

Abstract
The t(2;11)(q31;p15) chromosomal translocation results in a fusion between the NUP98 and HOXD13genes and has been observed in patients with myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML). We previously demonstrated that expression of the NUP98‐HOXD13 (NHD13) fusion gene in transgenic mice results in an invariably fatal MDS; approximately one third of mice die due to complications of severe pancytopenia, and about two thirds progress to a fatal acute leukemia. In the present study, we used retroviral insertional mutagenesis to identify genes that might collaborate with NHD13 as the MDS transformed to an acute leukemia. Newborn NHD13 transgenic mice and littermate controls were infected with the MOL4070LTR retrovirus. The onset of leukemia was accelerated, suggesting a synergistic effect between the NHD13 transgene and the genes neighbouring retroviral insertion events. We identified numerous common insertion sites located near protein‐coding genes, and confirmed dysregulation of a subset of these by expression analyses. Among these genes were Meis1, a known collaborator of HOX and NUP98‐HOX fusion genes, and Mn1, a transcriptional coactivator involved in human leukemia through fusion with the TEL gene. Other putative collaborators included Gata2, Erg and Epor. Of note, we identified a common insertion site that was >100 kb from the nearest coding gene, but within 20 kb of the miR29a/miR29b1 microRNA locus. Both of these miRNA were upregulated, demonstrating that retroviral insertional mutagenesis can target miRNA loci as well as protein‐coding loci. Our data provides new insights into NHD13 mediated leukemogenesis as well as retroviral insertional mutagenesis mechanisms.
Introduction
The NUP98‐HOXD13 (NHD13) fusion gene arises from the t(2;11)(q31;p15) translocation, which occurs in the malignant cells of patients with myeloid malignancies (1). We previously characterised a transgenic mouse model resulting from the expression of this fusion gene under the control of a pan‐haematopoietic promoter which caused a myelodysplastic syndrome (MDS) progressing to an acute leukemia (2). This phenotype is highly penetrant, however, the latency period preceding the onset of acute leukemia is consistently longer than six months. This latency period suggests the possibility that further genetic or epigenetic events are required for progression to acute leukemia. This is consistent with the hypothesis that mutations affecting multiple cellular pathways are required for oncogenesis. Of particular importance in leukemia are two types of mutations; those that lead to impaired differentiation, and those that result in inappropriate proliferation and/or apoptosis (3). NUP98‐HOX fusion genes have been shown to inhibit haematopoietic differentiation (2, 4, 5), therefore one might anticipate that a complementary event affecting one or more proliferation or apoptotic pathways would be required before the NHD13 transgenic cell could progress to leukemia.
Retroviral insertional mutagenesis is a powerful screening technique used for identifying genes that can lead to malignant transformation. Upon infection into newborn mice, the retrovirus inserts into the genome of the host cells (6–8), and in doing so can affect the expression of nearby genes (9). If the altered expression of these genes is oncogenic, clonal expansion of the cell in which that particular insertion occurred will ensure that that clone will be predominant in the resultant tumour tissue. One important advantage of insertional mutagenesis over chemical mutagenesis is that the introduction of foreign sequence into the genome “tags” the affected genes, simplifying the subsequent identification of the genes. The approach has been widely demonstrated as useful for identifying proto‐oncogenes (10–12) and, specifically, for identifying collaborating events in sensitised models such as transgenic or knockout mice (13).
In our initial studies (2, 14), we noted that the transgenic NHD13 model results in a spectrum of leukemic phenotypes, most commonly AML and pre‐T LBL. For the retroviral insertional mutagenesis experiment, we attempted to bias the system towards myeloid rather than lymphoid malignancies, because the NHD13 fusion gene has been observed only in patients with myeloid malignancies. For this reason we chose to use the MOL4070LTR retrovirus, created by replacing most of the U3 region of the Moloney murine leukemia virus (MMLV) long terminal repeat (LTR) with that of 4070A. Unlike the parental MMLV, which induces exclusively lymphoblastic T cell lymphomas in unsensitised FVB mice, MOL4070LTR produces predominantly myeloid neoplasms (15).
Materials and Methods
Retroviral Infection
The MOL4070LTR retrovirus was produced by seeding 105 NIH3T3 cells chronically infected with virus with an equal number of uninfected NIH3T3 cells, in a 100 mm dish. The cells were propagated for 4 days, the medium was replaced with fresh medium and, on day 5, the virus‐containing medium was harvested and titre was determined by the XC assay (16). Newborn mice were inoculated intraperitoneally with 4×104 infectious particles in 0.05 ml of culture medium.
Phenotype Analysis
Mice were under daily observation for early signs of leukemia. These signs included lethargy, laboured breathing, enlarged lymph nodes or abdominal masses. Mice were euthanised upon observation of symptoms, and blood, bone marrow and tissues were harvested for analysis. Hematoxylin‐eosin (H&E), CD3 (DAKO, Carpinteria, CA), B220 (CD45R; Pharmingen, San Diego, CA), anti‐myeloperoxidase (MPO; DAKO), and F4/80 (Caltag, San Francisco, CA) stained sections from tissues including the thymus, lymph nodes, spleen, liver, kidney, lung, and tibia were produced using conventional staining techniques. Bone marrow cells were harvested from femurs by flushing with Iscove’s modified minimal essential media and assessed microscopically by May‐Grunwald‐Giemsa stained cytospin preparations. Two‐color flow cytometry was used to determine the immunophenotype of a single‐cell suspension prepared from thymus, spleen, and/or bone marrow. The cells were stained with fluorescein isothiocyanate (FITC)‐conjugated anti‐mouse CD8, B220, Gr‐1, and c‐kit and phycoerythrin (PE)‐conjugated anti‐mouse CD4, IgM, Mac‐1, and ScaI (Pharmingen). Diseases were classified according to the Bethesda proposals (17, 18).
Inverse PCR
One microgram of spleen DNA from leukemic mice was digested for 16 hours with BamHI in a 20‐µl final volume. DNA fragments were ligated in a 500‐µl volume by using T4 DNA Ligase (Invitrogen, Carlsbad, CA) at 16°C for 16 hours, and were resuspended in 20 µl of water after precipitation. Two PCRs were performed from each template; one using the 5’ LTR as template, and the other using the 3’ LTR. The PCR mix included 2 µl of template DNA, 400 µM dNTP, 0.4 µM of each primer, and 2.5 units of Taq enzyme (Expand Long Template PCR, Roche), in a 50‐µl final volume. Primer sequences are available upon request. Primary PCRs were performed with a denaturation step (5 min at 94°C), followed by 30 cycles of amplification (each cycle included 30 sec of denaturing at 94°C, 1 min annealing at 60°C, and 10 min extension at 68°C), and a final extension step (5 min at 68°C). Secondary PCRs were performed using nested primers and 1 µl of the primary PCR as a template. Products from the secondary PCR were analyzed by electrophoretic separation in a 1% agarose gel and sequenced directly using the secondary PCR primers.
