

Abstract
To study the pathogenesis of chronic wasting disease (CWD) in deer and elk, transgenic (tg) mice were generated that expressed the prion protein (PrP) of deer containing a glycine at amino acid (aa) 96 and a serine at aa 225 under transcriptional control of the murine PrP promoter. This construct was introduced into murine PrP-deficient mice. As anticipated, neither non-tg mice nor PrP ko mice were susceptible when inoculated intracerebrally (i.c.) or orally with CWD brain material (scrapie pool from six mule deer) and followed for 600+ days (dpi). Deer PrP tg mice were not susceptible to i.c. inoculation with murine scrapie. In contrast, a fatal neurologic disease occurred accompanied by conversion of deer PrPsen to PrPres by western blot and immunohistochemistry after either i.c. inoculation with CWD brain into two lines of tg mice studied (312 ± 32 dpi [mean ± 2 standard errors] for the heterozygous tg line 33, 275 ± 46 dpi for the heterozygous tg line 39 and 210 dpi for the homozygous tg line 33) or after oral inoculation (381 ± 55 dpi for the homozygous tg line 33 and 370 ± 26 dpi for the homozygous tg line 39). Kinetically, following oral inoculation of CWD brain, PrPres was observed by day 200 when mice were clinically healthy in the posterior surface of the dorsum of the tongue primarily in serous and mucous glands, in the intestines, in large cells at the splenic marginal zone that anatomically resembled follicular dendritic cells and macrophages, and in the olfactory bulb and brain stem but did not occur in the cerebellum, cerebral cortex or hippocampus or in hearts, lungs and livers of infected mice. After 350 days when mice become clinically ill the cerebellum, cerebral cortex and hippocampus became positive for PrPres and displayed massive spongiosis, neuronal drop out, gliosis and florid plaques.
Introduction
Transmissible spongiform encephalopathies (TSE, scrapie, or prion) are a group of rare uniformly fatal neurodegenerative diseases of humans and animals (Chesebro, 1999; Prusiner, 2001). Prion diseases occur in various mammalian species, including humans with Kuru and Creutzfeldt-Jakob disease (CJD), cattle with bovine spongiform encephalopathy (BSE) or “mad cow disease”, sheep with scrapie and deer and elk with chronic wasting disease (CWD). Disease is associated with the conversion of the normal host PrP (PrPsen) found in most cells to abnormally folded beta sheath enriched protease resistant isoform PrPres (PrPsc). Although prion diseases dramatically damage the central nervous system (CNS), infection as well as the protein PrPres can be detected within peripheral tissues, including lymphoid organs (Bosque et al., 2002; Chesebro, 1999; Peden et al., 2006; Prusiner, 2001), skeletal muscle (Angers et al., 2006; Bosque et al., 2002; Peden et al., 2006), tongue, oral and nasal mucosa (DeJoia et al., 2006; Mulcahy et al., 2004), kidney1, pancreas1 (Heikenwalder et al., 2005), intestine1, blood and heart (Aguzzi and Glatzel, 2006; Hewitt et al., 2006; Houston et al., 2000; Jewell et al., 2006; Trifilo et al., 2006).
CWD represents both a major economic and a public health concern (Medicine, 2004). Deer raised on farms and in the wild have CWD. Spread of CWD from one infected deer to others in holding pens strongly suggests horizontal transmission of the disease. This is supported by the recent detection of infectious transmissible material in deer saliva (Mathiason et al., 2006). Although there is a species barrier that usually prevents passage of scrapie from one species to another, it is currently unknown whether CWD in deer and elk is transmissible to humans. Concern about this possibility stems from three observations. First, experimental results indicate that human PrPsen converts to human PrPres in vitro in the presence of CWD PrPres (Caughey, 2001; Caughey and Chesebro, 2001). Although conversion is of low efficiency, it is in the range observed when human PrPsen is changed to human PrPres in the presence of BSE PrPres, and BSE has been transmitted to humans (reviewed (Aguzzi and Glatzel, 2006; Chesebro, 1999; Prusiner, 1999)). Second, CWD has been transmitted to non-human primates (Marsh et al., 2005). Third, saliva and blood from deer with CWD produced disease after inoculation into uninfected deer hosts (Mathiason et al., 2006), demonstrating that materials which come in contact with humans harbors the infection. However, there is as yet no evidence that CWD can transmit disease to humans as tg mice expressing human PrP fail to develop disease when given deer/elk scrapie (Kong et al., 2005) and public health surveillance of humans in contact with deer has not revealed transmission of disease (Medicine, 2004).
CWD was identified in the late 1960s among captive mule deer at a Colorado research facility and designated as a TSE disease that affects white tailed and mule deer as well as elk (Williams and Young, 1980; Williams and Young, 1982). CWD bears many similarities to other TSE diseases, e.g., unremitting neurologic disease with PrPres accumulation, neuronal loss, widespread spongiform formation and gliosis within the CNS, and presence of PrPres in follicular dendritic cells in the spleen. CWD, scrapie in sheep and human vCJD, initially transmitted by BSE, display PrPres prominently in lymphoid tissues.
To understand the spread of infectious material, host genetic susceptibility and the pathogenesis of CWD we generated a tg mouse model (Meade-White et al., 2007). Here we report the utilization of such tg mice to study infectivity, spread and disease following oral inoculation with CWD brain. After infection, mice homologous for the deer PrP gene remain healthy up to 300 days but show expression of PrPres by both western blot and immunohistochemistry in the tongue, gut, spleen, olfactory bulb and brain stem, but not in the cerebellum, cerebral cortex or hippocampus. After 350 days clinical disease occurs and is associated with both PrPres deposits and spongiform patterns classic of scrapie disease throughout the cerebellum, cerebral cortex, entorhinal cortex and hippocampus. Progression of clinical signs of ataxia, tremor, lethargy and weakness leads to marked disease usually by 380 to 410 days post-infection. At this time the brain displays histopathologic evidence of advanced spongiosis, pyknotic neurons, neuronal drop out, florid plaques, and astrogliosis.
