The resulting C-2′ amino was selectively acylated with (R)-3-tetradecanoyloxy-hexadecanoic acid 733 using N,N′-dicyclohexyl carbodiimide (DCC) as the activating reagent to afford 12 in a yield of 78%. Reduction of the azido moiety of 12 was easily accomplished by reaction with propane-1,3-dithiol in a mixture of pyridine, triethylamine, and water, and the amine and hydroxyls of the resulting compound were acylated with (R)-3-benzyloxy-hexadecanoic acid (8) using DCC and 4-dimethylaminopyridine (DMAP) as activating reagents to afford 13 in an overall yield of 59%.
Hydrolysis of the thiophenyl moiety of 13 by treatment with NIS and a catalytic amount of TfOH in wet DCM proved problematic and two products were isolated namely the required lactol 15 and the 1,2-oxazoline derivative 14. The latter byproduct was formed by neighboring participation of the C-2 amide after activation of the thioglycoside of 13 with NIS followed by elimination of water. Fortunately, the 1,2-oxazoline could be converted into compound 15 by treatment with dibenzylphosphate in wet DCM34 in an 80% yield. The anomeric hydroxyl of 15 was oxidized using pyridinium chlorochromate (PCC) to give lactone 16 in high yield. Finally, the benzyl ethers and benzylidene acetal of 16 were removed by catalytic hydrogenation over Pd/C to afford target compound 3.
To avoid the formation of the unwanted 1,2-oxazoline observed during the preparation of 3, a different strategy was employed for the preparation of compounds 1 and 2. In the new approach, glycosyl acceptor 627, 28 was employed, which contains an 〈-instead of a β-anomeric thiophenyl moiety (Scheme 2). It was hoped that the hydrolysis of the α-anomeric thioglycoside would be less problematic because the C-2 amide cannot directly displace the anomeric leaving group. As a result, the hydrolysis will proceed through an oxa-carbenium ion, which can more easily be trapped by water to give a lactol then when the anomeric leaving group can be directly displaced by the C-2 amide leading to an 1,2-oxazoline. Furthermore, the acyloxyacyl moiety at C-2′ was introduced by first acylation with (R)-3-levulinoyloxy-hexadecanoic acid (9)35 followed by selective removal of the Lev ester and acylation of the resulting alcohol with an appropriate fatty acid. This approach is attractive because it provides an easy route to a variety of structural analogs. Thus, NIS/TMSOTf mediated glycosylation of glycosyl donor 4 with glycosyl acceptor 6 in DCM at −75 °C provided disaccharide 17 in 82% yield. Treatment of 17 with ethylenediamine in refluxing n-butanol followed by acylation of the resulting C-2′ amino with 9 using DCC as the activating reagent afforded 18 in a yield of 78%. The azido moiety of 18 was reduced with propane-1,3-dithiol and the amine and hydroxyls of the resulting compound were acylated with 8 using DCC and DMAP as activating reagents to provide 19 in an overall yield of 61%. Next, the Lev ester of 19 was selectively removed by treatment with hydrazine acetate, and the hydroxyl of the resulting compound was acylated with 27-benzyloxyoctacosanoic- and octacosanoic acid to give compounds 20 and 21, respectively. Gratifyingly, no 1,2-oxazoline was formed when the anomeric thioglycosides of 20 and 21 were hydrolyzed with NIS/TMSOTf and lactols 22 and 23 were isolated in high yields. Finally, 22 and 23 were oxidized using PCC to give lactones 24 and 25 which were globally deprotected using standard conditions to afford the target compounds 1 and 2, respectively.
Scheme 2.
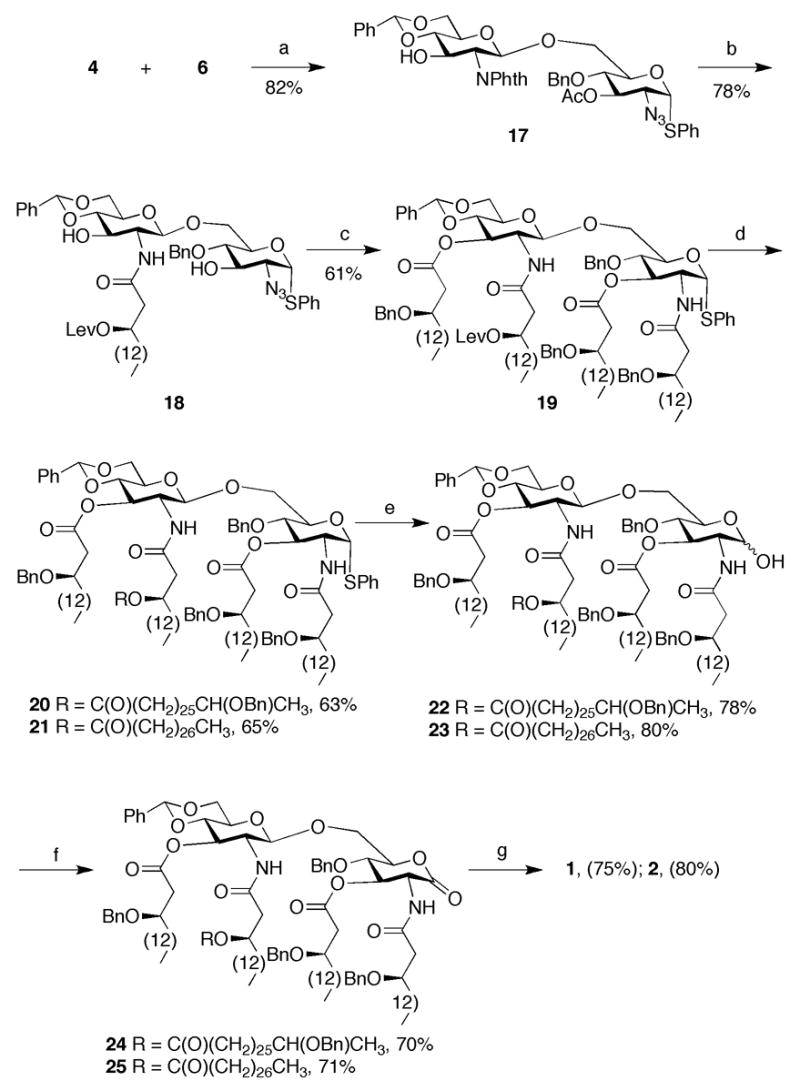
Reagents and conditions: (a) NIS, TfOH, MS 3 Å, DCM, −75 °C; (b) 1: H2N(CH2)2, n-BuOH, 90 °C; 2: 9, DCC, DCM; (c) 1: HS(CH2)3SH, pyridine, Et3N, H2O; 2: 8, DCC, DMAP, DCM; (d) 1: H2NNH2·HOAc, DCM; 2: ROH, DCC, DMAP, DCM; (e) NIS, TfOTMS, DCM, H2O; (f) PCC, MS 3 Å, DCM; (g) Pd/C, H2, t-BuOH, THF.
2.2. Biological evaluations
Compounds 1-3 were tested over a wide concentration range for their ability to activate a human monocytic cell line (Mono Mac 6) to produce TNF-α protein and the resulting values were compared with similar data obtained for E. coli LPS, R. sin-1 LPS, and R. sin-1 lipid A. Thus, incubation of Mono Mac 6 cells with E. coli LPS for 5.5 hours yielded a clear dose response effect of TNF-α production with maximal supernatant concentrations of TNF-α being caused by 10 ng/mL (0.093 nM) of E. coli LPS. Incubations of concentrations up to 10 μg/mL for R. sin-1 LPS (270 nM) and R. sin-1 lipid A (6.6 μM) and up to 100 μg/mL (approximately 50 μM) for the synthetic compounds 1-3 did not induce significant production of TNF-α (Figure 3).
Figure 3.
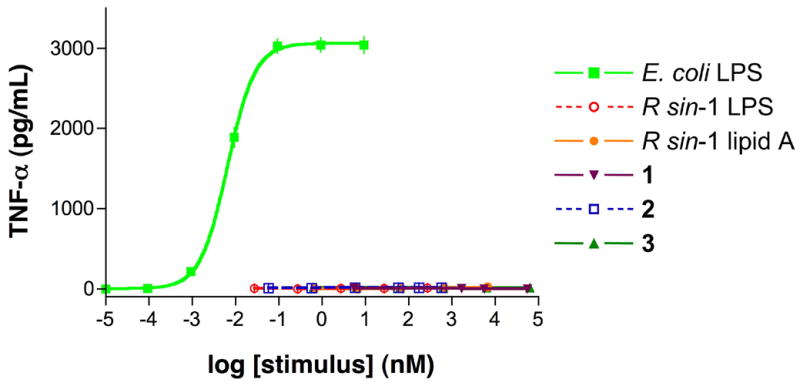
Concentration-response curves of E. coli LPS, R. sin-1 LPS, R. sin-1 lipid A, and synthetic compounds 1–3 in human monocytic cells. Mono Mac 6 cells were incubated for 5.5 h at 37 °C with increasing concentrations of E. coli LPS, R. sin-1 LPS, R. sin-1 lipid A, 1, 2, or 3 as indicated. TNF-α protein in cell supernatants was measured using ELISA. (Please note that R. sin-1 LPS, R. sin-1 lipid A, and 1–3 show background values and therefore overlap in the figure). Treatment with E. coli LPS, R. sin-1 LPS, R. sin-1 lipid A, and 1–3 did not affect cell viability, as judged by cellular exclusion of trypan blue.
Based on its lack of proinflammatory effects, compounds 1-3 were tested over a wide concentration range for its ability to antagonize TNF-α production by monocytic cells incubated with E. coli LPS (10 ng/mL). At the highest concentration tested, compound 1 antagonized the effect E. coli LPS by 86%, and an IC50 (concentration producing 50% inhibition) of 9.1 μM (16.4 μg/mL) was established (Figure 4). A similar inhibition experiment with compound 2 gave a similar IC50 value of 7.5 μM (13.2 μg/mL). However, compound 3 was only able to marginally inhibit the production of TNF-α. Thus, these data show that the hydroxyl of the 27-hydroxyoctacosanoic acid moiety of R. sin-1 lipid A is not important for antagonistic properties. However shortening the octacosanoic acid moiety as in compound 3 resulted in a significant reduction in inhibitory potential. Furthermore, the EC50 values for compounds 1 and 2 are similar to that of R. sin-1 lipid A, which was obtained by mild acid hydrolysis of the corresponding LPS. As expected, R. sin-1 LPS was a more potent inhibitor and an IC50 value of 6.5 nM (239 ng/mL) was determined when incubated with 10 ng/mL of E. coli LPS (Figure 4). Probably, the KDO moiety of R. sin-1 LPS accounts for the greater biological activity.
Figure 4.