Ligation‐Mediated PCR
Genomic DNA was digested with NlaIII or MseI and ligated to linkers constructed by annealing the oligonucleotides s 5’‐gtaatacgactcactatagggctccgcttaagggaccatg‐3’ and 5’‐ Phos‐gtcccttaagcggag‐C3spacer‐3’ for NlaIII‐digested DNA, and 5’‐ gtaatacgactcactatagggctccgcttaagggac‐3’ and 5’‐Phos‐tagtcccttaagcggag‐C3spacer‐3’ for MseI‐digested DNA (Integrated DNA Technologies, Coralville, IA). Primary PCR was performed using primers complementary to the linkers and the LTR of the MOL4070LTR retroviral sequence. Secondary PCR was performed using nested primers after 1:50 dilution of the primary PCR product. All primer sequences are available on request. Products were ligated into pGEM‐T Easy (Promega, Madison, WI) and transformed into DH5α cells (Invitrogen). DNA isolated from Ampicillin‐resistant colonies was sequenced using an SP6 primer with BigDye Terminator reagents and analysed on a 3730 DNA Analyzer (Applied Biosystems, Foster City, CA). Sequences were matched to the genomic build of March 2005 (NCBI Build 34) using the UCSC genome browser (http://genome.ucsc.edu/cgi-bin/hgBlat), and will be submitted to the Retrovirus Tagged Cancer Gene Database (RTCGD; http://RTCGD.ncifcrf.gov).
Southern Blot Analysis
Genomic DNA was isolated from mouse spleen tissue and digested with either BglII, EcoRI, EcoRV, or HindIII. Digested DNA was size‐fractionated on a 0.8% agarose gel, denatured, neutralized, and transferred to a nitrocellulose membrane. The nitrocellulose membrane was hybridized to a 32P‐labeled virus‐specific or locus‐specific probe generated by PCR. Primer sequences used to generate probes are available upon request.
Northern Blot miRNA Analysis
RNA was isolated from spleen using the Trizol reagent and protocols (Invitrogen). RNA samples (10 µg each) were run on 15% acrylamide denaturing (urea) precast gels (Invitrogen), and then transferred onto Nytran Supercharge membrane (Schleicher & Schuell, Dassel, Germany). Probes were labelled with [α‐32P]ATP using the Starfire oligo labelling kit (Integrated DNA Technologies). Probe hybridization was performed at 37°C in 7% SDS/0.2M Na2PO4 overnight. Membranes were washed twice with 2×SSPE/0.1% SDS at 37°C. Blots were reprobed with a 5S RNA probe as a loading control.
Reverse Transcription PCR
RNA was isolated from spleen using the Trizol reagent (Invitrogen), and 1 µg RNA was reverse transcribed using Superscript II reverse transcriptase (RT) with random hexamer primer (Invitrogen). A forward Mn1 primer (5’‐tacctcaacccctgacagctatgg‐3’) and a reverse Moloney pol primer (5’‐ctcccgatctccattggttacctc‐3’) were used for amplification of a fusion product. After 3 minutes at 94°C, 35 or 45 cycles (as noted) of 94°C for 30 seconds, 60°C for 30 seconds, and 72°C for 90 seconds were used, followed by a terminal 5‐minute extension at 72°C. PCR products were analyzed by agarose gel electrophoresis.
Real Time PCR
First‐strand cDNA templates were generated as described above. Real‐time RT‐PCR was carried out on an 7500 Fast Real‐Time Taqman PCR System (Applied Biosystems). Primer and probe sets were purchased from Applied Biosystems and used under recommended conditions. Primer details are available upon request. The expression of Gapdh was used as an endogenous control. As controls, cDNA from the spleen and bone marrow of three wild type mice were used, and the average value taken. All reactions were performed in triplicate and the ΔΔCT mean and standard error calculated for each sample (19). Values were normalised to the value generated for normal spleen.
Results
MOL4070LTR Infection Accelerates Leukemogenesis in NHD13 Mice
The recombinant MOL4070LTR virus retains the majority of the genomic sequence of MMLV, but the U3 LTR regions have been replaced with those from the 4070A virus. Infection of wild‐type FVB mice with this recombinant virus results in induction of leukemia with a mean latency of 31.7 weeks (15). NHD13 and wild type littermate mice were infected with MOL4070LTR on the day after birth, and a cohort of 34 transgenic and 41 wild type mice were then monitored for disease symptoms. Complete blood counts (CBCs) were obtained from a subset of 5 NHD13 and 5 wild‐type littermates at the age of two months to confirm that these mice showed an MDS phenotype consistent with that observed in non‐infected NHD13 (Supplemental Table 1). Leukemia developed more rapidly in NHD13 infected mice than either wild type infected mice (p=5×10−11) or NHD13 non‐infected mice (p=2×10−5) (Figure 1a; Supplemental Table 1). The median latency of disease in NHD13 infected mice was 19.4 weeks (mean 19.9). Mice were euthanized when leukemic symptoms were identified, and tissues were harvested for molecular and histological analysis to confirm and classify the leukemia.
Figure 1. Acceleration of leukemic transformation in NHD13 mice by MOL4070LTR infection.
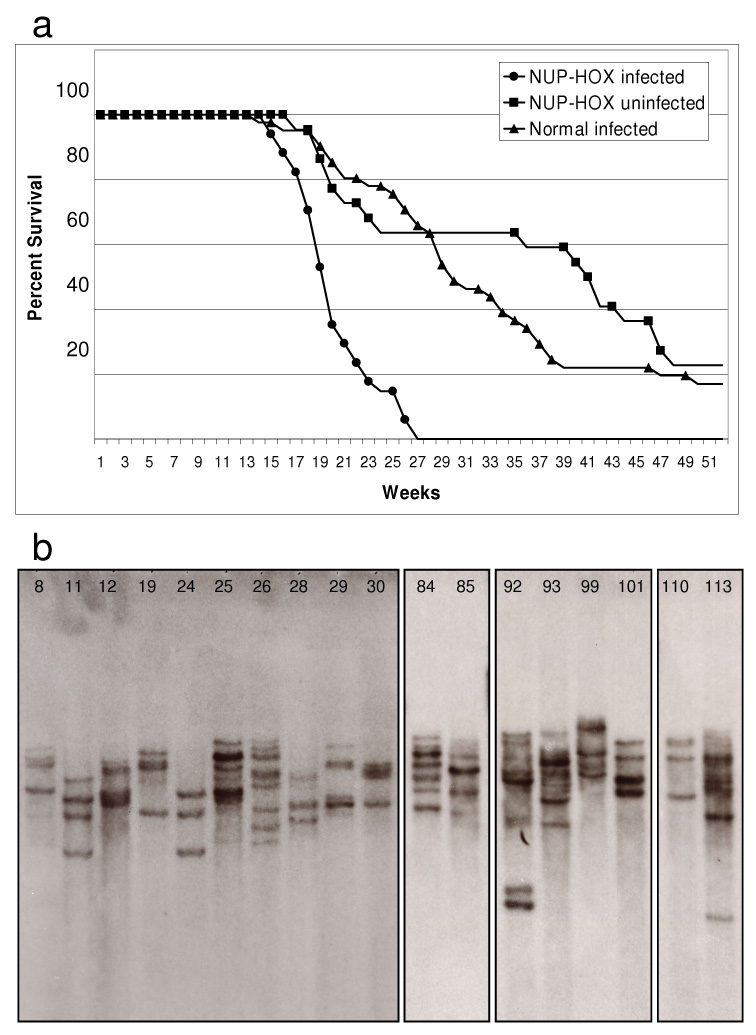
a) Survival plot of MOL4070LTR‐infected NHD13 transgenic mice (circles; n = 34), MOL4070LTR‐infected wild‐type mice (triangles; n = 41) and non‐infected NHD13 transgenic mice (squares; n = 22). b) Southern analysis of a representative panel of NHD13 MOL4070LTR infected mice. An EcoRI‐digested DNA membrane was hybridised with a MOL4070LTR envelope‐region probe. Note the variable intensity of bands within samples; such variation is likely attributable to oligoclonality of the tumour.