Results
Generation and phenotyping of deer PrP tg mice
Reports that deer PrP allelic variation correlated with susceptibility of deer to natural disease suggested a greater susceptibility with glycine instead of a serine at residue 96 of deer PrP and a serine instead of a phenylalanine at position 225 (Jewell et al., 2005; Johnson et al., 2006; O’Rourke et al., 2004) lead to the construction of a plasmid containing deer PrP 96 gly/225 ser under transcriptional control of murine PrP (Meade-White et al., 2007). The resultant DNA fragment was inoculated into fertilized eggs from PrP ko mice (129/Ola back-crossed to C57Bl/6 mice for over six generations) at The Scripps Research Institute transgenic facility. Eggs were then transplanted into pseudo-ovulating foster mothers using standard techniques (Race et al., 1995; Trifilo et al., 2006). Due to the low efficiency employing eggs from PrP ko donors, 642 eggs were transplanted which yielded 14 founder mice, an efficiency slightly over 2% as compared to the usual efficiency of 20 to 30% processing eggs from C57Bl/6 mouse donors. Of the 14 founder mice, two were selected, numbers 33 and 39, to generate lines for subsequent studies because first, they passed the transgene with an expression level roughly equivalent to that of murine PrP in C57Bl/6 mice and, second, bred robustly. Initial we bred heterologous mice (+/−) by crossing line 33 or line 39 to PrP ko mice. Thereafter homologous (+/+) breeding was done.
Phenotyping to judge the utility of the two deer PrP tg lines was accomplished by inoculating heterologous tg mice from both lines 33 and 39 intracerebrally (i.c.) with a 2% cleared brain homogenate from a pool of CWD (Meade-White et al., 2007) (pool 2). As shown in Table 1, both lines 33 and 39 developed disease with a mean incubation time for line 33 of 312 ± 32 days and for line 39 of 275 ± 46 days. Clinically ill mice displayed ataxia, tremors, reduced movement, kyphosis and leg weakness which was first detected at day 225 in some mice with continuous progression involving all mice until severity of disease required sacrifice. When homologous mice were used these signs appeared approximately 70 days earlier. Histologic examination of brains of severely ill mice displayed classic spongiosis, neuronal drop out and gliosis, similar to but in several mice more intense than the picture usually observed with other TSE diseases. Immunohistochemistry revealed deposition of PrPres but staining with either Thioflavin-S or Congo red failed to document amyloid positive plaques. Western blots, as shown in Figure 1, documented the conversion of deer PrPsen to deer PrPres. PrPres was glycosylated with diglycosylated PrPres as the predominant species, although the nonglycosylated form was also present. The glycosylated pattern varied among individual mice studied similar to observations for CWD infected deer and elk (Race et al., 2002). By contrast, neither heterologous nor homologous line 33 or 39 deer PrP tg mice when inoculated with diluent of sterile phosphate buffered saline free of CWD brain or either non-tg C57Bl/6 mice or PrP ko mice inoculated with pooled CWD brain developed disease or showed conversion of deer PrPsen to deer PrPres after 600 days of observation. These results indicated that lines 33 and 39 expressing deer PrP with a glycine at position 96 and a serine at position 225 were susceptible to CWD brain challenge and were suitable for oral transmission experiments.
Table 1.
Clinical disease following inoculation of CWD brain into deer PrP tg micea
| Inoculate | Route of inoculation | Recipient mice | Infected mice totalb | Mean ± SD when mice were sacrificed |
|---|---|---|---|---|
| CWD brain | i.c. | PrPko | 0/5 | ≥ 700 |
| +/− line 33 | 13/13 | 312 ± 32 | ||
| +/− line 39 | 15/15 | 275 ± 46 | ||
| +/+ line 39 | 5/5 | 210 ± 15 | ||
| C57Bl/6 | 0/6 | ≥ 500 | ||
| PBS | i.c. | PrPko | 0/5 | ≥ 600 |
| +/− line 33 | 0/4 | ≥ 600 | ||
| +/− line 39 | 0/4 | ≥ 600 | ||
| +/+ line 39 | 0/4 | ≥ 600 | ||
| C57Bl/6 | 0/4 | ≥ 600 | ||
| CWD brain | oral | PrPko | 0/5 | ≥ 600 |
| +/+ line 33 | 3/3 | 381 ± 55 | ||
| +/+ line 39 | 9/9 | 370 ± 26 | ||
| C57Bl/6 | 0/4 | ≥ 600 | ||
| PBS | oral | PrPko | 0/2 | ≥ 640 |
| +/+ line 33 | 0/2 | ≥ 640 | ||
| +/+ line 39 | 0/2 | ≥ 640 | ||
| C57Bl/6 | 0/2 | ≥ 640 |
PrPko, heterozygous (+/−) or homozygous (+/+) deer PrP tg, or C57Bl/6 mice were inoculated i.c. or orally with homogenate made from pooled brains obtained from mule deer dying from CWD. Recipient mice were followed for signs of disease that included ataxia, tremors, weakness, lethargy, and kyphosis.
Numbers of mice having clinical signs and on sacrifice showing PrPres by both Western blot and immunohistochemistry over the total inoculated. Those not showing clinical signs (PrPko and C57Bl/6 mice) were sacrificed at times indicated. Mice displaying clinical signs were sacrificed either early in disease or when moribund.
Figure 1.
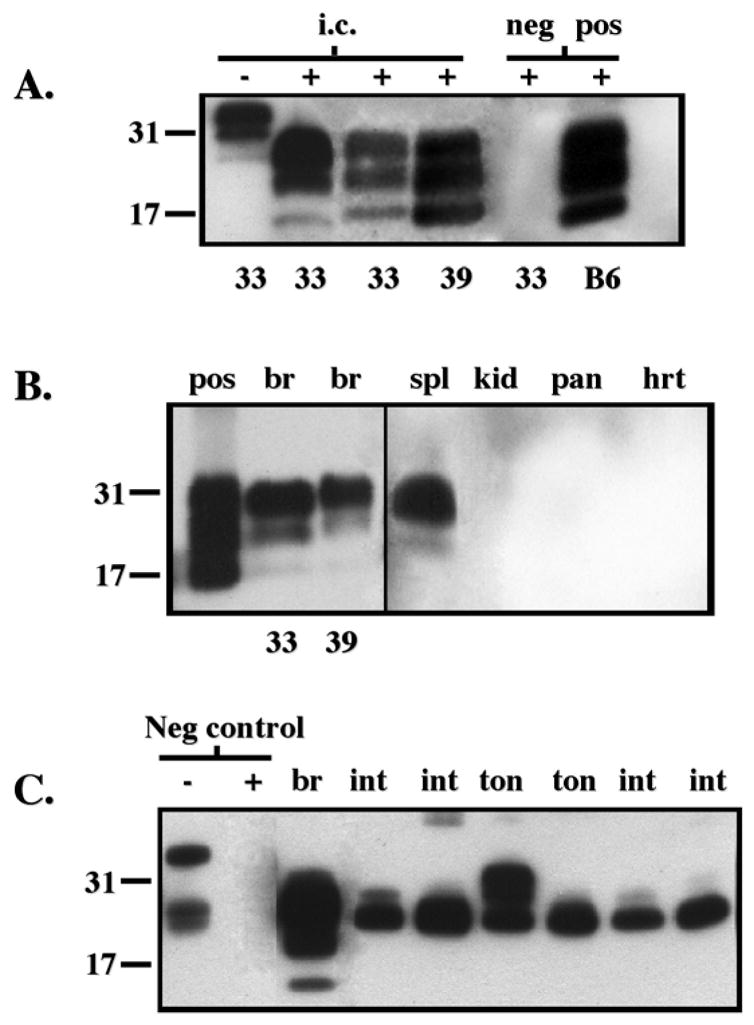