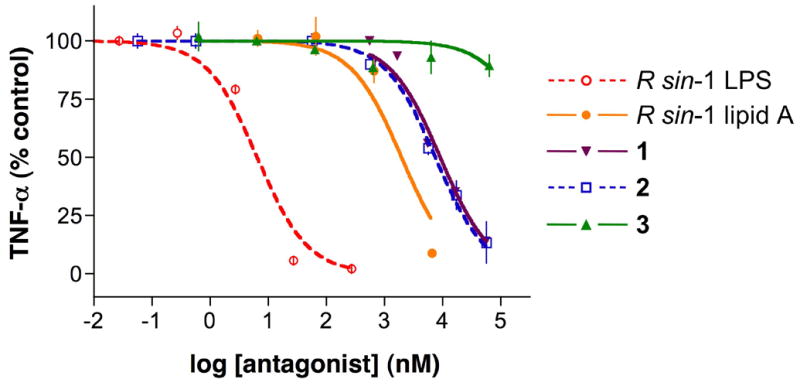
Antagonism of E. coli LPS by R. sin-1 LPS, R. sin-1 lipid A, and synthetic compounds 1–3 in human monocytic cells. TNF-α concentrations were measured after preincubation of Mono Mac 6 cells with increasing concentrations of R. sin-1 LPS, R. sin-1 lipid A, 1, 2, or 3 as indicated for 1 h at 37 °C, followed by 5.5 h of incubation with E. coli LPS (10 ng/mL). Results are expressed as percentage of cytokine concentration of control cells, which are incubated only with E. coli LPS.
3. Conclusions
Several studies have indicated that compounds that can antagonize cytokine production induced by enteric LPS may have the potential to be developed as therapeutics for the treatment of Gram-negative septicemia.36 Success in this area has been limited and most efforts have been directed towards the synthesis of analogs of lipid A of R.sphearoides18, 19 and derivatives of lipid X.15–17 Unique features of R. sin-1 lipid A is lack of phosphates, an 2-aminogluconolactone moiety, and a very long chain fatty acid 27-hydroxyoctacosanoic acid. Previously, we reported that the 2-aminogluconolactone is in equilibrium with the corresponding 2-aminogluconate.37 Furthermore, studies with a compound that was locked in the latter form displayed antagonistic activity. In this report, we have shown that acylation of the C-2′ acyloxyacyl moiety with octacosanoic acid is important for optimal antagonistic properties. The hydroxyl of the natural 27-hydroxyoctacosanoic moiety does, however, not account for inhibitory activity.
4. Experimental
4.1. Chemical synthesis
Column chromatography was performed on silica gel 60 (EM Science, 70-230 mesh). Reactions were monitored by thin-layer chromatography (TLC) on Kieselgel 60 F254 (EM Science), and the compounds were detected by examination under UV light and by charring with 10% sulfuric acid in MeOH. Solvents were removed under reduced pressure at <40 °C. CH2Cl2 was distilled from CaH2 and tetrahydrofuran (THF) was distilled from sodium directly prior to the application. CH3OH was dried by refluxing with mageniusm methoxide and then was distilled and stored under argon. Pyridine was dried by refluxing with CaH2 and then was distilled and stored over molecular sieves (3 Å). Molecular sieves (3 and 4 Å), used for reactions, were crushed and activated in vacuo at 390 °C during 8 h in the first instance and then for 2–3 h at 390 °C directly prior to application. Optical rotations were measured with a Jasco model P-1020 polarimeter. !H NMR and 13C NMR spectra were recorded with Varian spectrometers (model Inova500) equipped with Sun workstations. !H NMR spectra were recorded in CDCl3 and reference to residual CHCl3 at 7.24 ppm, and 13C NMR spectra were referenced to the center peak of CDCl3 at 77.0 ppm. Assignments were made by standard gCOSY and gHSQC. High resolution mass spectra were obtained on a Bruker model Ultraflex MALDI-TOF mass spectrometer. Signals marked with a subscript L symbol belong to the biantennary lipid at C-2′, whereas signals marked with subscript L′ symbol belong to the side chain. Signals marked with a subscript S symbol belong to the monoantennary lipids at C-2, C-3, and C-3′.
4.1.1. (R)-3-Tetradecanoyloxy-hexadecanoic Acid (7)
Myristoyl chloride (2.22 mL, 8.16 mmol) was added dropwise to a solution of 2-(4-bromophenyl)-2-oxoethyl (R)-3-hydroxyhexadecanoate (3.65 g, 7.77 mmol), pyridine (1.26 mL, 15.54 mmol) and DMAP (47 mg, 0.39 mmol) in DCM (70 mL). After stirring the reaction mixture at room temperature for 9 h, it was diluted with DCM (50 mL), and then washed with saturated aqueous NaHCO3 (2 x 60 mL) and brine (2 x 60 mL). The organic phase was dried (MgSO4) and filtered, and the filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography (eluent: toluene) to afford 2-(4-bromophenyl)-2-oxoethyl (R)-3-tetradecanoyloxy-hexadecanoate as a colorless syrup (4.95 g, 94%). Rf = 0.68 (DCM). 1H NMR (300 MHz, CDCl3): δ 7.74 (d, 2H, J = 8.7 Hz, aromatic), 7.64 (d, 2H, J = 8.7 Hz, aromatic), 5.29-5.25 (m, 1H, H-3), 5.25 (s, 2H, H-2), 2.78-2.65 (m, 2H, H-2a, 2b), 2.28 (t, 2H, J2′,3′ = 7.5 Hz, H-2a′, H-2b′), 1.63-1.56 (m, 4H, H-3a′, H-3b′, H-4a, H-4b), 1.23 [broad, 42H, 21 x CH2], 0.88-0.84 (m, 6H, 2 x CH3). HR MS (m/z) calcd for C38H63BrO5[M+Na]+, 701.3757; found, 701.3568. Zinc dust (719 mg, 11.1 mmol) was added portionwise to 2-(4-bromophenyl)-2-oxoethyl (R)-3-tetradecanoyloxyhexadecanoate (1.50 g, 2.21 mmol) in acetic acid (15 mL). The reaction mixture was stirred at 60 °C for 2 h and then diluted with DCM (20 mL). The solids were filtered off through a pad of Celite, and the residue was washed with DCM (3 x 5 mL). The combined filtrates were concentrated in vacuo, and the residue was purified by silica gel column chromatography (eluent: DCM/methanol, 100/1, v/v) to afford 7 as a white solid (1.04 g, 98%). Rf = 0.35 (toluene/ethyl acetate, 4/1, v/v); [〈]24.9D = −1.2° (c = 1.0, CHCl3). 1H NMR (300 MHz, CDCl3): δ 5.21-5.14 (m, 1H, H-3), 2.66-2.51 (m, 2H, H-2a, 2b), 2.26 (t, 2H, J2′,3′ = 7.8 Hz, H-2′), 1.60-1.56 (m, 4H, H-3a′, H-3b′, H-4a, H-4b), 1.23 (bs, 42H, 21 x CH2), 0.88-0.83 (m, 6H, 2 x CH3). 13C NMR (75 MHz, CDCl3): δ 176.28 (C=O), 173.28 (C=O), 69.96 (C-3), 38.87 (C-2), 34.34 (C-2′), 33.97-22.69 [C- (4-15), (3′-13′)], 14.11 (C-14′, 16). HR MS (m/z) calcd for C30H58O4[M+Na]+, 505.4233; found, 505.4313.
4.1.2. Phenyl 3-O-acetyl-6-O-(4,6-O-benzylidene-2-deoxy-2-phthalimido-β-D-glucopyranosyl)-2-azido-4-O-benzyl-2-deoxy-1-thio-β-D-glucopyanoside (11)
A suspension of glycosyl donor 4 (435 mg, 0.81 mmol), acceptor 5 (290 mg, 0.68 mmol) and activated molecular sieves (4 Å, 500 mg) in DCM (15 mL) was stirred under an atmosphere of argon at room temperature for 2 h. The reaction mixture was cooled (−75 °C), and NIS (191 mg, 0.85 mmol) and TfOH (5.8 μL, 0.07 mmol) were added. After being warmed up to −35 °C in 10 min, the reaction mixture was quenched by addition of pyridine (100 μL) and diluted with DCM (20 mL). The reaction mixture was washed with aqueous Na2S2O3 (15%, 2 x 40 mL) and water (2 x 50 mL). The organic phase was dried (MgSO4) and filtered, and the filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography (eluent: hexane/ethyl acetate, 4/1, v/v) to afford 6 as an amorphous solid (461 mg, 84 %). Rf = 0.45 (hexane/ethyl acetate, 5/2, v/v). [α]24.9D = −32.7° (c = 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ 6.94-7.76 (m, 19H, aromatic), 5.59 (s, 1H, CH, benzylidene), 5.41 (d, 1H, J1′,2′ = 8.7 Hz, H-1′), 5.02 (dd, 1H, J2,3 = J3,4 = 9.3 Hz, H-3), 4.71-4.63 (m, 1H, H-3′), 4.43-4.33 (m, 3H, H-1, H-2′, H-6′a), 4.25 (d, 1H, J = 10.8 Hz, CHH, Bn), 4.19 (d, 1H, CHH, Bn), 4.09 (d, 1H, H-6a), 3.88-3.82 (m, 1H, H-6′b), 3.74 (dd, J5,6a = 11.1 Hz, J6a,6b = 4.2 Hz, H-6b), 3.68-3.64 (m, 2H, H-4′, H-5′), 3.46-3.42 (m, 1H, H-5), 3.45 (dd, 1H, J4,5 = 9.6 Hz, H-4), 3.18 (t, 1H, H-2), 1.91 (s, 3H, COCH3). 13C NMR (75 MHz, CDCl3): δ 169.57 (C=O), 168.09 (C=O), 137.13-123.52 (aromatic), 102.01 (CH, benzylidene), 98.64 (C-1′), 85.46 (C-1), 82.25(C-4′), 75.66(C-3 or 4), 75.59 (C-3 or 4), 74.51 (CH2, Bn), 68.68 (C-3′, 6′), 67.64 (C-6), 66.20 (C-5′), 62.84 (C-2), 56.37 (C-2), 23.72 (CH3). HR MS (m/z) calcd for C42H40N4O11S [M+Na]+, 831.2312, found, 831.2519.