The number of retroviral insertions in each leukemic sample was determined by Southern blot analysis using a probe specific for the viral genome (Figure 1b). This allowed assessment of the number of insertions per tumour, and an estimation of the clonality of each tumour. The relative intensity of bands obtained by Southern analysis suggests that many of the tumours were oligoclonal. This observation is consistent with the immunohistochemistry and immunophenotyping data obtained, which in many cases indicated the presence of more than one leukemic clone (eg Supplemental Figure 1). All infected NHD13 mice died of an acute leukemia. The leukemic phenotype was determined by FACS or immunohistochemistry for 26 of the 31 mice. In total, there were 5 mice with acute lymphocytic leukemia (ALL) and 22 mice with acute non‐lymphocytic leukemia (ANLL) (one mouse had an immunohistologically apparent biclonal tumour; Supplemental Table 2). This represents a substantial decrease in the frequency of lymphocytic leukemias compared to the phenotype spectrum of the cohort of transgenic non‐infected mice, which featured 7 ALLs and 7 ANLLs. This is consistent with the expected non‐lymphoid bias of the MOL4070LTR virus which, in a wild‐type FVB background, induced 8 ALLs and 16 ANLLs in a cohort of 22 mice (3 mice had an immunhistologically apparent biclonal tumour) (15). In our study, survival of mice with ANLL was not significantly different from that of mice with ALL in either cohort (data not shown). In addition, when only mice with ANLL are considered, the acceleration of leukemia onset in the NHD13 infected mice compared to NHD13 non‐infected mice was highly significant (median age of onset 21 vs 39 weeks, p=0.005).
Cloning of Retroviral Insertion Sites
A total of 279 independent insertion sites were identified from the 31 NHD13 infected mice by anchored PCR techniques (Supplemental Table 3). The number of integration sites ranged from 6 to 16 insertions per mouse. This number is generally higher than the number of bands identified on the Southern blots, suggesting that some insertions identified may derive from non‐leukemic tissue, or from a minor leukemic clone. Fourteen Common Insertion Sites (CIS; defined as at least two insertion sites no more than 100kb apart (20)) were identified in these mice (Table 1). Additionally, some insertion events that were not recurrent are of interest based on the known function of the gene or the presence of similar insertion events in other models (Table 2).
Table 1.
Common Insertion Sites in NHD13 mice.
| Common Insertion Site | Incidence | Mouse Ids | Occurrence in RTCGD |
|---|---|---|---|
| Meis1 | 14 | 3, 12, 30, 89, 99, 101, 106, 112, 116, 118, 119, 121 | 27 |
| Mn1 | 8 | 19, 26, 28, 84, 110, 112, 113, 118 | 3 |
| Gata2 | 5 | 7, 12, 86, 99, 119 | 0 |
| Erg | 4 | 24, 85, 88, 92 | 2 |
| Runx2 | 3 | 8, 11, 106 | 7 |
| Rras2 | 2 | 106, 113 | 32 |
| Epor | 2 | 29, 89 | 1 |
| mir29a & mir29b | 2 | 3, 11 | 11 |
| Pim1 | 2 | 12, 76 | 34 |
| Gfi1B | 2 | 11, 25 | 1 |
| Rasgrp1 | 2 | 28, 76 | 31 |
| Ebf1 | 2 | 106, 113 | 3 |
| Evi1 | 2 | 11, 84 | 24 |
| 6qA3.3 | 2 | 106, 118 | 0 |
RTCGD (Retrovirus Tagged Cancer Gene Database) data derived from transposon insertion studies are not included.
Meis1
Meis1 was the most commonly detected insertion site in the present study, occuring in 12 mice as determined by PCR, consistent with previous findings by other investigators (21, 22). All 12 of the Meis1 insertion events detected by PCR occurred within 155kb of one another (Figure 2a). Nine were in the reverse orientation and within 8kb 5’ of the gene. The remaining three were in the forward orientation, in intron 7 (#89), the 3’ UTR (#99), or 8kb 3’ of Meis1 (#118). A screen for additional Meis1 insertions by Southern blot revealed insertions in two additional mice (#7 and #26) that were not detected by PCR (Figure 2b).
Figure 2. Meis1 insertions are frequent and result in overexpression of the Meis1 transcript.
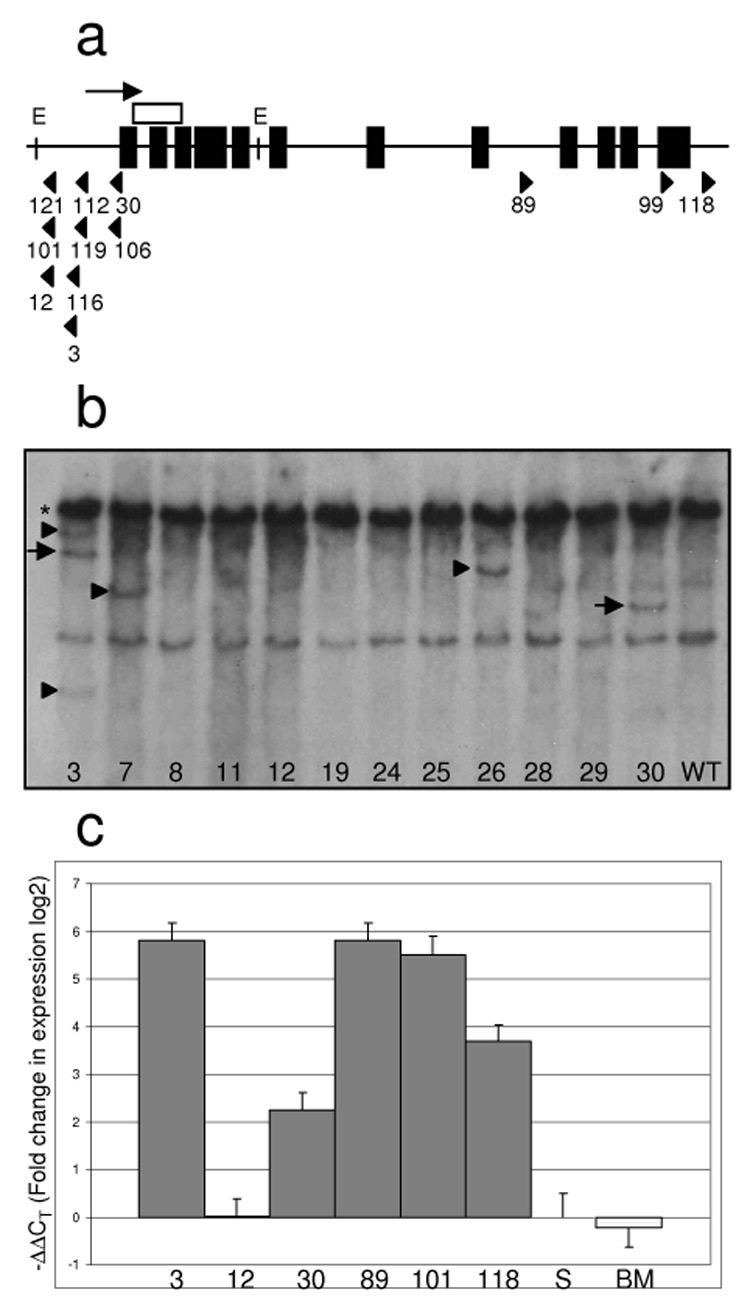
a) Exon/intron structure of the Meis1 locus and location and orientation of viral insertion sites. Black boxes are Meis1 exonsl. Direction of transcription is indicated with an arrow. Viral insertion site position and orientation are indicated with arrowheads. The white box indicates the position of the cDNA Southern probe. E indicates EcoRV restriction site. Numbers refer to the mouse ID. b) Southern analysis of Meis1 locus. The asterisk indicates the expected germline band. Arrows indicate insertion‐derived bands of predicted size based on LM‐PCR data. Arrowheads indicate bands resulting from viral insertion events not detected by LM‐PCR. c) Real‐time PCR analysis of Meis1 expression in spleens of mice with Meis1 insertion events. Expression was calculated using the ΔΔCT method (19) and is shown in log2 scale. Expression level was normalised to that of wild‐type spleen. Columns are labeled with mouse ID #. S = wild‐type spleen, BM = wild‐type bone marrow.