Detection of PrPres deposits in CWD-infected deer PrP tg mice by Western blot. Panel A shows the presence of PrPres in brain tissue after i.c. inoculation of CWD brain. Data from clinically ill tg+/− mice from lines 33 or 39 are displayed in lanes 2–4. Similar results were obtained from over 10 additional ill CWD-infected tg mice from lines 33 and 39 at 280 to 310 dpi. Lane 1 shows Western blot in the absence of protease K treatment (−) while all the remaining lanes were blots with materials treated with proteinase K (25 μg/ml) (+). Negative (neg) control was +/− tg mouse inoculated i.c. with PBS diluent and sacrificed 400 dpi while the positive (pos) control was a C57Bl/6 mouse inoculated i.c. with murine scrapie and sacrificed at 155 dpi. PrP material was detected with monoclonal antibody D13. Panel B displays PrPres within the brain (br), spleen (spl), kidney (kid), pancreas (pan) and heart (hrt) of orally inoculated tg+/+ mice at 370 dpi using monoclonal antibody D13. Spl, kid, pan and hrt are from mouse line 39 whose br is also shown. Similar results for all these tissues were obtained from six additional CWD-infected mice, three from tg33+/+ and three from tg39+/+ strains. Brain homogenate from clinically ill CWD-infected tg+/+ mice (pos) inoculated i.c. is shown for comparison. Panel C shows PrPres deposits within the brain (br), intestine (int), and tongue (ton) of several individual tg+/+ mice infected orally with CWD. Detection was with monoclonal antibody L42. Similar to D13 antibody, L42 antibody failed to detect PrPres in kid, pan or hrt (not shown) but in contrast to D13 antibody detected PrPres in ton and int. The first two lanes on the left show the absence (−) or presence (+) of protease K (25 μg/ml) digestion of deer PrP tg mouse brain (br) inoculated with murine scrapie (neg control). The remainder of the lanes are from protease K (25 μg/ml) digestions of tissues from deer PrP tg mice infected orally with CWD.
Oral transmission of CWD brain to deer PrP tg mice
Oral transmission was studied in homologous deer PrP tg lines 33 and 39 mice. Such mice received by nasal gastric tube 100 λ of a 5% brain homogenate first clarified by low-speed centrifugation. We used the same CWD brain pool as that employed for i.c. inoculations and performed i.c. inoculations as positive controls. All mice given CWD brain orally developed clinical signs of ataxia, lethargy, tremors and weakness, between 347 and 400 days post-inoculation. These signs similar to those present in the i.c. inoculated mice were, however, slower to appear and lasted longer in mice inoculated orally. Nevertheless, over time, disease progressed relentlessly leading to sacrifice of ill mice. As shown in Table 1, mean incubation time for disease was 381 ± 55 days for line 33 and 370 ± 26 for line 39.
Study of brains from both lines showed PrPres by both western blot (Fig. 1) and immunohistochemistry (Fig. 2, Panels C, D, and E). Figure 2 (Panels A and B) displays the spongiosis seen in the deer PrP tg mice infected orally with CWD brain and sacrificed early in the course of clinical disease. In addition, activation of astrocytes (gliosis) was observed. Figure 2, Panel E, shows activated processes from astrocytes surrounding a PrPres plaque. Such PrPres plaques were common and located in the olfactory bulb, entorhinal cortex, cerebral cortex, hippocampus, limbic system and cerebellar areas. As with the i.c. inoculation there was no evidence of amyloid deposits using Thioflavin-S or Congo red staining. As a positive control to detect amyloid, brain sections from murine scrapie infected tg mice that expressed the anchorless form of PrP and abundantly deposited Thioflavin-S or Congo red staining amyloid were used (Chesebro et al., 2005; Trifilo et al., 2006). Our inability to find amyloid in i.c. or orally inoculated deer PrP tg mice that develop disease contrasts with other reports for tg models of CWD and CWD-infected cervids (Browning et al., 2004; Guiroy et al., 1991; Sigurdson et al., 2006; Tamguney et al., 2006). A few mice sacrificed later when clinical disease was severe showed massive amounts of spongiosis, neuronal loss and gliosis throughout the brain. Remarkable was the susceptibility of granular cells in the olfactory bulb, hippocampus and cerebellum along with destruction of neurons and structure in the limbic system, cerebellum, entorhinal cortex and cerebral cortex often associated with enlargement of the lateral ventricle. Again, neither histologic disease nor conversion of deer PrPsen to deer PrPres occurred either in deer PrP tg mice inoculated with murine scrapie or deer PrP tg mice inoculated with the PBS diluent (Table 1; Fig. 1; Fig. 2, Panel F).
Figure 2.
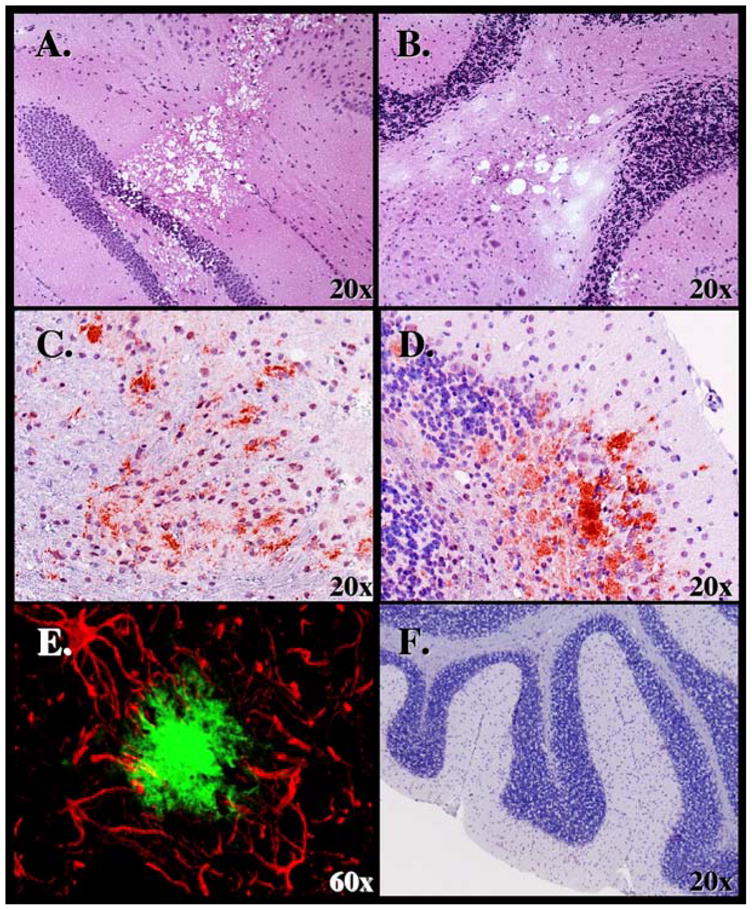
Analysis of CNS tissues from deer PrP tg+/+ mice given CWD brain orally. Presence of spongiform lesions characteristic of TSE lesion in the hippocampus is shown in Panel A and cerebellum in Panel B. H & E staining used and mice sacrificed at 340 dpi. Panel C shows deposition of PrPres within the frontal cortex at 370 dpi and Panel D in the cerebellum at 320 dpi. D13 monoclonal antibody was used to detect PrPres for immunohistochemical analysis. Panel E displays double-immunochemical staining of an area in the cortex displaying astrocytic processes with antibody to GFAP (red) that surround a PrPres plaque (greenish/blue) stained with D13 antibody. Mouse sacrificed at 380 dpi. Panel F shows the absence of disease in cerebellum of deer PrP tg mouse inoculated with PBS diluent and sacrificed 400 dpi. Neither gliosis nor PrPres staining occurred in similar mice or in deer PrP tg mice inoculated with murine scrapie or non-tg mice inoculated with CWD.