4.1.3. Phenyl 2-azido-4-O-benzyl-6-O-{4,6-O-benzylidene-2-deoxy-2[(R)-3-tetradecanoyloxy-hexadecanoylamino]-β-D-glucopyranosyl}-1-thio-β-D-glucopyranoside (12)
Compound 11 (250 mg, 0.31 mmol) was dissolved in a mixture of n-butanol (25 mL) and ethylenediamine (5 mL, 75 mmol). The reaction mixture was stirred at 90 °C for 20 h, after which it was concentrated in vacuo. The residue was dissolved in DCM (30 mL), and the solids were removed by filtration. The filtrate was concentrated in vacuo, and the residue was purified by silica gel column chromatography (eluent: DCM/methanol, 30/1, v/v) to afford phenyl 6-O-(2-amino-4,6-O-benzylidene-2-deoxy-β-D-glucopyranosyl)-2-azido-4-O-benzyl-2-deoxy-1-thio-β-D-glucopyranoside as a white solid (180 mg, 92%). Rf = 0.45 (DCM/methanol, 10/1, v/v). 1H NMR (300 MHz, CDCl3): δ 7.59-7.25 (m, 15H, aromatic), 5.54 (s, 1H, CH, benzylidene), 4.78 (d, 1H, J = 10.4 Hz, CHH, Bn), 4.69 (d, 1H, CHH, Bn), 4.50 (d, 1H, J1,2 = 10.2 Hz, H-1), 4.33-4.28 (m, 2H, H-1′, H-6′a), 4.14 (dd, 1H, J5,6a = 1.8 Hz, J6a,6b = 11.1 Hz, H-6a), 3.80-3.50 (m, 6H, H-3, H-3′, H-4′, H-5′, H-6b, H-6′b), 3.44-3.36 (m, 1H, H-5), 3.34-3.25 (m, 2H, H-3, H-4), 2.78 (t, 1H, J2,3 = 9.6 Hz, H-2), 1.91 (s, 3H, COCH3, acetyl); HR MS (m/z) calcd for C32H36N4O8S[M+Na]+, 659.2151; found, 659.2238. A mixture of DCC (45 mg, 0.218 mmol) and 7 (70 mg, 0.145 mmol) in DCM (2 mL) was stirred at room temperature for 10 min, and then the above mentioned amino derivative (58 mg, 91 μmol) in DCM (1 mL) was added. The reaction mixture was stirred at room temperature for 2 h, after which the solids were removed by filtration, and the residue was washed with DCM (2 x 3 mL). The combined filtrates were concentrated in vacuo, and the residue was purified by silica gel column chromatography (eluent: DCM/methanol, 50/1, v/v) to afford 12 as a white solid (85 mg, 85%). Rf = 0.60 (DCM/diethyl ether, 6/1, v/v); [α]24.8D = −18.3° (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.57-7.31 (m, 15H, aromatic), 5.50 (s, 1H, CH, benzylidene), 5.04 (m, 1H, H-3L), 4.76 (d, 1H, J = 11.5 Hz, CHH, Bn), 4.73 (d, 1H, J1′,2′ = 8.0 Hz, H-1′), 4.67(d, 1H, CHH, Bn), 4.56 (d, 1H, J1, 2 = 10.0 Hz, H-1), 4.46 (bs, 1H, OH′), 4.34 (dd, 1H, J5,6′a = 5.0 Hz, J6′a,6′b = 10.0 Hz, H-6′a), 4.16 (d, 1H, H-6a), 4.06 (dd, 1H, J2′,3′ = 8.5 Hz, J3′,4′ = 9.5 Hz, H-3′), 3.79 (dd, 1H, J5′,6′b = 10.5 Hz, H-6b′), 3.60-3.70 (m, 3H, H-3, H-5, H-6b), 3.58 (dd, 1H, J4′,5′ = 10.0 Hz, H-4′), 3.52 (m, 1H, H-2′), 3.45 (ddd, 1H, H-5′), 3.27 (dd, 1H, J2,3 = 9.5 Hz, H-2), 3.27 (dd, 1H, J3,4 = J4,5 = 9.5 Hz, H-4), 2.76( b, 1H, OH), 2,25-2.31 (m, 2H, H-2L), 1.53-1.58 (m, 4H, H-4L, H-3L′), 1.25 (m, 42H, 21 x CH2, lipid), 0.87-0.90 (m, 6H, 2 x CH3, lipid). 13C NMR (75 MHz, CDCl3): δ 174.31 (C=O), 171.72 (C=O), 137.65-126.36 (aromatic), 101.96 (CH, benzylidene), 101.13 (C-1′), 85.57 (C-1), 81.49 (C-4′), 78.59 (C-5), 77.86 (C-4), 77.21 (C-3), 74.84 (CH2, Bn), 71.72 (C-3′), 71.48 (C-3L), 68.78 (C-6), 68.58 (C-6′), 66.44 (C-2), 59.14 (C-2′). HR MS (m/z) calcd for C62H92N4O11S[M+Na]+, 1123.6381; found, 1123.9268.
4.1.4. Phenyl 4-O-benzyl-6-O-{4,6-O-benzylidene-3-O-[(R)-3-benzyloxy-hexadecanoyl]-2-deoxy-2-[(R)-3-tetradecanoyloxy-hexadecanoylamino]-β-D-glucopyranosyl}-2-[(R)-3-benzyloxy-hexadecanoylamino]-3-O-[(R)-3-benzyloxy-hexadecanoyl]-2-deoxy-1-thio-β-D-glucopyranoside (13)
1,3-Propanedithiol (0.18 mL, 1.75 mmol) was added to a stirred solution of 12 (95 mg, 0.086 mmol) and triethylamine (0.2 mL) in a mixture of pyridine and H2O (7 mL, 6/1, v/v). The reaction mixture was stirred at room temperature for 16 h and then concentrated in vacuo to dryness. The residue was purified by silica gel column chromatography (eluent: DCM/methanol, 60/1, v/v) to afford phenyl 2-amino-4-O-benzyl-6-O-{4,6-O-benzylidene-2-deoxy-2-[(R)-3-tetradecanoyloxy-hexadecanoylamino]-β-D-glucopyranosyl}-2-deoxy-1-thio-β-D-glucopyranoside as a white solid (85.5 mg, 92.5%). Rf = 0.40 (DCM/diethyl ether, 6/1, v/v). 1H NMR (500 MHz, CDCl3): δ 7.26-7.53 (m, 15H, aromatic), 6.08 (d, 1H, JNH,2 = 5.0 Hz, NH), 5.57 (s, 1H, CH, benzylidene), 5.06 (m, 1H, H-3L), 4.82 (d, 1H, J = 11.5 Hz, CHH, Bn), 4.72 (d, 1H, J1′,2′ = 9.0 Hz, H-1′), 4.66 (d, 1H, CHH, Bn), 4.53 (d, 1H, J1,2 = 9.5 Hz, H-1), 4.34 (dd, 1H, J5′,6′a = 5.0 Hz, J6′a,6′b = 10.5 Hz, H-6′a), 4.20 (d, 1H, H-6a), 4.05 (dd, 1H, J2′,3′ = 9.5 Hz, J3′,4′ = 9.0 Hz, H-3′), 3.79 (dd, 1H, J5′,6′b = 10.0 Hz, H-6′b), 3.64-3.71 (m, 2H, H-5, H-6b), 3.58 (dd, 1H, J4′5′ = 9.0 Hz, H-4′), 3.49-3.52 (m, 2H, H-2′, H-3), 3.45 (ddd, 1H, H-5′), 3.26 (dd, 1H, J3,4 = J4,5 = 9.0 Hz, H-4), 2.63 (dd, 1H, J2,3 = 10,0 Hz, H-2), 2.27-2.43 (m, 4H, H-2L, H-2L′), 1.50-1.58 (m, 4H, H-4L, H-3L′), 1.25 (m, 42H, 21 x CH2, lipid), 0.88-0.90 (m, 6H, 2 x CH3, lipid). 13C NMR (75 MHz, CDCl3): δ 126.60-138.18 (aromatic), 102.18 (CH, benzylidene), 101.45 (C-1′), 89.18 (C-1), 81.73 (C-4′), 78.88 (C-5), 78.55 (C-3), 77.45 (C-4), 74.71 (CH2, Bn), 72.04 (C-3′), 71.59 (C-3L), 69.48 (C-6), 68.83 (C-6′), 66.67 (C-5′), 59.49 (C-2′), 56.50 (C-2). HR MS (m/z) calcd for C62H94N2O11S[M+Na]+, 1097.6476; found, 1097.6069. A mixture of DCC (57.5 mg, 0.279 mmol) and (R)-3-benzyloxy-hexadecanoic acid 8 (85 mg, 0.232 mmol) in DCM (3 mL) was stirred at room temperature for 10 min, after which DMAP (6.2 mg, 0.051 mmol) and the above mentioned amino derivative (50 mg, 46.5 μmol) in DCM (1.5 mL) was added. The reaction mixture was stirred at room temperature for 5 h, and then the solids were removed by filtration and the residue was washed with DCM (2 x 2 mL). The combined filtrates were concentrated in vacuo, and the residue was purified by silica gel column chromatography (eluent: hexane/ethyl acetate, 5/1, v/v) to afford 13 as a white solid (62 mg, 63%). Rf = 0.60 (hexane/ethyl acetate, 3/1, v/v). [α]25.2D = +9.1° (c = 1.0, CHCl3). 1H NMR (300M Hz, CDCl3): δ 7.14-7.43 (m, 30H, aromatic), 6.46 (d, 1H, JNH,2 = 9.6 Hz, NH), 5.42 (s, 1H, CH, benzylidene), 5.36 (d, 1H, JNH,2′ = 9.0 Hz, NH′), 5.24 (dd, 1H, J2′.3′ = 9.6 Hz, J3′,4′ = 9.9 Hz, H-3′), 5.10 (dd, 1H, J2,3 = 8.7 Hz, J3,4 = 10.2 Hz, H-3), 4.96 (m, 1H, H-3L), 4.67 (d, J1′,2′ = 8.4 Hz, H-1′), 4.41-4.61 (m, 9H, H-1, 4 x CH2, Bn), 4.32 (dd, 1H, J5,6a = 4.8 Hz, J6a,6b = 10.5 Hz, H-6a), 4.04 (m, 1H, H-2), 3.96 (d, 1H, H-6′a), 3.59-3.84 (m, 7H, H-2′, H-4′, H-6b, H-6′b, 3 x H-3s), 3.36-3.49 (m, 3H, H-4, H-5, H-5′), 2.09-2.69 (m, 10H, H-2L, H-2L′, 3 x H-2S), 1.57-0.98 (m, 118H, 59 x CH2, lipid), 0.82 (m, 15H, 5 x CH3, lipid). 13C NMR (75Hz, CDCl3): δ 126.34-138.73 (aromatic), 101.62 (C-1′, CH, benzylidene,), 86.95 (C-1), 79.68 (C-5 or 5′), 79.13 (C-4′), 75.67-76.30 (C-3, 3*C-3s), 74.67 (C-4), 71.18-71.63 (C-3′,3L, 4 x CH2, Bn), 68.86 (C-6 or 6′), 68.33 (C-6 or 6′), 66.43 (C-5 or 5′), 55.24 (C-2′), 53.10 (C-2); HR MS (m/z) calcd for C131H202N2O17S[M+Na]+, 2130.4622; found, 2130.6140.