We examined the expression of Meis1 in six mice that had Meis1 insertions (3, 12, 30, 89, 101, 118). In 5 of the 6, Meis1 expression was increased more than 4‐fold compared to normal spleen and bone marrow (Figure 2c), the exception being mouse #12. However, as no abnormal Meis1 band was evident for this sample by Southern blot, the leukemic clone containing the Meis1 insertion in this mouse may well be a minor clone.
Mn1
We identified four insertion sites in the same intron of Mn1, all in the forward orientation with respect to Mn1 (Figure 3a). Southern blots demonstrated that the clone with the Mn1 insertion was a prominent clone in all four cases (Figure 3b, left and upper right). Since an MN1‐TEL fusion has been identified in human leukemia patients, and the insertions we detected were in the same intron as the MN1‐TEL breakpoints, we suspected that a Mn1‐viral fusion may be produced in these mice. We used RT‐PCR to amplify a Mn1‐viral fusion transcript in all four mice that had an intronic insertion (Figure 3b, lower right). Sequencing of this product revealed Mn1 exon 1 spliced to a cryptic splice acceptor in the viral pol gene, resulting in an in‐frame Mn1‐pol fusion (Figure 3c). This in‐frame fusion transcript encodes a protein that fuses the N terminal portion of MN1 to the final 115 residues of the integrase peptide encoded by the pol gene (accession number AAC98548).
Figure 3. Mn1 insertions contribute to the major leukemic clone and result in overexpression of the normal allele as well as a fusion transcript.
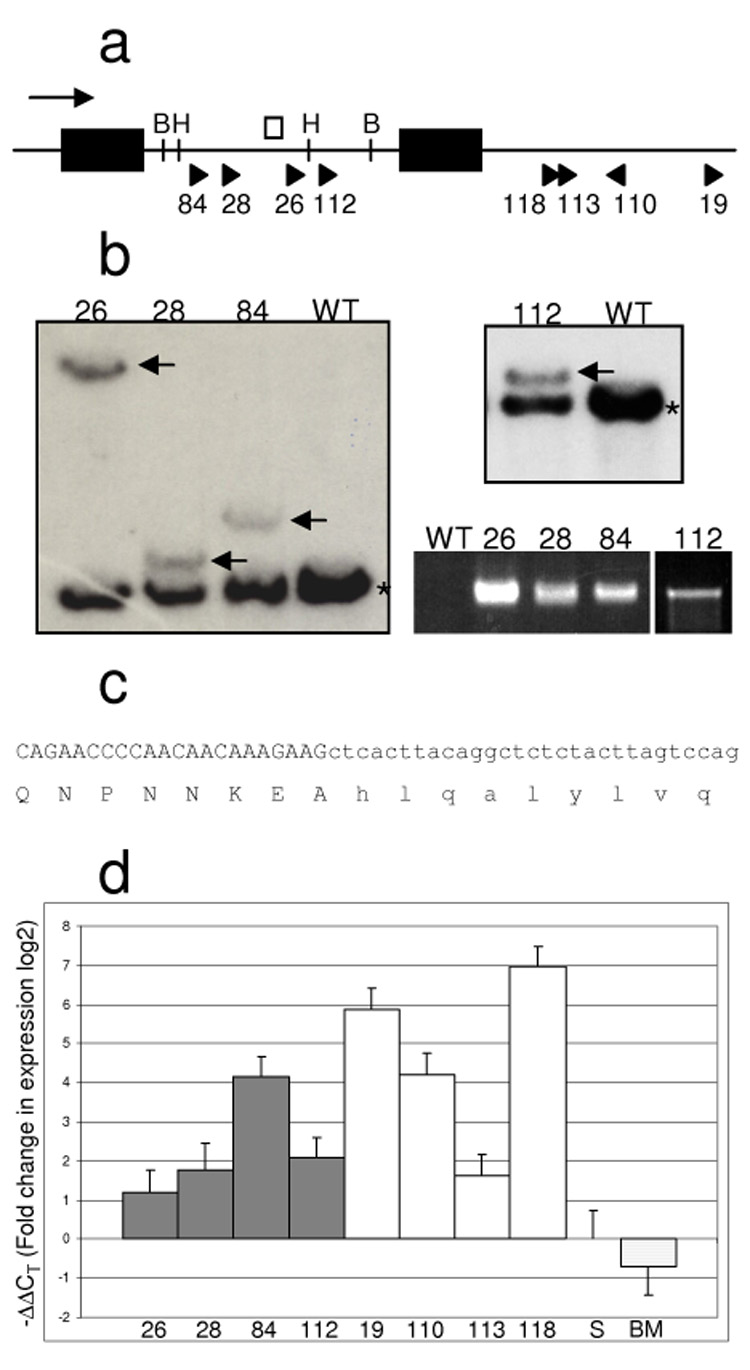
a) Exon/intron structure of the Mn1 locus and location and orientation of viral insertion sites. Black boxes are Mn1 exons. Direction of transcription is indicated with an arrow. Viral insertion site position and orientation are indicated with arrowheads. The white box indicates the position of the Southern probe. B indicates BglII restriction site. H indicates HindIII restriction site. Numbers refer to the mouse ID. b) Southern analysis of Mn1 locus and expression of fusion transcript. Samples were digested with HindIII (left) or BglII (upper right) and probed with the probe indicated in a). The black asterisk indicates the expected germline band. Black arrows indicate bands of predicted size based on LM‐PCR data. RT‐PCR was performed on these samples using an Mn1 forward primer and a MOL4070LTR viral reverse primer, detecting the presence of a fusion transcript (lower right). 35 cycles of PCR were performed, except for sample #112 for which 45 cycles were performed. c) Partial cDNA sequence with predicted protein translation of the band produced by RT‐PCR as in d), indicating the fusion is in‐frame. Black sequence derives from Mn1, grey sequence from MOL4070LTR virus. d) Real‐time PCR analysis of Mn1 expression in spleens of mice with Mn1 insertion events. Dark bars represent mice with intronic insertions. White bars represent mice with 3’ insertions. Expression was calculated using the ΔΔCT method (19)and is shown in log2 scale. Expression level was normalised to that of wild‐type spleen. Columns are labeled with mouse ID #. S = wild‐type spleen, BM = wild‐type bone marrow.
In addition, we found a second CIS 80–166 kb 3’ of Mn1 that was present in four additional mice (#19, #110, #113, #118; Figure 3a). These insertions were not all in the same orientation, excluding the possibility that a Mn1‐pol fusion gene (or at least a common fusion gene) could be formed. We designed a real time RT‐PCR assay to assess the expression levels of wild‐type Mn1 transcripts in all eight mice. All eight mice overexpressed wild‐type Mn1 transcripts, in one case by one hundred fold (Figure 3d). Therefore in the first set of four mice, the insertion appears to have two effects: the creation of a fusion transcript, and an overexpression of the wild‐type Mn1. In the second set of four mice, overexpression of wild‐type Mn1 is the only detected outcome.