Study of peripheral tissues revealed deer PrPres in the spleen, intestinal tract, oral cavity and tongue (Fig. 1, 3) but not in pancreas, heart, liver, thymus, lung, or skeletal muscle. For detection of PrPres by western blot (Fig. 1) we used both monoclonal antibodies D13 and L42 (Chesebro et al., 2005; Meade-White et al., 2007; Trifilo et al., 2006). For this assay L42 has a 10- to 15-fold higher affinity for detection of deer PrPres than does D13. However, for immunohistochemistry we employed D13 as L42 reacted poorly displaying unsatisfactory background noise. In the spleen, the majority of PrPres staining was within large cells surrounding the B and T zones, likely macrophages and/or dendritic cells according to their location and size (Fig. 3). Institutional biosafety concerns disallowed segregating and staining such cells for FACS analysis and thus a more precise definition of the cell(s) containing PrPres. Also, as shown in Figure 3, PrPres was identified immunohistologically on the posterior surface of the dorsum of the tongue, localized primarily to serous and mucosa glands (Fig. 3A and C). Double-staining with antibodies to detect PrPres and neuronal cell body and their processes did not colocalize the PrPres staining in the serous and mucosa glands with markers for neuronal cells. Occasionally the taste buds were noted to contain PrPres. In the intestinal track PrPres was noted in Peyer’s patches and neuronal ganglion cells in the lower intestinal track. In contrast, PrPres deposits were not found in these tissues when studying either PBS inoculated deer PrP tg mice (n = 8), CWD brain inoculated C57Bl/6 mice (n = 6) or in PrP ko mice (n = 5) when observed for up to 600 days post-inoculation.
Figure 3.
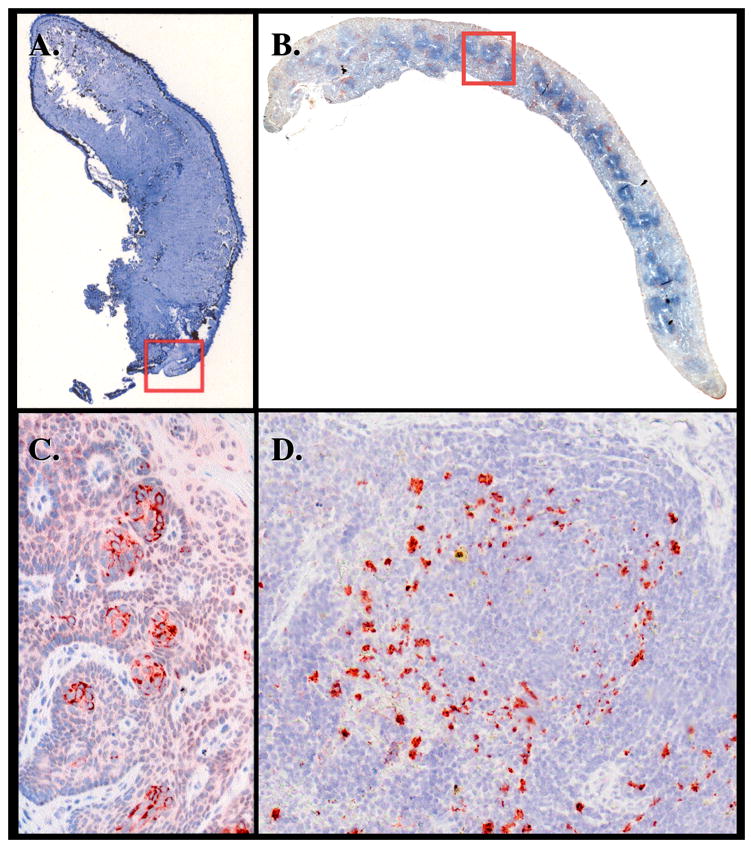
Analysis of peripheral tissues from deer PrP tg+/+ mice given CWD brain orally. Panels A and C (insert from Panel A) are from the tongue of a mouse sacrificed 380 dpi. Serous and mucous glands in the posterior part of the dorsum of the tongue contain PrPres material detected with D13 antibody. Staining of same or adjacent section with monoclonal antibody to neuron-specific nuclear protein NeuN or with antibody to neurofilament H failed to show colocalization to PrPres (data not shown). This observation was confirmed in five additional deer PrP tg mice infected orally with deer PrP. Age-matched tongue specimens from deer PrP tg mice infected with murine scrapie or inoculated with PBS diluent or from C57Bl/6 mice inoculated with CWD brain were all negative (n = 5 or more/group). Panels B and D (insert of Panel B) are from the spleen. Panel D shows PrPres deposits in large cells, likely macrophage and/or dendritic cells, around splenic granule.
Kinetics of deer PrPres expression in deer PrP tg mice following oral inoculation with CWD brain
The last series of studies compared deer PrP tg mice after oral inoculation with CWD brain for the expression of PrPres at day 200 to 300 when such mice were clinically asymptomatic with similarly inoculated littermates at 350 to 410 days when overt signs of disease occurred. Table 2 shows results of mice from line 39 which was similar to those observed with line 33. All mice analyzed when clinically well displayed PrPres in the tongue and spleen, while two of three also possessed PrPres in the lower intestine, brain stem and olfactory bulb, but not in the cerebellum, cerebral cortex, or hippocampus. Mice with clinical signs of disease retained PrPres expression in the tongue, spleen, intestine, olfactory bulb and brain stem but, in contrast, now showed abundant PrPres in the cerebellum, cerebral cortex, entorhinal cortex and hippocampus. PrPres did not occur in lung, liver, or heart.