4.1.5. 4-O-Benzyl-6-O-{4,6-O-benzylidene-3-O-[(R)-3-benzyloxy-hexadecanoyl]-2-deoxy-α-2-[(R)-3-tetradecanoyloxy-hexadecanoylamino]-β-D-glucopyranosyl}-2-[(R)-3-benzyloxy-hexadecanoylamino]-3-O-[(R)-3-benzyloxy-hexadecanoyl]-2-deoxy-α-D-glucopyranoside (15)
TfOH (0.5 μL, 4.4 μmol) was added to a stirred solution of 13 (31 mg, 14.7 μmol) and NIS (9.9 mg, 44 μmol) in a mixture of DCM and H2O (4 mL, 100/1, v/v) at 0 °C. The reaction mixture was vigorously stirred for 30 min until TLC analysis indicated that the reaction had gone to completion, and then it was diluted with DCM (10 mL) and washed with aqueous Na2S2O3 (15%, 15 mL) and water (2 x 10 mL). The organic phase was dried (MgSO4) and filtered, and the filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography (elulent: hexane/ethyl acetate, 6/1-4/1, v/v) to afford 15 as a white solid (10.5 mg, 35%) and its 1,2-oxazoline derivative 14 (15 mg, 51%). A mixture of the resulting 1,2-oxazoline and dibenzylphosphate (4 mg, 15 μmol) in wet 1,2-dichloroethane (1 mL) was stirred at room temperature for 1 h. The reaction mixture was diluted with DCM (10 mL) and wahsed with saturated aqueous NaHCO3 (2 x 8 mL) and brine (2 x 8 mL). The organic phase was dried (MgSO4) and filtered, and the filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography (eluent: hexane/ethyl acetate, 4/1, v/v) to afford 15 as a white solid (13.5 mg, 90%). The overall yield of 15 for the two steps was 80%. Rf = 0.2 (hexane/ethyl acetate, 2.5/1, v/v). 1H NMR (500 MHz, CDCl3): δ 7.18-7.40 (m, 25H, aromatic), 6.28 (d, 1H, JNH,2 = 9.0 Hz, NH), 5.95 (d, 1H, JNH′,2′ = 8.5 Hz, NH′), 5.44 (s, 1H, CH, benzylidene), 5.39-5.43 (m, 2H, H-3, H-3′), 5.17 (d, 1H, J1′,2′ = 8.5 Hz, H-1′), 5.09 (m, 1H, H-1), 4.95 (m, 1H, H-3L), 4.40-4.61 (m, 8H, 4 x CH2, Bn), 4.35 (dd, 1H, J5′,6′a = 5.0 Hz, J6′a,6′b = 10.0 Hz, H-6′a), 4.20 (m, 1H, H-2), 4.07 (m, 1H, H-5), 3.96 (d, 1H, H-6a), 3.74-3.85 (m, 4H, H-6′b, 3 x H-3S), 3.61-3.68 (m, 3H, H-2′, H-4′, H-6b), 3.52 (ddd, 1H, H-5′), 3.37 (t, 1H, J3,4 = J4,5 = 10.0 Hz, H-4), 2.20-2.66 (m, 10H, H-2L, H-2L′, 3 x H-2S), 1.59-1.10 (m, 118H, 59 x CH2, lipid), 0.84 (m, 15H, 5 x CH3, lipid). 13C NMR (75Hz, CDCl3): δ 126.0-138.6 (aromatic), 101.5 (CH, benzylidene), 100.4 (C-1′), 91.5 (C-1), 79.0 (C-4′), 76.4 (C-4), 75.5-76.4 (C-3x3S), 74.4 (CH2, Bn), 73.5 (C-3 or 3′), 71.6 (C-5), 71.0-71.4 (C-3L, 3 or 3′, 3 x CH2, Bn), 68.7 (C-6′), 67.5 (C-6), 66.5 (C-5′), HR MS (m/z) calcd for C125H198N2O18[M+Na]+, 2038.4537; found, 2038.3850.
4.1.6. 4-O-Benzyl-6-O-{4,6-O-benzylidene-3-O-[(R)-3-benzyloxy-hexadecanoyl]-2-deoxy-2-[(R)-3-tetradecanoyloxy-hexadecanoylamino]-β-D-glucopyranosyl}-2-[(R)-3-benzyloxy-hexadecanoylamino]-3-O-[(R)-3-benzyloxy-hexadecanoyl]-2-deoxy-D-glucono-1,5-lactone (16)
A suspension of 15 (8.0 mg, 4.0 μmol) and activated molecular sieves (3 Å, 15 mg) in DCM (2.0 mL) was stirred at room temperature under an atmosphere of argon for 1 h. PCC (4.3 mg, 20 μmol) was then added and the reaction mixture was stirred for 1 h until TLC analysis indicated the reaction had gone to completion. The reaction mixture was purified by Iatro beads column chromatography (eluent: hexane/ethyl acetate, 4/1, v/v) to afford 16 as a white solid (7 mg, 87%). [α]23D = −12.5° (c = 1, CHCl3). Rf = 0.65 (hexane/ethyl acetate, 2.5/1, v/v). 1H NMR (500 MHz, CDCl3): δ 7.20-7.38 (m, 25H, aromatic), 6.82 (d, 1H, JNH,2 = 8.5 Hz, NH), 6.61 (d, 1H, JNH′,2′ = 7.5Hz, NH′), 5.60 (t, 1H, J2′,3′ = J3′,4′ = 10.0 Hz, H-3′), 5.39 (s, 1H, CH, benzylidene), 5.34 (dd, 1H, J2,3 = J3,4 = 10.0 Hz, H-3), 5.07 (m, 1H, H-3L), 4.95 (d, 1H, J1′,2′ = 8.0 Hz, H-1′), 4.79 (t, 1H, H-2), 4.37-4.60 (m, 9H, H-5, 4 x CH2, Bn), 4.28 (dd, 1H, J5′,6′a = 3.5 Hz, J6′a,6′b = 9.5 Hz, H-6′a), 4.02-4.06 (m, 2H, H-4, H-6a), 3.80-3.85 (3H, 3 x 3s), 3.75 (t, 1H, J5′,6′b = 10.0 Hz, H-6′b), 3.52-3.60 (m, 4H, H-2′, H-4′, H-5′, H-6a), 2.25-2.68 (m, 10H, H-2L, H-2L′, 3 x H-2S), 1.60-1.03 (m, 118H, 59 x CH2, lipid), 0.86 (m, 15H, 5 x CH3, lipid). HR MS(m/z) calcd for C125H196N2O18[M+Na]+, 2036.4381; found, 2036.8009.
4.1.7. 2-Deoxy-6-O-{2-deoxy-3-O-[(R)-3-hydroxy-hexadecanoyl]-2-[(R)-3-tetradecanoyloxy-hexadecanoylamino-β-D-glucopyranosyl}-2-[(R)-3-hydroxy-hexadecanoylamino]-3-O-[(R)-3-hydroxy-hexadecanoyl]-D-glucono-1,5-lactone (3)
Pd/C (10 mg) was added to the solution of lactone 16 (7 mg, 3.5 μmol) in a mixture of THF and t-BuOH (3 mL, 1/1, v/v ). The reaction mixture was shaken under an atmosphere of H2 (15 psi) at room temperature for 24 h, after which the catalyst was removed by filtration and washed with THF (3 x 0.5 mL). The combined filtrates were concentrated in vacuo to afford 3 as a white solid (5 mg, 81%). 1H NMR (500 MHz, CDCl3): δ 5.34 (t, 1H, J2,3 = J3,4 = 10.0 Hz, H-3), 5.10 (m, 1H, H-3L), 5.00 (t, 1H, J2′,3′ = J3′,4′ = 10.0 Hz, H-3′), 4.56 (d, 1H, J1′,2′ = 8.5 Hz, H-1′), 4.26 (m, 1H, H-5), 4.21 (d, 1H, H-2), 4.19 (d, 1H, J6a,6b = 11.0 Hz, H-6a), 3.74-3.92 (m, 7H, H-4, H-2′, H-6′a, H-6b, 3 x H-3s), 3.63 (m, 1H, H-6′b), 3.47 (dd, 1H, J4′,5′ = 9.0 Hz, H-4′), 3.27 (m, 1H, H-5′), 2.27-2.18 (m, 10H, H-2L, 2L′, 3 x H-2S), 1.63-0.96 (m, 118H, 59 x CH2, lipid), 0.84 (m, 15H, 5 x CH3, lipid). HR MS (m/z) calcd for C90H168N2O18[M+Na]+, 1588.2190; found, 1588.4548.
4.1.8. Phenyl 3-O-acetyl-6-O-(4,6-O-benzylidene-2-deoxy-2-phthalimido-β-D-glucopyranosyl)-2-azido-4-O-benzyl-2-deoxy-1-thio-β-D-glucopyanoside (17)
The glycosylation of 4 (87 mg, 0.20 mmol) and 6 (58 mg, 0.136 mmol) was performed similar to the synthesis of 11 using NIS (47 mg, 0.21 mmol) and TfOH (1.7 μL, 0.02 mmol) in DCM (2 mL) to afford 13 as an amorphous solid (90 mg, 82%). Rf = 0.41 (hexane/ethyl acetate, 5/2, v/v). [α]24.9D = −40.5° (c = 1.0, CHCl3). 1H NMR (300 MHz, CDCl3): δ 7.70-6.88 (m, 19H, aromatic), 5.56-5.55 (m, 2H, H-1, CH, benzylidene), 5.32-5.23 (m, 2H, H-1′, H-3), 4.71-4.63 (m, 1H, H-3′), 4.40-4.27 (m, 3H, H-2′, H-5, H-6′a), 4.25 (d, 1H, J = 10.5 Hz, CHH, Bn), 4.03-3.96 (m, 2H, H-6a, CHH, Bn), 3.85-3.77 (m, 3H, H-2, H-6b, H-6′b), 3.66-3.61 (m, 2H, H-4′, H-5′), 3.47 (dd, 1H, J3,4 = J4,5 = 9.6 Hz, H-4), 1.90 (s, 3H, COCH3, acetyl). 13C NMR (75 MHz, CDCl3): δ 169.30 (C=O), 136.89-123.38 (aromatic), 101.79 (CH, benzylidene), 98.59 (C-1′), 86.92 (C-1), 81.75 (C-4′), 75.98 (C- 4), 74.26 (CH2, Bn), 73.01 (C-3 ), 70.51 (C-5), 68.36 (C-3′, C-6′), 67.26 (C-6), 66.17 (C-5′), 61.93 (C-2), 56.28 (C-2), 20.59 (CH3, acetyl). HR MS (m/z) calcd for C42H40N4O11S [M+Na]+, 831.2312; found, 831.1761.