Gata2, Erg, and Epor
We assayed expression levels of Gata2, Erg and Epor in mice from which these common insertion sites were cloned. Five mice (#7, #12, #86, #99 and #119) had insertions that were thought to potentially affect the expression of Gata2. The location of these insertions was widely varied, however, from 130 kb 5’ of the gene (#119) to within intron 3 of the gene (#12). We found a consistent increase in expression levels in the leukemic spleen compared to normal spleen (1.7x – 65x; Supplemental Figure 2a). Four mice (#24, #85, #88, #92) had insertions in the Erg gene, three in intron 2 and one in intron 4. Erg expression in the leukemic spleens was upregulated compared to normal spleen in all four samples, between 2.5‐fold and 14‐fold (Supplemental Figure 2b). Two mice (#29 and #89) had insertions near the Epor gene; one in intron 2 and the other immediately 3’ of the gene. We examined expression of the Epor transcript in these mice and found that the level of expression was substantially increased over the levels of expression in both normal bone marrow and normal spleen, suggesting that the impact of these insertions is to upregulate the expression level of the Epor in these mice (Supplemental Figure 2c). Interestingly, one of these mice (#29) had ANLL with marked basophilia, and FACS analysis demonstrated that the major leukemic clone was positive for CD41 and CD117 and negative for Mac1 and ter119 (data not shown), indicating that Epor overexpression was associated with characteristics of “trilineage” (erythroid, megakaryocyte, mast) precursor cells (23–25).
microRNA insertions
Two mice (#3 and #11) had insertion sites on chromosome 6qA3.3 separated by only 250 bp. This locus is not near any characterised protein‐coding genes, but is 20kb from a microRNA cluster containing miR29a and miR29b1. We examined the expression of these microRNAs by Northern blot. Both miR29a and miR29b1 are expressed at high levels in the spleens of both #3 and #11. We compared the expression levels of the microRNAs in some of the other retrovirally‐induced tumours of similar phenotype, in which retroviral insertions at this locus were not detected. The expression level of both miR29a and miR29b1 in five other tumours was very low by comparison, demonstrating upregulation associated with retroviral insertion near the locus (Figure 4a and Figure 4b).
Figure 4. Retroviral insertion results in overexpression of mature microRNAs.
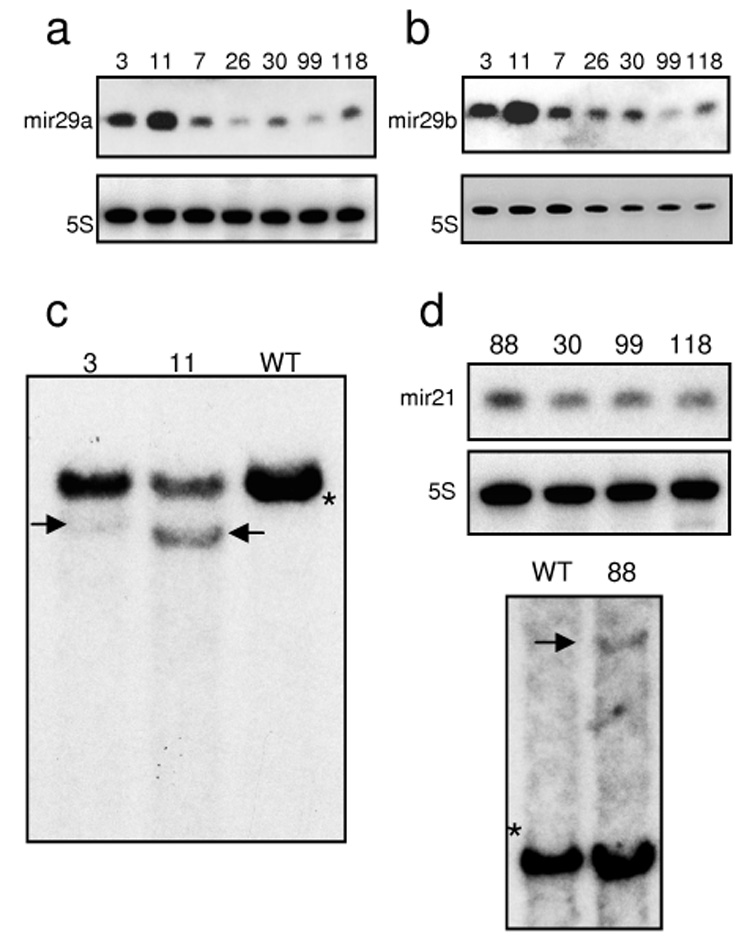
a) Northern blot showing the expression of miR29a in spleen tissue of two mice with an insertion near the miR29 locus and five leukemic mice without an insertion near the locus. Lanes are labeled with mouse ID #. 5S RNA was used as a loading control. b) Northern blot showing the expression of miR29b1 on duplicate membrane to that in a). c) Southern blot showing the insertion alleles of the miR29 locus in mice 3 and 11. The black asterisk indicates the expected genomic band. Black arrows indicate bands of predicted size based on LM‐PCR data. The intensity of the insertion bands indicates that the miR29 insertions contribute to the major leukemic clone. d) Northern blot (upper) showing the expression of miR21 in spleen tissue of one mouse with an insertion near the miR21 locus and three leukemic mice without an insertion near the locus. Lanes are labeled with mouse ID #. 5S RNA was used as a loading control. Southern blot (lower) showing the insertion allele of the miR21 locus in mouse #88. The black asterisk indicates the expected genomic band. Black arrows indicate bands of predicted size based on LM‐PCR data.
We examined the predominance of the clone containing this insertion site in the spleens of each of these mice by Southern blot. A non‐germline band was evident in both mice, although it was fairly faint in mouse #3, suggesting that the leukemic clone with the miR29 insertion was a minor clone in this mouse (Figure 4c), consistent with the more dramatic upregulation of miR29a and miR29b1 in mouse #11 (Figure 4a and Figure 4b).
Because the upregulation of a microRNA locus by a retroviral insertional event has not been previously described, we sought other potential examples of microRNA upregulation in our data set. We identified five instances of retroviral insertions within 100 kb of known microRNA loci (Table 2), and performed expression analyses for microRNAs at each of these insertion loci. The only locus which showed any suggestion of microRNA overexpression was miR21 in mouse #88 (Figure 4d, upper). We performed a Southern blot to determine the relative contribution of the miR21 containing clone to the spleen tissue used for expression analysis (Figure 4d, lower); this experiment confirmed that the miR21 insertion derived from an expanded clone but also that the clone comprised only a minor portion of the spleen tissue. Therefore the relatively modest upregulation observed by Northern blot is consistent with the Southern blot indicating that the clone with the miR21 insertion is a relatively minor clone.
Table 2.
Selected Single Insertion Sites in NHD13 Leukemia.
| Insertion Sites Present in RTCGD | |
|---|---|
| Signal Transduction | Flt3, Gfra1, Ifnar2, Kdr, Nf1, Rasa3, Stat5b |
| Cell Cycle | Ccnd1, Ccnd2, Ccnd3 |
| Differentiation | Fli1, Gfi1, Myb, Sfpi1 |
| Other | Ccr1, Mef2c, Rad51, Trp53, Tspan5 |
| Insertion Sites Not Present in RTCGD | |
| Signal Transduction | Als2, Akt2, Fgrf1, Flt1, Ksr1, Ksr2, Pdgfrb, Pik3ap1, Pitpnm1, Pitpnm2, Tie1, Tcra |
| Cell Cycle | Ccrk, Cdkn2d, Plk2, Ptpn1 |
| Differentiation | Elf1, Esrrb, Fzd9, Hbp1, Hoxb8, Hoxc9, Nfe2, Samsn1, Zfpm2 |
| MicroRNA | mir21, mir142, mir148b, mir297, mir92‐2 cluster |
| Other | Adrbk1, Adrbk2, Bcl2l14, Bin1, Irs1, Mllt3, Ncf1, Nkd2, Plcb2, Prkar2b, Pthlh, Ptp4a2, Ptpn18, Ranbp10, Rpsa, Sdc1, Smarca4, Smndc1, Sox6, Tcf3, Tmem49, Tmem16f, Tspan4, Vil2 |
RTCGD data derived from transposon insertion sites are not included.