Table 2.
PrPres distribution following oral infection of deer PrP+/+ tg mice with CWD braina
| Sac. date | Clinical signs | Mouse # | Non-neuronal tissues | Neuronal tissues | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tongue | Spleen | Lung | Liver | Heart | Intestine | Olf. bulb | Br. stem | Cereb | Cortex | Hippo. | |||
| 200–300 dpi | No | 1 | ++ | ++ | nil | nil | nil | ++ | ++ | ++ | nil | nil | nil |
| No | 2 | ++ | ++ | nil | nil | nil | ++ | ++ | ++ | nil | nil | nil | |
| No | 3 | ++ | nil | nil | nil | nil | nil | nil | nil | nil | nil | nil | |
| 350–410 dpi | Yes | 1 | ++ | ++ | nil | nil | nil | ++ | ++ | ++ | ++ | ++ | ++ |
| Yes | 2 | ++ | ++ | nil | nil | nil | ++ | ++ | ++ | ++ | ++ | ++ | |
| Yes | 3 | ++ | ++ | nil | nil | nil | ++ | ++ | ++ | ++ | ++ | ++ | |
Mice were inoculated orally with a 5% brain homogenate stock made from mule deer dying of CWD. Neural and non-neural tissues were isolated and stored in 4% paraformaldehyde and processed for immunohistochemistry.
Discussion
Here we record a murine tg model of CWD in which deer scrapie is transmitted orally, thus mimicking the suspected natural transmission route of this TSE disease. Although several murine tg models of CWD have been reported (Browning et al., 2004; LaFauci et al., 2006; Sigurdson et al., 2006; Tamguney et al., 2006) they described transmission by the i.c. route while to our knowledge this is the first description of CWD disease transmitted by the oral route.
Recently Mathiason (Mathiason et al., 2006) reported that saliva and blood of deer with CWD transmitted infectivity when transferred i.c. or transfused i.v. to naïve deer. In our tg model we noted that prior to and during clinical disease serous and mucous glands found in the dorsum of the tongue which produce saliva contained PrPres. These findings both support and extend the findings of Mathiason (Mathiason et al., 2006) in deer and indicate the utility of the murine tg model for more detailed and complex studies in pathogenesis and for studies of transmission which are difficult to do in deer due to size and housing, experimental numbers required and cost. We have initiated studies to test the transmission of CWD in orally inoculated tg mice through saliva by biting or via shared water supply. Further, our finding that following oral ingestion of CWD the deposition of PrPres is not initially found throughout the brain in a near symmetrical pattern that would follow blood supply to that organ suggests that spread of infection is unlikely via the circulation. This conclusion is further strengthened by not finding evidence of PrPres in or around blood vessels or in the heart or kidney. The initial expression of PrPres in the CNS occurs in the brain stem and olfactory bulb and not in the cerebellum, cerebral cortex or hippocampus suggesting early transmission to the CNS is likely via nerves connecting the oral cavity and gut to the CNS (Sigurdson et al., 2001).
The distribution of spongiosis, gliosis and PrPres although earliest in the olfactory bulb and brain stem, progressed over time to involve dramatically the cerebral cortex, entorhinal cortex, olfactory bulb, cerebellum, hippocampus and limbic system. Although neuronal injury was observed throughout, the susceptibility of granular cells at several sites, i.e., olfactory bulb, hippocampus and cerebellum was remarkable. At end stage, brains analyzed after i.c. or oral inoculations were generally indistinguishable from each other. While florid plaques were easily observed, we failed to detect, by either Thioflavin-S or Congo red staining, amyloid deposits. Why amyloid fibrils failed to occur in our tg mouse model despite the heavy accumulation of PrPres is not presently known. However, in our model such insoluble amyloid protein aggregates are not required for tissue destruction and disease.
The finding of PrPres in spleen and lymph nodes of deer PrP tg mice following oral inoculation of CWD brain mimics what has been reported in deer developing CWD either through natural or experimental infection (Fox et al., 2006; Sigurdson et al., 2002; Sigurdson et al., 1999). We were not able to identify with precision those lymphoid cells that contain PrPres. Nevertheless, anatomic study of the spleen strongly suggested that PrPres was contained in macrophages and/or dendritic cells, cells that have been previously identified as infected by scrapie in other systems (Brown et al., 1999; Heikenwalder et al., 2004).
In addition to the study of pathogenesis, the deer PrP tg model will be a valuable bioassay tool to investigate select PrP polymorphism of residues 96 and 225, to determine whether or not there are unique deer scrapie strains, to determine what materials in TSE deer carry infectious material and to quantitate the amounts of infectivity found in materials transmitting infection.
In collaboration with Chesebro’s group (Meade-White et al., 2007), it was recently noted that deer PrP tg mice expressing glycine at residue 96 were significantly more susceptible to infection and disease after CWD brain inoculation than deer PrP tg mice expressing a serine at this position. These findings in tg mice support suggestive field epidemiologic findings with several deer populations (Johnson et al., 2006; Miller and Williams, 2004; O’Rourke et al., 2004) and suggest that genotyping and selecting deer with serine 96 for commercial breeding might result in a deer population more resistant to CWD.
Finally, utilizing deer PrP glycine 96/serine 225 tg mice we have inoculated urine, feces, blood, white and red blood cells, serum, and saliva obtained from six distinct cases of deer with CWD. Samples were collected kinetically at three or six month intervals from onset of experimental inoculation of deer with CWD until clinical illness 2. At the time of autopsy such samples as well as lymph nodes, spleens and brains harvested from these deer were also collected and inoculated into our tg mice. Currently at over 250 days of observation, none of our deer PrP tg mice have shown signs of CWD disease. Western blots of brains from two deer PrP tg mice sacrificed 140 days after receiving saliva from deer with CWD have not shown conversion of deer PrPres from deer PrPsen while lymph node from the same deer inoculated into PrP tg mice have. These ongoing observations should provide leads into detecting when and which samples contain infectious material, point to those samples that should be titered to quantitate amounts of infectivity, offer insights following time of incubation to illness for location of PrPres, better our understanding of pathogenes and determine whether there may be distinct strains of CWD as has been observed in other TSE diseases.