4.1.9. Phenyl 2-azido-4-O-benzyl-6-O-{4,6-O-benzylidene-2-deoxy-2-[(R)-3-levulinoyloxy-hexadecanoylamino]-β-D-glucopyranosyl}-2-deoxy-1-thio-α-D-glucopyranoside (18)
The phthalimido and acetyl group of 17 (210 mg, 24.7 mmol) were removed similar to the deprotection of 11 in a mixture of n-butanol (15 mL) and ethylenediamine (3 mL, 45 mmol) to afford phenyl 6-O-(2-amino-4,6-O-benzylidene-2-deoxy-β-D-glucopyranosyl)-2-azido-4-O-benzyl-2-deoxy-1-thio-α-D-glucopyranoside as a colorless syrup (142 mg, 91%). Rf = 0.52 (DCM/methanol, 1/9, v/v). DCC (67 mg, 32.4 mmol) was added to a solution of (R)-3-levulinoyloxyhexadecanoic acid 9 (100 mg, 27.0 mmol) in DCM (5 mL) and the resulting solution was stirred for 10 min. Next, the above described amino derivative (142 mg, 22.5 mmol) in DCM (2 mL) was added and the reaction mixture was stirred for 12 h at room temperature. The solids were filtered-off and the residue was washed with DCM (2 × 3 mL). The combined filtrates were concentrated in vacuo and the residue was purified by silica gel column chromatography (eluent: DCM/methanol, 50/1, v/v) to afford 18 as a white solid (189 mg, 86%). Rf = 0.55 (DCM/diethyl ether, 6/1, v/v). [α]23D = +4.9° (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.53-7.18 (m, 15H, aromatic), 6.34 (d, 1H, JNH′,2′ = 7.0 Hz, NH′), 5.61 (d, 1H, J1,2 = 5.0 Hz, H-1), 5.56 (s, 1H, CH, benzylidene), 5.07-5.02 (m, 1H, H-3L), 4.91 (d, 1H, J1′,2′ = 8.5 Hz, H-1′), 4.84 (d, 1H, J = 11.5 Hz, CHH, Bn), 4.74 (d, 1H, CHH, Bn), 4.37 (d, 1H, J = 10.0 Hz, H-5), 4.32 (dd, 1H, J5′,6′a = 5.0 Hz, J6′a,6′b = 10.5 Hz, H-6′a), 4.23 (t, 1H, J2′,3′ = J3′,4′ = 9.5 Hz, H-3′), 4.13 (d, 1H, J6a,6b = 10.0 Hz, H-6a), 3.98 (dd, 1H, J = 10.0 Hz, J = 9.0 Hz, H-3), 3.89-3.86 (m, 2H, H-2, H-6′b), 3.80 (t, 1H, J5,6b = 10.0 Hz, H-6b), 3.63 (t, 1H, J3,4 = J4,5 = 9.5 Hz, H-4), 3.58 (dd, 1H, J4′,5′ = 9.0 Hz, H-4′), 3.54-3.48 (m, 2H, H-2′, H-5′), 2.97-2.24 (m, 6H, H-2L, 2 x CH2, Lev), 2.17 (s, 3H, CH3, Lev), 1.59-1.51 (m, 2H, H-4L), 1.24 (broad, 22H, 11 x CH2, lipid), 0.92-0.90 (m, 3H, CH3, lipid). 13C NMR (75 MHz, CDCl3): δ 137.76-123.74 (aromatic), 102.02 (CH, benzylidene), 100.86 (C-1′), 87.64 (C-1), 81.62(C-4′), 78.43 (C-4), 75.26 (CH2, Bn), 74.31 (C-3), 72.6 4(C-3L), 71.62 (C-3, 5), 71.45 (C-3′), 69.02 (C-6′), 68.33 (C-6), 66.71 (C-5′), 64.19 (C-2), 59.41 (C-2′). HR MS (m/z) calcd for C53H72N4O11S[M+Na]+: calcd, 1012.2143; found, 1012.3649.
4.1.10. Phenyl 4-O-benzyl-6-O-{4,6-O-benzylidene-3-O-[(R)-3-benzyloxy-hexaadecanoyl-2-deoxy-2-[(R)-3-levulinoyloxy-hexadecanoylamino]-β-D-glucopyranosyl}-2-[(R)-3-benzyloxy-hexadecanoylamino]-3-O-[(R)-3-benzyloxy-hexadecanoyl]-2-deoxy-1-thio-α-D-glucopyranoside (19)
Triethylamine (1.0 mL) was added to a stirred solution of 18 (450 mg, 0.455 mmol) and propanedithiol (491 mg, 45.5 mmol) in pyridine (15 mL) and H2O (1.5 mL). The reaction mixture was stirred at room temperature for 14 h, after which it was concentrated in vacuo to dryness. The residue was co-evaporated with toluene (2 x 30 mL) and ethanol (2 x 20 mL) and then purified by silica gel column chromatography (eluent: DCM/methanol, 30/1, v/v) to afford phenyl 2-amino-4-O-benzyl-6-O-{4,6-O-benzylidene-2-deoxy-2-[(R)-3-levulinoyloxy-hexadecanoylamino]-β-D-glucopyranosyl}-2-deoxy-1-thio-α-D-glucopyranoside as a colorless syrup (397 mg, 91 %). Rf = 0.45 (DCM/methanol, 15/1, v/v). A mixture of (R)-3-benzyloxy-hexadecanoic acid 8 (226 mg, 624 μmol) and DCC (154 mg, 749 μmol) was stirred for 10 min, and then the above described amine (150 mg, 156 μmol) in DCM (2 mL) and DMAP (3.0 mg, 24.7 μmol) were added. After stirring the reaction mixture for 16 h, the solids were filtered off and the residue was washed with DCM (2 x 1 mL). The combined filtrates were concentrated in vacuo, and the residue was purified by silica gel column chromatography (eluent: toluene/ethyl acetate, 10/1, v/v) to afford 19 as a white solid (209 mg, 67%). Rf = 0.56 (DCM/methanol, 60/1, v/v). [α]23D = +11.5° (c = 1.0, CHCl3). 1H NMR (600 MHz, CDCl3): δ 7.38-7.20 (m, 30H, aromatic), 6.55 (d, 1H, JNH′,2′ = 9.6 Hz, NH′), 6.45 (d, 1H, JNH,2 = 8.4 Hz, NH), 5.71 (d, 1H, J1,2 = 4.8 Hz, H-1), 5.33-5.31 (m, 2H, H-3′, CH, benzylidene), 5.24 (t, 1H, J2,3 = J3,4 = 9.6 Hz, H-3), 4.88 (m, 1H, H-3L), 4.83 (d, 1H, J1′,2′ = 8.4 Hz, H-1′ ), 4.62-4.41 (m, 9H, H-2, 4 x C H2, Bn), 4.41-4-27 (m, 2H, H-5, H-6′a), 4.17 (m, 1H, H-2′ ), 4.01 (d, 1H, J6a,6b = 10.8 Hz, H-6a), 3.92 (d, 1H, H-6b), 3.80-3.76 (m, 4H, H-4, 3 x H-3S), 3.69 (t, 1H, J 5′,6′b = J6′a,6′b = 10.6 Hz, H-6′b), 3.62 (t, 1H, J 3′,4′ = J4′,5′ = 9.6 Hz, H-4′), 3.47 (m, 1H, H-5′), 2.87-2.21 (m, 12H, H-2L, 3 x H-2S, 2 x CH2, Lev), 2.07(s, 3H, CH3, Lev), 1.59-1.51 (m, 8H, H-4L, 3 x H-4S), 1.23 (broad, 88H, 44 x CH2, lipid), 0.88-0.86 (m, 12H, 4 x CH3, lipid). HR MS (m/z) calcd for C122H182N2O18S[M+Na]+, 2018.3006; found, 2018.2588.
4.1.11. Phenyl 4-O-benzyl-6-O-{4,6-O-benzylidene-3-O-[(R)-3-benzyloxy-hexadecanoyl-2-deoxy-2-[(R)-3-(27-benzyloxy-octacosanoyloxy)-hexadecanoylamino]-β-D-glucopyranosyl}-2-[(R)-3-benzyloxy-hexadecanoylamino]-3-O-[(R)-3-benzyloxy-hexadecanoyl]-2-deoxy-1-thio-α-D-glucopyranoside (20)
To a solution of 19 (50.0 mg, 25 μmol) in DCM (2 mL) was added dropwise hydrazine acetate (2.4 mg, 26 μmol) in methanol (0.2 mL). The resulting mixture was stirred at room temperature for 3 h after which TLC indicated completion of the reaction. The reaction mixture was concentrated in vacuo, and the residue was purified by preparative silica gel TLC chromatography (eluent: DCM/methanol, 60/1, v/v) to afford the alcohol as a white solid (43 mg, 90%). Rf = 0.48 (DCM/methanol, 60/1, v/v). 1H NMR (600 MHz, CDCl3): δ 7.37-7.21 (m, 30H, aromatic), 6.48 (d, 1H, JNH,2 = 7.8 Hz, NH), 5.68-5.65 (m, 2H, H-1, NH′ ), 5.46 (t, 1H, J2′,3′ = J3′,4′ = 9.6 Hz, H-3′), 5.41 (s, 1H, CH, benzylidene), 5.25 (t, 1H, J2,3 = J3,4 = 10.2 Hz, H-3), 4.79 (d, 1H, J1′,2′ = 7.8 Hz, H-1′ ), 4.62-4.41 (m, 9H, H-2, 4 x CH2, Bn), 4.32 (m, 1H, H-5), 4.29 (dd, 1H, J5′,6′a = 4.8 Hz, J6′a,6′b = 10.2 Hz, H-6′a), 3.98 (d, 1H, J6a,6b = 9.6 Hz, H-6a), 3.84-3.69 (m, 8H, H-H-2′, H-4, H-6b, H-6′b, 4 x H-3S), 3.63 (t, 1H, J3′,4′ = J4′,5′ = 9.6 Hz, H-4′), 3.48 (m, 1H, H-5′ ), 2.62 -2.56 (m, 2H, H-2S), 2.49 (dd, 1H, J = 5.4 Hz, J = 15.0 Hz, H-2Sa), 2.43( dd, 1H, J = 5.4 Hz, J = 15.6 Hz, H-2Sb), 2.32-2.27 (m, 2H, H-2S), 1.96-1.88 (m, 2H, H-2S), 1.57-1.55 (m, 8H, 4 x H-4S), 1.24 (broad, 88H, 44 x CH2, lipid), 0.87-0.85 (m, 12H, 4 x CH3, lipid). 