Single Insertion Sites
Several loci that were only identified once in our cohort are nonetheless of interest because of the known or expected function of the gene. A selection of these loci are listed in Table 2, and includes a large number of genes implicated in leukemia, such as Flt3, Stat5b, Ccnd1, Ccnd2, Ccnd3, Fli1, Gfi1, Myb, Trp53, Pdgfrb, Fgfr1, Flt1, Akt2, and Mllt3. Comparison of this list with the retroviral insertion mutagenesis data contained within the RTCGD (26) indicated that a large number of integration sites had not previously been identified.
Discussion
Previously, we demonstrated that the NHD13 fusion protein, generated by the t(2;11)(q31;p15) chromosomal translocation associated with MDS and AML, leads to impaired haematopoietic differentiation and MDS in transgenic mice (2). However, it is likely that this fusion protein is insufficient to induce leukemia, as the NHD13 mice develop overt leukemia only following a long latent period. An evolving paradigm suggests that AML results from one mutation that impairs differentiation, and at least one complementary mutation that increases cell proliferation and/or decreases apoptotic cell death (27). We sought to identify events that could complement the NHD13 fusion during leukemic transformation by using retroviral insertional mutagenesis.
Leukemia was induced with a latency of 3–6 months by injecting the MOL4070LTR retrovirus into newborn NHD13 pups. The efficiency of induction was much greater than either injecting the retrovirus into wild type pups (3–10 months) or NHD13 pups which did not receive virus (4–14 months). This provided evidence that the NHD13 transgene and the retroviral insertions were indeed collaborating leukemogenic events. We identified a total of 14 CIS, defined as the occurrence of two insertions within 100 kb (20). Among the CIS, several had been observed in other retroviral insertion screens for proto‐oncogenes or tumor suppressor genes, but others have not been previously reported as CIS in retroviral tagging experiments (eg Mn1, Gata2; see Table 1).
To verify that insertional mutagenesis altered the transcription of nearby genes, we chose a subset of CIS genes and examined the level of expression in the leukemic spleen tissues from which the insertions were cloned. We investigated five genes (Meis1, Mn1, Gata2, Erg and Epor), and in most cases examined, the gene was overexpressed with respect to normal spleen. This suggests that upregulation of the gene is a consequence of the insertion, and therefore is likely to be contributing to the oncogenicity of the leukemic clone. For those mice in which the gene at the CIS was not markedly overexpressed, it is possible that the clone in question is a minor clone.
Meis1 was first identified as a CIS in a spontaneously recombinant retrovirus (BXH2) leukemogenic model (28), and has since been well‐established as a collaborator of overexpressed HOX genes in leukemogenesis (29–31). Meis1 is also known to collaborate with NUP98‐HOX fusion genes (22, 32), including NHD13 (21). It was therefore not surprising that Meis1 would be a CIS identified by this screen. We identified Meis1 insertions in a total of 14 mice, more than for any other gene. Almost all of these insertions were near the 5’ end of the gene; therefore, the mechanism by which this insertion contributes to leukemic transformation is most likely overexpression of the normal Meis1 transcript, consistent with previous findings. By quantitative real‐time PCR analysis of mice known to contain Meis1 insertions, the majority of tumours showed clear upregulation of Meis1 expression. For the minority that did not, it is possible that the Meis1 clone was a minor clone in an oligoclonal tumour, such as mouse #12.
The second most frequent CIS identified in our screen was Mn1. MN1 is a transcriptional coactivator known to synergise with retinoic acid receptor mediated transcription (33), and has been shown to be involved in leukemia by at least two mechanisms. MN1 is involved in formation of a fusion transcript with the TEL oncogene in AML (34), and high levels of MN1 expression have recently been shown to be a poor prognostic marker for AML patients with normal cytogenetics (35). Mn1 has not been identified as a CIS in any other retroviral mutagenesis experiment contained in the RTCGD, supporting the possibility that Mn1 is a specific collaborator with NHD13. The insertion sites in Mn1 identified in our study fell into two distinct groups. Four mice had an insertion in the forward orientation in the homologous intron of Mn1 in which the MN1‐TEL breakpoint occurs. These mice expressed a fusion transcript between Mn1 and the integrase viral gene, perhaps recapitulating the MN1‐TEL fusion. Of note, a recent study showed that MN1‐TEL collaborated with HOXA9 to induce myeloid leukemia in mice (36), so collaboration of an Mn1‐integrase fusion gene with NHD13 may occur via a similar mechanism. If these mechanisms are indeed similar, then the TEL and integrase portions of the respective fusions may not contribute any required function to the fusion protein, and the amino terminal portion of MN1 may be sufficient to promote leukemic transformation. Alternatively, it may be that some non‐sequence‐specific scaffolding function is required of the MN1 fusion partner; for instance, the retained portions of TEL and integrase each encode a DNA‐binding domain (37). Four other mice had insertions 3’ of Mn1, and all of these mice, as well as the mice with intronic insertions, overexpressed wild‐type Mn1 transcripts by real time PCR.
Gata2, identified as the third most frequent CIS in our screen, was not present in the RTCGD. This was somewhat surprising, as GATA2 is an important regulator of haematopoietic differentiation and has been implicated in leukemia by overexpression, especially in patients with 3q21 aberrations (38, 39). Gata2 expression was strongly upregulated in those mice that had nearby insertions, suggesting that it is indeed contributing to the leukemic phenotype.
The insertions at the miR29a and miR29b1 microRNA cluster result in overexpression of the mature form of both of these microRNAs. The likely mechanism is transcriptional upregulation of the pri‐microRNA transcript, followed by processing to the mature form. Whether a high level of expression of both microRNAs contributes to leukemic transformation, or whether upregulation of either miR29a or miR29b1 alone would be sufficient, is unknown. Microarray studies have shown that miR29b2, the mature sequence of which is identical to miR29b1, is frequently upregulated in breast, colon, pancreatic and prostate cancer (40), and modestly upregulated in papillary thyroid carcinoma(41). Little is known about the normal function of miR29a or miR29b1, although both have been shown to be expressed in a subset of haematopoietic cell lines (42).
To our knowledge, the insertions near the miR29a and miR29b1 microRNA cluster are the first demonstration of microRNA transcript upregulation by retroviral insertion. Of interest, a previous study (43) observed a common retroviral integration site near this locus in brain tumours, reinforcing insertion at this locus (and subsequent upregulation of these microRNAs) as oncogenic events. This finding also suggests that miR29a/b1 upregulation may not be a specific collaborator of NHD13, nor even specific to leukemia. Because upregulation of a microRNA by retroviral insertion has not been previously demonstrated, we sought to uncover a second instance of this phenomenon in our cohort. We identified five candidate single insertion sites and analysed expression of a corresponding mature microRNA in the appropriate mouse. Only one additional microRNA (miR21) was observed to be overexpressed with respect to other leukemia samples of similar phenotype, and the overexpression was fairly subtle. miR21 has been previously implicated in oncogenesis (40, 44–46), supporting the evidence for its overexpression having a role in the outgrowth of this clone.