Materials and methods
Generation and genotyping of deer PrP tg mice
DNA encoding the half genomic mouse PrP was modified to encode the deer PrP gene with glycine at residue 96 and a serine residue 225 as described (Meade-White et al., 2007). This construct was inoculated into eggs obtained from PrP ko mice. Inoculated eggs were transplanted in pseudo-ovulated C57Bl/6 mothers (Chesebro et al., 2005; Trifilo et al., 2006). DNA obtained from tail biopsy of pups was analyzed to select founders and establish lines using the following primers:
CWD 2U 5′ ACC AAA ACC TGG AGG AGG A 3′
CWD 4L 5′ TCG CTC CAT CAT CTT GAT GTC A 3′
Scrapie materials, mouse strains and inoculations
Two sources of TSE infectivity were used. First, a pool of six CWD brain isolates from free-ranging mule deer was obtained by Dr. Elizabeth Williams, Department of Veterinary Science, University of Wyoming, Laramie, Wyoming (Meade-White et al., 2007) (pool 2) and supplied by Rick Race, Laboratory of Persistent Viral Diseases, Rocky Mountain Laboratory, NIAID, NIH, Hamilton, Montana, and second, the RML (Chandler strain) murine scrapie was originally obtained from Rick Race, Laboratory of Persistent Viral Diseases, Rocky Mountain Laboratory, NIAID, NIH, Hamilton, Montana. The RML scrapie brain material has been maintained at Scripps by i.c. passage using C57Bl/6 mice. Six- to 8-week-old mice received either 30 λ of a 2% or 5% low-speed centrifugation cleared brain homogenate administered i.c. through the skull in the fronto-parietal region of the brain using a 27 gauge needle or 100 μl of a 5% low-speed centrifuge cleared brain homogenate given orally via a flexible polypropylene catheter inserted over the tongue about 1 to 2 cm into the esophagus (Race, Oldstone, and Chesebro, 2000; Trifilo et al., 2006). Mice used were bred and handled at The Scripps Research Institute vivarium in accord with AAALAC and NIH guidelines. Employed were C57Bl/6 mice, PrP ko mice and deer PrP tg mice bred onto C57Bl/6 background. Inoculated mice were observed daily for signs of clinical disease that included lethargy, tremors, ataxia, weakness and kyphosis.
Histologic and Western blot analysis
Tissue collected was immediately either fixed in neutral buffered 4% formaldehyde for 3 to 5 days or stored at −80°C until used. Tissues fixed in 4% formaldehyde solution were dehydrated, embedded in paraffin, cut into 4 to 5 μ sections and placed on glass slides. Histologic examination utilized hematoxylin and eosin stain. Immunohistochemical staining was performed as described (Chesebro et al., 2005; Trifilo et al., 2006). Briefly, slides were deparaffinized and rehydrated in xylene, alcohol, PBS rinses, and autoclaved for 15 min. at 20 psi/120°C for retrieval of antigen. Samples were then blocked in 2% BSA and stained for PrPres using either 3 μg/ml of D13 monoclonal antibody (Chesebro et al., 2005; Trifilo et al., 2006), astrocyte activation using a 1:200 dilution of rabbit antibody to glial fibrillary acid protein (GFAP) (Dako Cytomation, Denmark) or neuronal markers using monoclonal antibody to neuronal nuclei (NeuN) and rabbit anti-neurofilament H antibody (both from Chemicon International, USA, and used according to the manufacturers recommendations). After an overnight incubation at 4°C, slides were rinsed with buffer and stained with the appropriate secondary biotinylated goat anti-human (for D 13), goat anti-rabbit (for GFAP and neurofilament H) or goat anti-mouse (NeuN) antibody. Streptavidin conjugated rhodamine or fluorescence or horseradish peroxidase (HRP) followed by ventana aminoethylcarbazol. Thioflavin S and Congo red staining was done as reported (Chesebro et al., 2005; Trifilo et al., 2006). Microscopy was performed with an Axiovert S100 Zeiss microscope.
For Western blot analysis, individual tissues selected for study were placed separately into 2 ml polypropylene screw cap tubes, filled up to 25% with sterilized glass beads (1.0 mm diameter beads from Biospec Products, Inc., Bartlesville, OK) containing 1 ml sterile 0.01M Tris-HCl, pH 7.4, with 0.005M MgCl2. Tubes were placed into the Mini Beadbeater 8 (Biospec) and homogenized for 1 min. Homogenates were transferred to sterile 1.5 ml Eppendorf tubes and incubated with DNase (RNase-free DNase I; Roche Diagnostics, Indianapolis, IN; 1 mg/g starting tissue) for 1 h at 37°C. Samples were then transferred to polyallomer Bell-top Quick Seal centrifuge tubes and an equal volume of 20% Sarkosyl in Tris-HCl buffer added. The suspension was vortexed, incubated for 1 h at room temperature and topped off with 10% Sarkosyl (final volume, 3.5 ml per tube). Tissue homogenate samples were clarified by centrifugation at 10,000g for 30 min at 10°C; the supernatant collected and centrifuged at 70,000g for 2 h at 10°C in a Beckman TL-100 Ultracentrifuge with a TLA 100.3 rotor. The resultant pellet was resuspended in 1 ml of sterile water (1 ml/200 mg starting tissue), sonicated until disrupted in a Sonic Dismembrator 60, setting 1 (Fisher Scientific, Pittsburgh, PA), and digested with Proteinase K, 25 μg/ml (Roche Diagnostics, Indianapolis, IN) for 30 min at 37°C. The reaction was stopped by addition of 0.1M phenylmethylsulfonylfluoride (PMSF) and cooling on ice for 15 min. After centrifugation at 70,000g for 1 h at 10°C, pellets were resuspended in 25 μl sample buffer, sonicated, boiled for 5 min and run on SDS-PAGE gels (Chesebro et al., 2005; Trifilo et al., 2006). Gels were blotted onto PVDF membranes (Invitrogen, Carlsbad, CA) and blocked for 1 h at room temperature in 5% powdered skim milk in sterile water. PrPres bands were detected by adding anti-PrP monoclonal antibody D13 (3 μg/ml) or anti-PrP monoclonal antibody L42 (180 ng/ml) (Chesebro et al., 2005; Trifilo et al., 2006) to membranes and incubating overnight at 4°C. Membranes were rinsed with buffer and stained with a HRP-conjugated goat anti-human antiserum for detection of D13 antibody or HRP-coated sheep anti-mouse antibody. Bands were identified with either the Pico or Phemto chemiluminescent detection kit (Pierce Biotechnology, Rockford, IL) (Trifilo et al., 2006).