13C NMR (75 MHz, CDCl3): δ 172.63 (C=O), 172.10 (C=O), 171.44 (C=O), 171.26 (C=O), 138.45-126.26 (aromatic), 101.49 (CH, benzylidene), 100.03 (C-1), 87.53 (C-1), 78.70 (C-4′), 77.20-75.96 (C-4, 3 x C-3S), 75.96 (C-4), 75.63 (CH2, Bn), 74.56 (C-3), 74.56, 73.46, 71.41, 71.19, 70.95 (C-3′, 5, 3 x CH2, Bn), 68.54 (C-6′), 68.19 (C-3S), 66.94 (C-6), 66.43 (C-5′), 55.40 (C-2′), 52.72 (C-2). A mixture of 27-hydroxyoctacosanoic acid 10 (22 mg, 42 μmol) and DCC (13 mg, 65 μmol) in DCM (1.5 mL) was stirred at room temperature for 10 min, and then the above alcohol (50 mg, 26 μmol) in DCM (1 mL) and DMAP (2.5 mg, 21 μmol) were added. The reaction mixture was stirred at room temperature for 15 h, the solids were filtered off and the residue was washed with DCM (2 x 1 mL). The combined filtrates were concentrated in vacuo, and the residue was purified by preparative silica gel TLC chromatography (eluent: DCM/methanol, 60/1, v/v) to afford 20 as a white fluffy solid (35.7 mg, 63%). Rf = 0.65 (DCM/methanol, 60/1, v/v). 1H NMR (500 MHz, CDCl3): δ = 7.40-7.18 (m, 30H, aromatic), 6.48 (d, 1H, JNH,2 = 8.5 Hz, NH), 5.72 (d, 1H, J 1,2 = 4.5 Hz, H-1), 5.61 (d, 1H, JNH′,2′ = 8.5 Hz, NH′ ), 5.39 (s, 1H, CH, benzylidene), 5.39 (t, 1H, J2′,3′ = J3′,4′ = 9.5 Hz, H-3′), 5.27 (dd, 1H, J = 9.5 Hz, J = 10.0Hz, H-3), 4.97 (m, 1H, H-3L), 4.89 (d, 1H, J 1′,2′ = 8.5 Hz, H-1′ ), 4.62-4.42 (m, 11H, H-2, 5 x CH2, Bn), 4.38 (m, 1H, H-5), 4.31 (dd, 1H, J 5′,6′a = 5.0 Hz, J6′a,6′b = 11.0 Hz, H-6′a), 3.98 (d, 1H, J6a,6b = 10.5 Hz, H-6a), 3.89-3.83 (m, 2H, H-6b, H-3S), 3.80-3.79 (m, 2H, 2 x H-3S), 3.73-3.66 (m, 3H, H-2′, H-4, H-6′b), 3.63 (t, 1H, J4′,5′ = 9.5 Hz, H-4′), 3.52-3.47 (m, 2H, H-5′, H-27L′), 2.65 -2.57 (m, 2H, H-2S), 2.51 (dd, 1H, J = 5.5 Hz, J = 15.0 Hz, H-2Sa), 2.45 (dd, 1H, J = 5.5 Hz, J = 15.5 Hz, H-2Sb), 2.33-2.19 (m, 4H, 2 x H-2 S, H-2La, H-2La′), 2.09-1.98 (m, 2H, H-2Lb, H-2Lb′), 1.59-1.40 (m, 12H, H-4L, H-3L′, H-26L′, 3 x H-4S), 1.25 (broad, 176H, 88 x CH2, lipid), 1.18 (d, 3H, J27L′,28L′ = 6.0 Hz, 28L′), 0.89-0.85 (m, 12H, 4 x CH3, lipid). 13C NMR (75 MHz, CDCl3): δ 173.98 (C=O), 172.45 (C=O), 171.40 (C=O), 171.26 (C=O), 169.96 (C=O), 139.65-126.31 (aromatic), 101.96 (CH, benzylidene), 101.01 (C-1′), 87.5 8 (C-1), 79.05 (C-4′), 77.26 (C-3S), 76.68 (C-3S), 75.96 (C-3S), 75.26 (C-27L′), 74.51 (CH2, Bn), 73.88 (C-3), 71.86 (C-5), 70.96 (C-3′), 70.68-70.42 (C-4, C-3L, 3 x CH2, Bn), 68.85 (C-6′ ), 67.78 (C-6), 66.56 (C-5′), 55.86 (C-2′), 53.12 (C-2). HR MS (m/z) calcd for C152H236N2O18S[M+Na]+, 2432.7232; found, 2432.9846.
4.1.12. Phenyl 4-O-benzyl-6-O-{4,6-O-benzylidene-3-O-[(R)-3-benzyloxy-hexadecanoyl]-2-deoxy-2-[(R)-3-octacosanoyloxy-hexadecanoylamino]-β-D-glucopyranosyl}-2-[(R)-3-benzyloxy-hexadecanoylamino]-3-O-[(R)-3-benzyloxy-hexadecanoyl]-2-deoxy-1-thio-α-D-glucopyranoside (21)
The above alcohol intermediate (40 mg, 20 μmol) was acylated similar to the synthesis of 20 with octacosanoic acid (12 mg, 30 μmol), using DCC (9.3 mg, 45 μmol) and DMAP (2 mg, 16.5 μmol) as activating agents, to afford 21 as a white fluffy solid (30 mg, 65%). Rf = 0.65 (DCM/methanol, 60/1, v/v). [α]26D = +14.8° (c = 1.0, CHCl3). 1H NMR (300 MHz, CDCl3): δ 7.41-7.11 (m, 30H, aromatic), 6.46 (d, 1H, JNH,2 = 8.4 Hz, NH), 5.70 (d, 1H, J1,2 = 5.3 Hz, H-1), 5.59 (d, 1H, JNH′,2′ = 8.1 Hz, NH′), 5.37 (s, 1H, CH, benzylidene), 5.37 (dd, 1H, J3′,4′ = 9.3 Hz, H-3′), 5.24 (t, 1H, J2,3 = J3,4 = 9.0 Hz, H-3), 4.95 (m, 1H, H-3L), 4.80 (d, 1H, J1′,2′ = 8.4 Hz, H-1′), 4.37-4.61 (m, 9H, H-2, 4 x CH2, Bn), 4.33 (m, 1H, H-5), 4.29 (dd, 1H, J5′,6′a = 5.4 Hz, J6′a,6′b = 11.1 Hz, H-6′a), 3.64-4.00 (m, 8H, H-2′, H-4, H-6a, H-6b, H-6′b, 3 x H-3S), 3.60 (dd, 1H, J4′,5′ = 9.3 Hz, H-4′), 3.48 (m, 1H, H-5′), 1.90-2.64 (m, 10H, H-2L, H-2L′, 3 x H-2S), 1.63-1.05 ((m, 146H, 73 x CH2, lipid), 0.85 (m, 15H, 5 x CH3, lipid). 13C NMR (75 MHz, CDCl3): δ 173.91 (C=O), 172.35 (C=O), 171.45 (C=O), 171.28 (C=O), 169.91 (C=O), 135.59-125.00 (aromatic), 101.23 (CH, benzylidene), 100.92 (C-1′), 87.03 (C-1), 78.08 (C-4′), 75.93 (C-4, 2 x C-3S), 75.21 (C-3S), 74.52 (CH2, Bn), 73.00 (C-3), 71.2 (C-5), 70.91 (C-3′), 70.42-70.64(3 x CH2, Bn), 70.33 (C-3L), 68.26 (C-6′), 67.41 (C-6), 66.03 (C-5′), 55.52 (C-2′), 52.66 (C-2). HR MS (m/z) calcd for C145H230N2O17S[M+Na]+, 2326.6847; found, 2326.7550.
4.1.13. 4-O-Benzyl-6-O-{4,6-O-benzylidene-3-O-[(R)-3-benzyloxy-hexadecanoyl]-2-deoxy-2-[(R)-3-(27-benzyloxy-octacosanoyloxy)-hexadecanoylamino]-β-D-glucopyranosyl}-2-[(R)-3-benzyloxy-hexadecanoylamino]-3-O-[(R)-3-benzyloxy-hexadecanoyl]-2-deoxy-D-glucopyranose (22)
NIS (10.0 mg, 44.4 μmol) and TMSOTf (0.3 μL, 1.5 μmol) were added to a stirred solution of 20 (25 mg, 10.4 μmol) in DCM/H2O (3 mL, 100/1, v/v) at 0 °C. The reaction mixture was vigorously stirred at room temperature for 20 min until TLC analysis indicated that reaction had gone to completion. The reaction mixture was diluted with DCM (5 mL), and then washed with aqueous Na2S2O3 (10%, 5 mL) and water (2 x 5 mL). The organic phase was dried (MgSO4) and filtered. The filtrate was concentrated in vacuo, and the residue was purified by silica gel column chromatography (eluent: toluene/ethyl acetate, 20/1, v/v) to afford lactol 22 as a white solid (18 mg, 78%). Rf = 0.45 (DCM/methanol, 50/1, v/v). 1H NMR (600 MHz, CDCl3): δ 7.39-7.16 (m, 25H, aromatic), 6.28 (d, 1H, JNH,2 = 9.0 Hz, NH), 5.92 (d, 1H, JNH′,2′ = 7.8 Hz, NH′), 5.43 (s, 1H, CH, benzylidene), 5.42-5.39 (m, 2H, H-3, H-3′), 5.20 (d, 1H, J1′,2′ = 8.5 Hz, H-1′), 5.09 (m, 1H, H-1), 4.96 (m, 1H, H-3L), 4.61-4.40 (m, 8H, 4 x CH2, Bn), 4.35 (dd, 1H, J5′,6′a = 5.4 Hz, J6′a,6′b = 10.8 Hz, H-6′a), 4.20 (m, 1H, H-2), 4.06 (m, 1H, H-5), 3.95 (d, 1H, J6a,6b = 12.0Hz, H-6a), 3.84-3.74 (m, 4H, H-6′b, 3 x H-3S), 3.69-3.58 (m, 3H, H-2′, H-4′, H-6b), 3.54-3.48 (m, 2H, H-5′, H-27L′), 3.37 (t, 1H, J4,3 = J4,5 = 9.6 Hz, H-4), 2.65-2.56 (m, 2H, H-2S), 2.50 (dd, 1H, J = 5.4 Hz, J = 15.0 Hz, H-2Sa), 2.41 (dd, 1H, J = 4.8 Hz, J = 15.6 Hz, H-2Sb), 2.35-2.20 (m, 4H, 2 x H-2S, H-2La, H-2La′), 2.12-2.02 (m, 2H, H-2Lb, 2Lb′), 1.59-1.40 (m, 12H, H-4L, H-3L′,26L′, 3 x H-4S), 1.25 (broad, 176H, 88 x CH2, lipid), 1.18 (d, 3H, J27L′,28L′ = 6.0 Hz, H-28L′), 0.89-0.87 (m, 12H, 4 x CH3, lipid). HR MS (m/z) calcd for C146H232N2O19[M+Na]+, 2340.7147; found, 2340.7925.