Many of the leukemias generated in this study contained more than one CIS, suggesting the possibility that a single retroviral integration is insufficient to complement NHD13, and that multiple integrations are required. However, since many of the leukemias appeared to be oligoclonal by Southern blot, it is also possible that each individual clone in the oligoclonal sample was generated by a single leukemogenic insertion (discounting bystander events). This question will be addressed by functional complementation studies.
Several loci that were identified only once in our cohort are nonetheless of interest because of the known or expected function of the gene. The list was searched against the RTCGD database to determine whether these events were common in retroviral insertion screening, A surprisingly high number of these events were not listed in the RTCGD suggesting that they are unique to our screen, possibly because of either the sensitising background (NHD13) or the virus used (MOL4070LTR). It is also possible that these insertion sites were not selected for but were merely bystander events.
The findings of the present study are likely to be relevant for human AML. Although translocations involving homeobox genes are relatively rare in patients with AML, translocations involving the MLL gene are thought to exert their oncogenic effect through activation of homeobox genes(47), and HOXA cluster genes are among the genes most commonly upregulated in patients with AML (48). In this context, it is important to note that recent reports have implicated overexpression of MN1 (35) or ERG (49), 2 of the 4 genes most frequently identified in our screen, to be important prognostic factors for patients with AML.
Our data shows that NHD13‐induced leukemia can be accelerated using the MOL4070LTR retrovirus. Some of the genes that we identified as potential collaborators (Mn1, Gata2, Erg) have not previously been reported as CIS in retroviral mutagenesis screens, and are potentially specific NHD13 collaborators in the same mode as Meis1, rather than more general collaborators associated with malignant transformation, such as Stat5b, Ccnd1, or Trp53. Indeed, some may be even more specific than Meis1, given their absence from the NHA9 BXH2 screen performed previously. Of particular interest was the upregulation of two microRNAs in response to retroviral insertion. Because insertion at this locus has been previously observed in retrovirally‐induced brain tumours, upregulation of either mir29a or mir29b1 may well be a more general oncogenic event. Our findings present novel implications for mechanisms of NHD13 leukemogenesis, and again demonstrate the utility of retroviral inertional mutagenesis as a means of identifying such information.
Supplementary Material
Acknowledgments
We would like to acknowledge the expert technical assistance of Christine Perella. We thank our colleagues Yang Jo Chung, Chul Won Choi, Richard Koller, David Caudell and Yue Cheng for discussion. This research was supported by the Intramural Research Program of the NIH, NCI.
References
- 1.Raza‐Egilmez SZ, Jani‐Sait SN, Grossi M, Higgins MJ, Shows TB, Aplan PD. NUP98‐HOXD13 gene fusion in therapy‐related acute myelogenous leukemia. Cancer Res. 1998;58:4269–4273. [PubMed] [Google Scholar]
- 2.Lin YW, Slape C, Zhang Z, Aplan PD. NUP98‐HOXD13 transgenic mice develop a highly penetrant, severe myelodysplastic syndrome that progresses to acute leukemia. Blood. 2005;106:287–295. doi: 10.1182/blood-2004-12-4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gilliland DG, Tallman MS. Focus on acute leukemias. Cancer Cell. 2002;1:417–420. doi: 10.1016/s1535-6108(02)00081-8. [DOI] [PubMed] [Google Scholar]
- 4.Takeda A, Goolsby C, Yaseen NR. NUP98‐HOXA9 induces long‐term proliferation and blocks differentiation of primary human CD34+ hematopoietic cells. Cancer Res. 2006;66:6628–6637. doi: 10.1158/0008-5472.CAN-06-0458. [DOI] [PubMed] [Google Scholar]
- 5.Pineault N, Abramovich C, Humphries RK. Transplantable cell lines generated with NUP98‐Hox fusion genes undergo leukemic progression by Meis1 independent of its binding to DNA . Leukemia. 2005;19:636–643. doi: 10.1038/sj.leu.2403696. [DOI] [PubMed] [Google Scholar]
- 6.Mitchell Rs, Beitzel BF, Schroder AR, et al. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004;2:E234. doi: 10.1371/journal.pbio.0020234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bushman FD. Targeting survival: integration site selection by retroviruses and LTR‐retrotransposons. Cell. 2003;115:135–138. doi: 10.1016/s0092-8674(03)00760-8. [DOI] [PubMed] [Google Scholar]
- 8.Wu X, Li Y, Crise B, Burgess SM. Transcription start regions in the human genome are favored targets for MLV integration. Science. 2003;300:1749–1751. doi: 10.1126/science.1083413. [DOI] [PubMed] [Google Scholar]
- 9.Uren AG, Kool J, Berns A, van Lohuizen M. Retroviral insertional mutagenesis: past, present and future. Oncogene. 2005;24:7656–7672. doi: 10.1038/sj.onc.1209043. [DOI] [PubMed] [Google Scholar]
- 10.Li J, Shen H, Himmel KL, et al. Leukaemia disease genes: large‐scale cloning and pathway predictions. Nat Genet. 1999;23:348–353. doi: 10.1038/15531. [DOI] [PubMed] [Google Scholar]
- 11.Lund AH, Turner G, Trubetskoy A, et al. Genome‐wide retroviral insertional tagging of genes involved in cancer in Cdkn2a‐deficient mice. Nat Genet. 2002;32:160–165. doi: 10.1038/ng956. [DOI] [PubMed] [Google Scholar]
- 12.Castilla LH, Perrat P, Martinez NJ, et al. Identification of genes that synergize with Cbfb‐MYH11 in the pathogenesis of acute myeloid leukemia. Proc Natl Acad Sci U S A . 2004;101:4924–4929. doi: 10.1073/pnas.0400930101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolff L. Retroviral insertional mutagenesis and its application to genetically engineered mice. In: Thebault S, editor. Progress in virus research. Nova Science Publishers; 2006. pp. 267–299. [Google Scholar]
- 14.Lin YW, Nichols RA, Letterio JJ, Aplan PD. Notch1 mutations are important for leukemic transformation in murine models of precursor‐T leukemia/lymphoma. Blood. 2006;107:2540–2543. doi: 10.1182/blood-2005-07-3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wolff L, Koller R, Hu X, Anver MR. A Moloney murine leukemia virus‐based retrovirus with 4070A long terminal repeat sequences induces a high incidence of myeloid as well as lymphoid neoplasms. J Virol. 2003;77:4965–4971. doi: 10.1128/JVI.77.8.4965-4971.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rowe WP, Pugh WE, Hartley JW. Plaque assay techniques for murine leukemia viruses. Virology. 1970;42:1136–1139. doi: 10.1016/0042-6822(70)90362-4. [DOI] [PubMed] [Google Scholar]
- 17.Kogan SC, Ward JM, Anver MR, et al. Bethesda proposals for classification of nonlymphoid hematopoietic neoplasms in mice. Blood. 2002;100:238–245. doi: 10.1182/blood.v100.1.238. [DOI] [PubMed] [Google Scholar]