Acknowledgments
This is Publication Number 18384 from The Scripps Research Institute. The authors thank Professor Eliezer Masliah, Department of Pathology, UCSD School of Medicine, La Jolla, CA; Gerald Bordin, Division of Pathology, Scripps Clinic and Green Hospital, La Jolla, CA, and both Bruce Chesebro and John Portis from the Laboratory of Persistent Viral Diseases, Rocky Mountain Laboratories, NIH, NIAID, Hamilton, MT, for helpful discussions concerning the neuropathologic and pathologic analysis and biochemical analysis (BC); Arineh Vartanian, Talya Bordin-Wosk, and Abegail Andaya for expert technical assistance, and Gay Wilkins-Blade for manuscript preparation. This research was supported by US Public Health Service grant AG004342 (MBAO) and NIH postdoctoral training grant NS041219 (MT).
Footnotes
Trifilo, M., Oldstone, M.B.A., 2005. Unpublished observations, manuscript in preparation.
Williams, E., Jewell, J. Unpublished observations.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguzzi A, Glatzel M. Prion infections, blood and transfusions. Nat Clin Pract Neurol. 2006;2:321–329. doi: 10.1038/ncpneuro0214. [DOI] [PubMed] [Google Scholar]
- Angers RC, Browning SR, Seward TS, Sigurdson CJ, Miller MW, Hoover EA, Telling GC. Prions in skeletal muscles of deer with chronic wasting disease. Science. 2006;311:1117. doi: 10.1126/science.1122864. [DOI] [PubMed] [Google Scholar]
- Bosque PJ, Ryou C, Telling G, Peretz D, Legname G, DeArmond SJ, Prusiner SB. Prions in skeletal muscle. Proc Natl Acad Sci USA. 2002;99:3812–3817. doi: 10.1073/pnas.052707499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KL, Stewart K, Ritchie DL, Mabbott NA, Williams A, Fraser H, Morrison WI, Bruce ME. Scrapie replication in lymphoid tissues depends on prion protein-expressing follicular dendritic cells. Nature Medicine. 1999;5:1308–1312. doi: 10.1038/15264. [DOI] [PubMed] [Google Scholar]
- Browning SR, Mason GL, Seward TS, Green M, Eliason GA, Mathiason C, Miller MW, Williams ES, Hoover E, Telling GC. Transmission of prions from mule deer and elk with chronic wasting disease to transgenic mice expressing cervid PrP. J Virol. 2004;78:13345–13350. doi: 10.1128/JVI.78.23.13345-13350.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B. Prion protein interconversions. Philosophical Trans of the Royal Society of London. 2001;356:197–200. doi: 10.1098/rstb.2000.0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B, Chesebro B. Transmissible spongiform encephalopathies and prion protein interconversions. Adv Virus Res. 2001;56:277–311. doi: 10.1016/s0065-3527(01)56031-5. [DOI] [PubMed] [Google Scholar]
- Chesebro B. Prion protein and the transmissible spongiform encephalopathy diseases. Neuron. 1999;24:503–506. doi: 10.1016/s0896-6273(00)81105-8. [DOI] [PubMed] [Google Scholar]
- Chesebro B, Trifilo MJ, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola SA, Caughey B, Masliah E, Oldstone M. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science. 2005;308:1435–1439. doi: 10.1126/science.1110837. [DOI] [PubMed] [Google Scholar]
- DeJoia C, Moreaux B, O’Connell K, Bessen RA. Prion infection of oral and nasal mucosa. J Virol. 2006;80:4546–4556. doi: 10.1128/JVI.80.9.4546-4556.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox KA, Jewell JE, Williams ES, Miller MW. Patterns of PrP CWD accumulation during the course of chronic wasting disease infection in orally inoculated mule deer (Odocoileus hemionus) J Gen Virol. 2006;87:3451–3461. doi: 10.1099/vir.0.81999-0. [DOI] [PubMed] [Google Scholar]
- Guiroy DC, Williams ES, Yanagihara R, Gajdusek DC. Immunolocalization of scrapie amyloid (PrP27-30) in chronic wasting disease of Rocky Mountain elk and hybrids of captive mule deer and white-tailed deer. Neurosci Letters. 1991;126:195–198. doi: 10.1016/0304-3940(91)90552-5. [DOI] [PubMed] [Google Scholar]
- Heikenwalder M, Prinz M, Heppner FL, Aguzzi A. Current concepts and controversies in prion immunopathology. J Mol Neurosci. 2004;23:3–12. doi: 10.1385/JMN:23:1-2:003. [DOI] [PubMed] [Google Scholar]
- Heikenwalder M, Zeller N, Seeger H, Prinz M, Klohn PC, Schwarz P, Ruddle NH, Weissmann C, Aguzzi A. Chronic lymphocytic inflammation specifies the organ tropism of prions. Science. 2005;307:1107–1110. doi: 10.1126/science.1106460. [DOI] [PubMed] [Google Scholar]
- Hewitt PE, Llewelyn CA, Mackenzie J, Will RG. Creutzfeldt-Jakob disease and blood transfusion: results of the UK Transfusion Medicine Epidemiological Review study. Vox Sang. 2006;91:221–230. doi: 10.1111/j.1423-0410.2006.00833.x. [DOI] [PubMed] [Google Scholar]
- Houston F, Foster JD, Chong A, Hunter N, Bostock CJ. Transmission of BSE by blood transfusion in sheep. Lancet. 2000;356:999–1000. doi: 10.1016/s0140-6736(00)02719-7. [DOI] [PubMed] [Google Scholar]
- Jewell JE, Brown J, Kreeger T, Williams ES. Prion protein in cardiac muscle of elk (Cervus elaphus nelsoni) and white-tailed deer (Odocoileus virginianus) infected with chronic wasting disease. J Gen Virol. 2006;87:3443–3450. doi: 10.1099/vir.0.81777-0. [DOI] [PubMed] [Google Scholar]
- Jewell JE, Conner MM, Wolfe LL, Miller MW, Williams ES. Low frequency of PrP genotype 225SF among free-ranging mule deer (Odocoileus hemionus) with chronic wasting disease. J Gen Virol. 2005;86:2127–2134. doi: 10.1099/vir.0.81077-0. [DOI] [PubMed] [Google Scholar]
- Johnson C, Johnson J, Vanderloo JP, Keane D, Aiken JM, McKenzie D. Prion protein polymorphisms in white-tailed deer influence susceptibility to chronic wasting disease. J Gen Virol. 2006;87:2109–2114. doi: 10.1099/vir.0.81615-0. [DOI] [PubMed] [Google Scholar]