4.1.14. 4-O-Benzyl-6-O-{4,6-O-benzylidene-3-O-[(R)-3-benzyloxy-tetradecanoyl-2-deoxy-2-[(R)-3-octacosanoyloxy-hexadecanoylamino]-β-D-glucopyranosyl}-2-[(R)-3-benzyloxy-hexadecanoylamino]-3-O-[(R)-3-benzyloxy-tetradecanoyl]-2-deoxy-D-glucopyranose (23)
Compound 21 (30 mg, 13 μmol) was hydrolyzed similar to the synthesis of 22 with NIS (12 mg, 52 μmol) and TMSOTf (1.0 μL, 2.7 μmol) in DCM/H2O (4 mL, 100/1) to afford 23 as a white solid (23.4 mg, 80%). Rf = 0.45 (DCM/methanol, 50/1, v/v). 1H NMR (300 MHz, CDCl3): δ 7.38-7.12 (m, 26H, aromatic), 6.26 (d, 1H, JNH,2 = 9.6 Hz, NH), 5.90 (d, 1H, JNH′,2′ = 8.1 Hz, NH′), 5.41 (s, 1H, CH, benzylidene), 5.40-5.36 (m, 2H, H-3, H-3′), 5.18 (d, 1H, J1′,2′ = 8.4 Hz, H-1′), 5.10 (d, 1H, J1,2 = 3.3 Hz, H-1), 4.95 (m, 1H, H-3L), 4.59-4.37 (m, 9H, H-2, 4 x CH2, Bn), 4.36 (m, 1H, H-6′a), 4.17 (m, 1H, H-2), 4.03 (m, 1H, H-5), 3.92 (m, 1H, H-6a), 3.83-3.47 (m, 8H, H-2′, H-4′, H-5′, H-6b, H-6′b, 3 x H-3S), 3.35 (t, 1H, J4,5 = 9.6 Hz, H-4), 1.64-1.99 (m, 10H, H-2L, H-2L′, 3 x H-2S), 1.63-1.05 (m, 146H, 73 x CH2, lipid), 0.88-0.84 (m, 15H, 5 x CH3, lipid). HR MS (m/z) calcd for C139H226N2O18[M+Na]+, 2234.6728; found, 2234.6657.
4.1.15. 4-O-Benzyl-6-O-{4,6-O-benzylidene-3-O-[(R)-3-benzyloxy-tetradecanoyl-2-deoxy-2-[(R)-3-(27-benzyloxy-octacosanoyloxy)-hexadecanoylamino]-β-D-glucopyranosyl}-2-[(R)-3-benzyloxy-hexadecanoylamino]-3-O-[(R)-3-benzyloxy-tetradecanoyl]-2-deoxy-D-glucono-1,5-lactone (24)
A suspension of 22 (10 mg, 4.3 μmol) and activated molecular sieves (3 Å, 25 mg) in DCM (2 mL) was stirred at room temperature under an atmosphere of argon for 1 h. PCC (9.3 mg, 43 μmol) was then added and the reaction mixture was stirred for another 1.5 h until TLC analysis indicated completion of the reaction. The reaction mixture was purified by Iatro beads column chromatography (eluent: hexane/ethyl acetate, 5/1-3/1, v/v) to afford lactone 24 as a colorless film (7.0 mg, 70%). Rf = 0.35 (hexane/ethyl acetate, 3/1, v/v). 1H NMR (600 MHz, CDCl3): δ = 7.39-7.18 (m, 25H, aromatic), 5.60 (t, 1H, J2′,3′ = J3′,4′ = 9.6 Hz, H-3′), 5.34 (s, 1H, CH, benzylidene), 5.32 (t, 1H, J2,3 = J3,4 = 9.6 Hz, H-3), 5.06 (m, 1H, H-3L), 4.94 (d, 1H, J 1′,2′ = 7.8 Hz, H-1′), 4.78 (t, 1H, J1,2 = J2,3 = 10.2 Hz, H-2), 4.59-4.36 (m, 8H, 4 x CH2, Bn), 4.40 (m, 1H, H-5), 4.26 (dd, 1H, J5′,6′a = 4.8 Hz, J6′a,6′b = 9.6 Hz, H-6′a), 4.04-4.01 (m, 2H, H-4, H-6a), 3.83 (m, 1H, H-3S), 3.80-3.76 (m, 2H, 2 x H-3S), 3.69 (t, 1H, J5′,6′b = 10.2 Hz, H-6′b), 3.56-3.47 (m, 5H, H-2′, H-4′, H-5′, H-6b, H-27L′), 2.64 (dd, 1H, J = 6.0 Hz, J = 14.4 Hz, H-2Sa), 2.52 (dd, 1H, J = 7.2 Hz, 15.6 Hz, H-2Sb), 2.46 (dd, 1H, J = 6.0 Hz, J = 18.0 Hz, H-2Sa), 2.39-2.23 (m, 7H, H-2S x 3, H-2L, 2L′), 1.59-1.40 (m, 12H, H-4L, H-3L′,26L′, 3 x H-4S), 1.24 (broad, 176H, 88 x CH2, lipid), 1.17 (d, 3H, J27L′,28L′ = 6.0 Hz, H-28L′), 0.88-0.86 (m, 12H, 4 x CH3, lipid). HR MS (m/z) calcd for C146H230N2O19[M+Na]+, 2338.6990; found, 2338.8489.
4.1.16. 4-O-Benzyl-6-O-{4,6-O-benzylidene-3-O-[(R)-3-benzyloxy-tetradecanoyl-2-deoxy-2-[(R)-3-octacosanoyloxy-hexadecanoylamino]-β-D-glucopyranosyl}-2-[(R)-3-benzyloxy-hexadecanoylamino]-3-O-[(R)-3-benzyloxy-tetradecanoyl]-2-deoxy-D-glucono-1,5-lactone (25)
Compound 23 (12 mg, 5.4 μmol) was oxidized similar to the synthesis of 24 with PCC (9.4 mg, 43.4 μmol) to afford lactone 25 as a colorless film (8.5 mg, 71%). Rf = 0.35 (hexane/ethyl acetate, 3/1, v/v). 1H NMR (500 MHz, CDCl3): δ 7.38-7.17 (m, 25H, aromatic), 5.60 (t, 1H, J2′,3′ = J3′,4′ = 9.5 Hz, H-3′), 5.39 (s, 1H, CH, benzylidene), 5.31 (dd, 1H, J2,3 = J3,4 = 9.5 Hz, H-3), 5.04 (m, 1H, H-3L), 4.93 (d, 1H, J1′,2′ = 8.5 Hz, H-1′), 4.78 (t, 1H, J2,3 = 10.5 Hz, H-2), 4.59-4.36 (m, 9H, H-5, 4 x CH2, Bn), 4.27 (dd, 1H, J5′,6′a = 5.5 Hz, J6′a,6′b = 10.5 Hz, H-6′a), 4.04-4.01 (m, 2H, H-4, H-6a), 3.87-3.75 (m, 3H, 3 x H-3S), 3.69 (t, 1H, J5,6′b = 9.5 Hz, H-6′b), 3.57-3.50 (m, 4H, H-2′, H-4′, H-5′, H-6b), 2.30-1.93 (m, 10H, H-2L, H-2L′, 3 x H-2S), 1.00-1.75 (m, 146H, 73 x CH2, lipid), 0.82 (m, 15H, 5 x CH3, lipid). HR MS (m/z) calcd for C139H224N2O18[M+Na]+, 2232.6572; found, 2232.6703.
4.1.17. 2-Deoxy-6-O-{2-deoxy-3-O-[(R)-3-hydroxy-tetradecanoyl]-2-[(R)-3-(27-hydroxy-octacosanoyloxy)-hexadecanoylamino]-β-D-glucopyranosyl}-2-[(R)-3-hydroxy-hexadecanoylamino]-3-O-[(R)-3-hydroxy-hexadecanoyl]-D-glucono-1,5-lactone (1)
Compound 24 (7.0 mg, 3.0 μmol) was dissolved in THF/t-BuOH (2 mL, 1/3, v/v) and Pd/C (10 mg) was added. The reaction mixture was shaken under an atmosphere of H2 (15 psi) at room temperature for 36 h, and the catalyst was filtered off and the residue was washed with THF (2 x 1 mL). The combined filtrates were concentrated to afford 1 as a colorless film (4.2 mg, 75%). 1H NMR (600 MHz, CDCl3/CD3OD, 1/1, v/v): δ = 5.40 (t, 1H, J2,3 = J3,4 = 9.6 Hz, H-3), 5.12 (bs, 1H, H-3L), 4.98 (t, 1H, J2′,3′ = J3′,4′ = 9.6 Hz, H-3′), 4.62 (bs, 1H, H-1′), 4.30-3.95 (m, H-2, H-4, H-5, H-6′a, H-6′b), 3.81-3.33 (m, H-2′, H-4′, H-5′, H-6a, H-6b, H-27L′), 2.44-2.24 (m, 10H, H-2L, H-2L′, 3 x H-2S,). HR MS (m/z) calcd for C104H196N2O19[M + Na]+, 1800.4330; found, 1800.6962.
4.1.18. 2-Deoxy-6-O-{2-deoxy-3-O-[(R)-3-hydroxy-tetradecanoyl]-2-[(R)-3-octacosanoyloxy-hexadecanoylamino]-β-D-glucopyranosyl}-2-[(R)-3-hydroxy-hexadecanoylamino]-3-O-[(R)-3-hydroxy-hexadecanoyl]-D-glucono-1,5-lactone (2)
Compound 25 (7.5 mg, 3.4 μmol) was deprotected similar to the synthesis of 1 by hydrogenation (H2, 15 psi) over Pd/C (10 mg) in THF/t-BuOH (2 mL, 1/3, v/v) to afforded 2 as a colorless film (4.8 mg, 80%). 1H NMR (600 MHz, CDCl3/CD3OD, 1/1, v/v): δ 5.39 (t, 1H, J3,4 = J3,4 = 9.6 Hz, H-3), 5.09 (bs, 1H, H-3L), 4.97 (t, 1H, J2′,3′ = J3′,4′ = 9.6 Hz, H-3′), 4.55 (bs, 1H, H-1′), 4.32 (m, 1H, H-5), 4.17 (m, 1H, H-2), 4.13 (m, 1H, H-6′a), 3.95 (m, 1H, H-4), 3.85-3.81 (m, 2H, H-2′, H-6′b), 3.76-3.70 (m, 2H, H-6a, H-6b), 3.54 (t, 1H, J4′,5′ = 9.3 Hz, H-4′), 3.33 (m, 1H, H-5′), 2.55-2.13 (m, 10H, H-2L, H-2L′, 3 x H-2S), HR MS (m/z) calcd for C104H196N2O18[M+Na]+, 1784.4381; found, 1784.4586.
4.2. Biological experiments
4.2.1. Reagents
E. coli 055:B5 LPS was obtained from List Biologicals, and R. sin-1 LPS and lipid A were kindly provided by Dr. R. Carlson (CCRC, Athens, GA). All data presented in this study were generated using the same batches of E. coli 055:B5 LPS and R. sin-1 LPS. Synthetic compounds 1–3 were stored lyophilized at −80 °C and reconstituted in dry THF on the day of the experiment; final concentrations of THF in the biological experiments never exceeded 0.5% to avoid toxic effects.