- 18.Morse HC, 3rd, Anver MR, Fredrickson TN, et al. Bethesda proposals for classification of lymphoid neoplasms in mice. Blood. 2002;100:246–258. doi: 10.1182/blood.v100.1.246. [DOI] [PubMed] [Google Scholar]
- 19.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 20.Hwang HC, Martins CP, Bronkhorst Y, et al. Identification of oncogenes collaborating with p27Kip1 loss by insertional mutagenesis and high‐throughput insertion site analysis. Proc Natl Acad Sci U S A. 2002;99:11293–11298. doi: 10.1073/pnas.162356099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pineault N, Buske C, Feuring‐Buske M, et al. Induction of acute myeloid leukemia in mice by the human leukemia‐specific fusion gene NUP98‐HOXD13 in concert with Meis1. Blood. 2003;101:4529–4538. doi: 10.1182/blood-2002-08-2484. [DOI] [PubMed] [Google Scholar]
- 22.Iwasaki M, Kuwata T, Yamazaki Y, et al. Identification of cooperative genes for NUP98‐HOXA9 in myeloid leukemogenesis using a mouse model. Blood. 2005;105:784–793. doi: 10.1182/blood-2004-04-1508. [DOI] [PubMed] [Google Scholar]
- 23.Ghinassi B, Sanchez M, Martelli F, et al. The hypomorphic Gata1low mutation alters the proliferation/differentiation potential of the common megakaryocytic‐erythroid progenitor. Blood. 2007;109:1460–1471. doi: 10.1182/blood-2006-07-030726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Migliaccio AR, Rana RA, Sanchez M, et al. GATA‐1 as a regulator of mast cell differentiation revealed by the phenotype of the GATA‐1low mouse mutant. J Exp Med. 2003;197:281–296. doi: 10.1084/jem.20021149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin DI, Zon LI, Mutter G, Orkin SH. Expression of an erythroid transcription factor in megakaryocytic and mast cell lineages. Nature. 1990;344:444–447. doi: 10.1038/344444a0. [DOI] [PubMed] [Google Scholar]
- 26.Akagi K, Suzuki T, Stephens RM, Jenkins NA, Copeland NG. RTCGD: retroviral tagged cancer gene database. Nucleic Acids Res. 2004;32:D523–D527. doi: 10.1093/nar/gkh013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelly LM, Gilliland DG. Genetics of myeloid leukemias. Annu Rev Genomics Hum Genet. 2002;3:179–198. doi: 10.1146/annurev.genom.3.032802.115046. [DOI] [PubMed] [Google Scholar]
- 28.Moskow JJ, Bullrich F, Huebner K, Daar IO, Buchberg AM. Meis1, a PBX1‐related homeobox gene involved in myeloid leukemia in BXH‐2 mice. Mol Cell Biol. 1995;15:5434–5443. doi: 10.1128/mcb.15.10.5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakamura T, Largaespada DA, Lee MP, et al. Fusion of the nucleoporin gene NUP98 to HOXA9 by the chromosome translocation t(7;11)(p15;p15) in human myeloid leukaemia. Nat Genet. 1996;12:154–158. doi: 10.1038/ng0296-154. [DOI] [PubMed] [Google Scholar]
- 30.Kroon E, Krosl J, Thorsteinsdottir U, Baban S, Buchberg AM, Sauvageau G. Hoxa9 transforms primary bone marrow cells through specific collaboration with Meis1a but not Pbx1b. Embo J. 1998;17:3714–3725. doi: 10.1093/emboj/17.13.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thorsteinsdottir U, Kroon E, Jerome L, Blasi F, Sauvageau G. Defining roles for HOX and MEIS1 genes in induction of acute myeloid leukemia. Mol Cell Biol. 2001;21:224–234. doi: 10.1128/MCB.21.1.224-234.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kroon E, Thorsteinsdottir U, Mayotte N, Nakamura T, Sauvageau G. NUP98‐HOXA9 expression in hemopoietic stem cells induces chronic and acute myeloid leukemias in mice. Embo J. 2001;20:350–361. doi: 10.1093/emboj/20.3.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Wely KH, Molijn AC, Buijs A, et al. The MN1 oncoprotein synergizes with coactivators RAC3 and p300 in RAR‐RXR‐mediated transcription. Oncogene. 2003;22:699–709. doi: 10.1038/sj.onc.1206124. [DOI] [PubMed] [Google Scholar]
- 34.Buijs A, Sherr S, van Baal S, et al. Translocation (12;22) (p13;q11) in myeloproliferative disorders results in fusion of the ETS‐like TEL gene on 12p13 to the MN1 gene on 22q11. Oncogene. 1995;10:1511–1519. [PubMed] [Google Scholar]
- 35.Heuser M, Beutel G, Krauter J, et al. High meningioma 1 (MN1) expression as a predictor for poor outcome in acute myeloid leukemia with normal cytogenetics. Blood. 2006 doi: 10.1182/blood-2006-04-014845. [DOI] [PubMed] [Google Scholar]
- 36.Kawagoe H, Grosveld GC. Conditional MN1‐TEL knock‐in mice develop acute myeloid leukemia in conjunction with overexpression of HOXA9. Blood. 2005;106:4269–4277. doi: 10.1182/blood-2005-04-1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roth MJ, Tanese N, Goff SP. Gene product of Moloney murine leukemia virus required for proviral integration is a DNA‐binding protein. J Mol Biol. 1988;203:131–139. doi: 10.1016/0022-2836(88)90097-6. [DOI] [PubMed] [Google Scholar]
- 38.Wieser R, Volz A, Vinatzer U, et al. Transcription factor GATA‐2 gene is located near 3q21 breakpoints in myeloid leukemia. Biochem Biophys Res Commun. 2000;273:239–245. doi: 10.1006/bbrc.2000.2947. [DOI] [PubMed] [Google Scholar]
- 39.Ohyashiki JH, Ohyashiki K, Shimamoto T, et al. Ecotropic virus integration site‐1 gene preferentially expressed in post‐myelodysplasia acute myeloid leukemia: possible association with GATA‐1, GATA‐2, and stem cell leukemia gene expression. Blood. 1995;85:3713–3718. [PubMed] [Google Scholar]
- 40.Volinia S, Calin GA, Liu CG, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He H, Jazdzewski K, Li W, et al. The role of microRNA genes in papillary thyroid carcinoma. Proc Natl Acad Sci U S A. 2005;102:19075–19080. doi: 10.1073/pnas.0509603102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu J, Wang F, Yang GH, et al. Human microRNA clusters: genomic organization and expression profile in leukemia cell lines. Biochem Biophys Res Commun. 2006;349:59–68. doi: 10.1016/j.bbrc.2006.07.207. [DOI] [PubMed] [Google Scholar]
- 43.Johansson FK, Brodd J, Eklof C, et al. Identification of candidate cancer‐causing genes in mouse brain tumors by retroviral tagging. Proc Natl Acad Sci U S A. 2004;101:11334–11337. doi: 10.1073/pnas.0402716101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Si ML, Zhu S, Wu H, Lu Z, Wu F, Mo YY. miR‐21‐mediated tumor growth. Oncogene. 2006 doi: 10.1038/sj.onc.1210083. [DOI] [PubMed] [Google Scholar]
- 45.Chan JA, Krichevsky AM, Kosik KS. MicroRNA‐21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65:6029–6033. doi: 10.1158/0008-5472.CAN-05-0137. [DOI] [PubMed] [Google Scholar]
- 46.Iorio MV, Ferracin M, Liu CG, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 47.Hess JL. MLL: a histone methyltransferase disrupted in leukemia. Trends Mol Med. 2004;10:500–507. doi: 10.1016/j.molmed.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 48.Golub TR, Slonim DK, Tamayo P, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 49.Marcucci G, Baldus CD, Ruppert AS, et al. Overexpression of the ETS‐related gene, ERG, predicts a worse outcome in acute myeloid leukemia with normal karyotype: a Cancer and Leukemia Group B study. J Clin Oncol. 2005;23:9234–9242. doi: 10.1200/JCO.2005.03.6137. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.