- Kong Q, Huang S, Zou W, Vanegas D, Wang M, Wu D, Yuan J, Zheng M, Bai H, Deng H, Chen K, Jenny AL, O’Rourke K, Belay ED, Schonberger LB, Petersen RB, Sy MS, Chen G, Gambetti P. Chronic wasting disease of elk: transmissibility to humans examined by transgenic mouse models. J Neurosci. 2005;25:7944–7949. doi: 10.1523/JNEUROSCI.2467-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFauci G, Carp RI, Meeker HC, Ye X, Kim JI, Natelli M, Cedeno M, Petersen RB, Kascsak R, Rubenstein R. Passage of chronic wasting disease prion into transgenic mice expressing Rocky Mountain elk (Cervus elaphus nelsoni) PrPC. J Gen Virol. 2006;87:3773–3780. doi: 10.1099/vir.0.82137-0. [DOI] [PubMed] [Google Scholar]
- Marsh RF, Kincaid AE, Bessen RA, Bartz JC. Interspecies transmission of chronic wasting disease prions to squirrel monkeys (Saimiri sciureus) J Virol. 2005;79:13794–13796. doi: 10.1128/JVI.79.21.13794-13796.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathiason CK, Powers JG, Dahmes SJ, Osborn DA, Miller KV, Warren RJ, Mason GL, Hays SA, Hayes-Klug J, Seelig DM, Wild MA, Wolfe LL, Spraker TR, Miller MW, Sigurdson CJ, Telling GC, Hoover EA. Infectious prions in the saliva and blood of deer with chronic wasting disease. Science. 2006;314:133–136. doi: 10.1126/science.1132661. [DOI] [PubMed] [Google Scholar]
- Meade-White K, Race B, Trifilo M, Bossers A, Favara C, LaCasse R, Miller MW, Williams ES, Oldstone M, Race R, Chesebro B. Resistance to chronic wasting disease infection in transgenic mice exposing a naturally occurring allelic variant of deer prion protein. J Virol. 2007 doi: 10.1128/JVI.02762-06. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medicine Io. Advancing Prion Science: Guidance for the National Prion Research Program. National Academy Press; Washington, D.C: 2004. [Google Scholar]
- Miller MW, Williams ES. Chronic wasting disease of cervids. Current Topics in Microbiology and Immunology. 2004;284:193–214. doi: 10.1007/978-3-662-08441-0_8. [DOI] [PubMed] [Google Scholar]
- Mulcahy ER, Bartz JC, Kincaid AE, Bessen RA. Prion infection of skeletal muscle cells and papillae in the tongue. J Virol. 2004;78:6792–6798. doi: 10.1128/JVI.78.13.6792-6798.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke KI, Spraker TR, Hamburg LK, Besser TE, Brayton KA, Knowles DP. Polymorphisms in the prion precursor functional gene but not the pseudogene are associated with susceptibility to chronic wasting disease in white-tailed deer. J Gen Virol. 2004;85:1339–1346. doi: 10.1099/vir.0.79785-0. [DOI] [PubMed] [Google Scholar]
- Peden AH, Ritchie DL, Head MW, Ironside JW. Detection and localization of PrPSc in the skeletal muscle of patients with variant, iatrogenic, and sporadic forms of Creutzfeldt-Jakob disease. Am J Pathol. 2006;168:927–935. doi: 10.2353/ajpath.2006.050788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB. Prion Biology and Disease. Cold Spring Harbor Press; Cold Spring Harbor, NY: 1999. [Google Scholar]
- Prusiner SB. Prions. In: Knipe DM, Howley PM, editors. Fields Virology. 4. Lippincott Williams and Wilkins; Philadelphia: 2001. pp. 3063–3087. [Google Scholar]
- Race R, Oldstone M, Chesebro B. Entry versus blockade of brain infection following oral or intraperitoneal scrapie administration: Role of prion protein expression in peripheral nerves and spleen. J Virol. 2000;74:828–833. doi: 10.1128/jvi.74.2.828-833.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Race RE, Priola SA, Bessen RA, Ernst D, Dockter J, Rall GF, Mucke L, Chesebro B, Oldstone MBA. Neuron-specific expression of a hamster prion protein minigene in transgenic mice induces susceptibility to hamster scrapie agent. Neuron. 1995;15:1183–1191. doi: 10.1016/0896-6273(95)90105-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Race RE, Raines A, Baron TG, Miller MW, Jenny A, Williams ES. Comparison of abnormal prion protein glycoform patterns from transmissible spongiform encephalopathy agent-infected deer, elk, sheep, and cattle. J Virol. 2002;76:12365–12368. doi: 10.1128/JVI.76.23.12365-12368.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdson CJ, Barillas-Mury C, Miller MW, Oesch B, van Keulen LJ, Langeveld JP, Hoover EA. PrP (CWD) lymphoid cell targets in early and advanced chronic wasting disease of mule deer. J Gen Virol. 2002;83:2617–2628. doi: 10.1099/0022-1317-83-10-2617. [DOI] [PubMed] [Google Scholar]
- Sigurdson CJ, Manco G, Schwarz P, Liberski P, Hoover EA, Hornemann S, Polymenidou M, Miller MW, Glatzel M, Aguzzi A. Strain fidelity of chronic wasting disease upon murine adaptation. J Virol. 2006;80:12303–12311. doi: 10.1128/JVI.01120-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdson CJ, Spraker TR, Miller MW, Oesch B, Hoover EA. PrP (CWD) in the myenteric plexus, vagosympathetic trunk and endocrine glands of deer with chronic wasting disease. J Gen Virol. 2001;82:2327–2334. doi: 10.1099/0022-1317-82-10-2327. [DOI] [PubMed] [Google Scholar]
- Sigurdson CJ, Williams ES, Miller MW, Spraker TR, O’Rourke K, Hoover EA. Oral transmission and early lymphoid tropism of chronic wasting disease PrPres in mule deer fawns (Odocoileus hemionus) J Gen Virol. 1999;80:2757–2764. doi: 10.1099/0022-1317-80-10-2757. [DOI] [PubMed] [Google Scholar]
- Tamguney G, Giles K, Bouzamondo-Bernstein E, Bosque PJ, Miller MW, Safar J, DeArmond SJ, Prusiner SB. Transmission of elk and deer prions to transgenic mice. J Virol. 2006;80:9104–9114. doi: 10.1128/JVI.00098-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifilo MJ, Yajima T, Gu Y, Dalton N, Peterson KL, Race RE, Meade-White K, Portis JL, Masliah E, Knowlton KU, Chesebro B, Oldstone MBA. Prion-induced amyloid heart disease with high blood infectivity in transgenic mice. Science. 2006;313:94–97. doi: 10.1126/science.1128635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams ES, Young S. Chronic wasting disease of captive mule deer: a spongiform encephalopathy. J Wildlife Dis. 1980;16:89–98. doi: 10.7589/0090-3558-16.1.89. [DOI] [PubMed] [Google Scholar]
- Williams ES, Young S. Spongiform encephalopathy of Rocky Mountain elk. J Wildlife Dis. 1982;18:465–471. doi: 10.7589/0090-3558-18.4.465. [DOI] [PubMed] [Google Scholar]