4.2.2. Cell maintenance
Mono Mac 6 (MM6) cells, provided by Dr. H.W.L. Ziegler-Heitbrock (Institute for Inhalationbiology, Munich, Germany), were cultured in RPMI 1640 medium with L-glutamine (BioWhittaker) supplemented with penicillin (100 u/mL)/streptomycin (100 μg/mL; Mediatech, OPI supplement (1%; Sigma; containing oxaloacetate, pyruvate and bovine insulin) and fetal calf serum (FCS; 10%; HyClone). New batches of frozen cell stock were grown up every 2 months and growth morphology evaluated. Before each experiment, MM6 cells were incubated with calcitriol (10 ng/mL; Sigma) for 2 days to differentiate into macrophage like cells. The cells were maintained in a humid 5% CO2 atmosphere at 37 °C.
4.2.3. Cytokine induction and TNF-α ELISA
On the day of the exposure assay differentiated MM6 cells were harvested by centrifugation and gently suspended (106 cells/mL) in prewarmed (37 °C) medium. MM6 cells were then incubated with different combinations of stimuli for 5.5 hours. Culture supernatants were then collected and stored frozen (−80 °C) until assayed for TNF-α production. Concentrations of human TNF-α protein in culture supernatants were determined by a solid phase sandwich ELISA. Plates (96-well MaxiSorp plates; Nalge Nunc International) were coated with purified mouse anti-human TNF-α antibody (Pharmingen). TNF-α in standards and samples was allowed to bind to the immobilized antibody. Biotinylated mouse anti-human TNF-α antibody (Pharmingen) was then added, producing an antibody-antigen-antibody “sandwich”. After addition of avidin-horseradish peroxidase conjugate (Pharmingen) and ABTS peroxidase substrate (Kirkegaard & Perry Laboratories), a green color was produced in direct proportion to the amount of TNF-α present in the sample. The reaction was stopped by adding peroxidase stop solution (Kirkegaard & Perry Laboratories) and the absorbance was measured at 405 nm using a microplate reader (BMG Labtech). TNF-α values are presented as the means ± SD of triplicate measurements, with each experiment being repeated three times.
4.2.4. Data analysis
Concentration-response and inhibition data were analyzed using nonlinear least-squares curve fitting in Prism (GraphPad Software, Inc.). Concentration-response data were fit with the following four parameter logistic equation: Y = Emax/(1 + (EC50/X)Hill slope), where Y is the cytokine response, X is logarithm of the concentration of the stimulus, Emax is the maximum response, and EC50 is the concentration of the stimulus producing 50% stimulation. Inhibition data were fit with the following logistic equation: Y = Bottom + (Top − Bottom)/(1 + 10(X − Log IC50)), where Y is the TNF-α response, X is the logarithm of the concentration of the inhibitor, and IC50 is the concentration of the inhibitor that reduces the TNF-α response by half.
Acknowledgments
This research was supported by the Institute of General Medicine of the National Institutes of Health (GM061761).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pittet D, Tarara D, Wenzel RP. JAMA, J Am Med Assoc. 1994;271:1598. doi: 10.1001/jama.271.20.1598. [DOI] [PubMed] [Google Scholar]
- 2.Roberts FJ, Geere IW, Coldman A. Rev Infect Dis. 1991;13:34. doi: 10.1093/clinids/13.1.34. [DOI] [PubMed] [Google Scholar]
- 3.Rietschel ET, Schletter J, Weidemann B, El-Samalouti V, Mattern T, Zahringer U, Seydel U, Brade H, Flad HD, Kusumoto S, Gupta D, Dziarski R, Ulmer AJ. Microbial Drug Resistance. 1998;4:37. doi: 10.1089/mdr.1998.4.37. [DOI] [PubMed] [Google Scholar]
- 4.Rietschel ET, Kirikae T, Schade FU, Mamat U, Schmidt G, Loppnow H, Ulmer AJ, Zahringer U, Seydel U, Di Padova F, Schreier M, Brade H. Faseb J. 1994;8:217. doi: 10.1096/fasebj.8.2.8119492. [DOI] [PubMed] [Google Scholar]
- 5.Gupta D, Kirkland TN, Viriyakosol S, Dziarski R. J Biol Chem. 1996;271:23310. doi: 10.1074/jbc.271.38.23310. [DOI] [PubMed] [Google Scholar]
- 6.Poltorak A, He X, Smirnova I, Liu MY, Huffel CV, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Science. 1998;282:2085. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 7.Lien E, Means TK, Heine H, Yoshimura A, Kusumoto S, Fukase K, Fenton MJ, Oikawa M, Qureshi N, Monks B, Finberg RW, Ingalls RR, Golenbock DT. J Clin Invest. 2000;105:497. doi: 10.1172/JCI8541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akashi S, Shimazu R, Ogata H, Nagai Y, Takeda K, Kimoto M, Miyake K. J Immunol. 2000;164:3471. doi: 10.4049/jimmunol.164.7.3471. [DOI] [PubMed] [Google Scholar]
- 9.Takeda K, Kaisho T, Akira S. Annu Rev Immunol. 2003;21:335. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 10.van Amersfoort ES, van Berkel TJC, Kuiper J. Clin Microbiol Rev. 2003;16:379. doi: 10.1128/CMR.16.3.379-414.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raetz CR. Ann Rev Biochem. 1990;59:129. doi: 10.1146/annurev.bi.59.070190.001021. [DOI] [PubMed] [Google Scholar]
- 12.Raetz CR, Whitfield C. Annu Rev Biochem. 2002;71:635. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caroff M, Karibian D, Cavaillon JM, Haeffner-Cavaillon N. Microbes Infect. 2002;4:915. doi: 10.1016/s1286-4579(02)01612-x. [DOI] [PubMed] [Google Scholar]
- 14.Suda Y, Ogawa T, Kashihara W, Oikawa M, Shimoyama T, Hayashi T, Tamura T, Kusumoto S. J Biochem (Tokyo) 1997;121:1129. doi: 10.1093/oxfordjournals.jbchem.a021705. [DOI] [PubMed] [Google Scholar]
- 15.Lam C, Hildebrandt J, Schutze E, Rosenwirth B, Proctor RA, Liehl E, Stutz P. Infect Immun. 1991;59:2351. doi: 10.1128/iai.59.7.2351-2358.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawata T, Bristol JR, Rossignol DP, Christ WJ, Asano O, Dubuc GR, Gavin WE, Hawkins LD, Lewis MD, McGuinness PD, Mullarkey MA, Perez M, Robidoux AL, Wang Y, Kishi Y, Kobayashi S, Kimura A, Hishinima I, Katayama K, Yamatsu I. In: Novel Therapeutic Strategies in the Treatment of Sepsis. Morrison DC, Ryan JL, editors. Marcel Dekker; New York: 1995. p. 171. [Google Scholar]
- 17.Golenbock DT, Hampton RY, Qureshi N, Takayama K, Raetz CR. J Biol Chem. 1991;266:19490. [PubMed] [Google Scholar]
- 18.Christ W, McGuinness P, Asano O, Wang Y, Mullarkey M, Perez M, Hawkins L, Blythe T, Dubuc G, Robidoux A. J Am Chem Soc. 1994;116:3637. [Google Scholar]
- 19.Christ WJ, Asano O, Robidoux AL, Perez M, Wang Y, Dubuc GR, Gavin WE, Hawkins LD, McGuinness PD, Mullarkey MA, Lewis MD, Kishi Y, Kawata T, Bristol JR, Rose JR, Rossignol DP, Kobayashi S, Hishinuma I, Kimura A, Asakawa N, Katayama K, Yamatsu I. Science. 1995;268:80. doi: 10.1126/science.7701344. [DOI] [PubMed] [Google Scholar]
- 20.Asai Y, Nozu Y, Ikeuchi T, Narazaki R, Iwamoto K, Watanabe S. Biol Pharm Bull. 1999;22:432. doi: 10.1248/bpb.22.432. [DOI] [PubMed] [Google Scholar]
- 21.Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. J Biol Chem. 1999;274:10689. doi: 10.1074/jbc.274.16.10689. [DOI] [PubMed] [Google Scholar]
- 22.Demchenko AV, Wolfert MA, Santhanam B, Moore JN, Boons GJ. J Am Chem Soc. 2003;125:6103. doi: 10.1021/ja029316s. [DOI] [PubMed] [Google Scholar]
- 23.Santhanam B, Wolfert MA, Moore JN, Boons GJ. Chem-Eur J. 2004;10:4798. doi: 10.1002/chem.200400376. [DOI] [PubMed] [Google Scholar]
- 24.Vandenplas ML, Carlson RW, Jeyaretnam B, McNeill B, Barton MH, Norton N, Murray TF, Moore JN. J Biol Chem. 2002;277:41811. doi: 10.1074/jbc.M205252200. [DOI] [PubMed] [Google Scholar]
- 25.Jeyaretnam B, Glushka J, Kolli VSK, Carlson RW. J Biol Chem. 2002;277:41802. doi: 10.1074/jbc.M112140200. [DOI] [PubMed] [Google Scholar]
- 26.Mehta S, Pinto BM. J Org Chem. 1993;58:3269. [Google Scholar]
- 27.Xue J, Guo ZW. Tetrahedron Lett. 2001;42:6487. [Google Scholar]
- 28.Sakairi N, Hayashida M, Amano A, Kuzuhara H. J Chem Soc Perkin Trans 1. 1990:1301. [Google Scholar]
- 29.Zuurmond HM, van der Klein PAM, de Wildt J, van der Marel GA, van Boom JH. J Carbohyd Chem. 1994;13:323. [Google Scholar]
- 30.Duffels A, Ley SV. J Chem Soc Perkin Trans 1. 1999:375. [Google Scholar]
- 31.Mehta S, Pinto BM. Tetrahedron Lett. 1991;32:4435. [Google Scholar]
- 32.Kanie O, Crawley SC, Palcic MM, Hindsgaul O. Carbohydr Res. 1993;243:139. doi: 10.1016/0008-6215(93)84087-m. [DOI] [PubMed] [Google Scholar]
- 33.Fukase K, Fukase Y, Oikawa M, Liu WC, Suda Y, Kusumoto S. Tetrahedron. 1998;54:4033. [Google Scholar]
- 34.Nakabayashi S, Warren CD, Jeanloz RW. Carbohyd Res. 1988;174:279. doi: 10.1016/0008-6215(88)85097-3. [DOI] [PubMed] [Google Scholar]
- 35.Santhanam B, Boons GJ. Org Lett. 2004;6:3333. doi: 10.1021/ol048746f. [DOI] [PubMed] [Google Scholar]
- 36.Rossignol DP, Hawkins LD, Christ WJ, Kobayashi S, Kawata T, Lynn M, Yamatsu I, Kishi Y. In: Endotoxin in Health and Disease. Helmut B, Opal SM, Vogel SN, Morrison DC, editors. Vol. 1. Marcel Dekker, Inc; New York: 1999. p. 699. [Google Scholar]
- 37.Lee HS, Wolfert MA, Zhang YH, Boons GJ. Chembiochem. 2006;7:140. doi: 10.1002/cbic.200500298. [DOI] [PubMed] [Google Scholar]