

Abstract
We assessed the temporal and spatial characteristics of PN-induced oxidative damage and its relationship to calpain-mediated cytoskeletal degradation and neurodegeneration in a severe unilateral controlled cortical impact (CCI) traumatic brain injury (TBI) model. Quantitative temporal time-course studies were performed to measure two oxidative damage markers: 3-nitrotyrosine (3NT) and 4-hydroxynonenal (4HNE) at 30 min, 1, 3, 6, 12, 24, 48, 72 hrs, 7 days after injury in ipsilateral cortex of young adult male CF-1 mice. Secondly, the time course of Ca++-activated, calpain-mediated proteolysis was also analyzed using quantitative western-blot measurement of breakdown products of the cytoskeletal protein α-spectrin. Finally, the time course of neurodegeneration was examined using de Olmos silver staining. Both oxidative damage markers increased in cortical tissue immediately after injury (30 min) and elevated for the first 3–6 hrs before returning to baseline. In the immunostaining study, the PN-selective marker, 3NT, and the lipid peroxidation marker, 4HNE, were intense and overlapping in the injured cortical tissue. α-Spectrin breakdown products, which was used as biomarker for calpain-mediated cytoskeletal degradation, were also increased after injury, but the time course lagged behind the peak of oxidative damage and did not reach its maximum until 24 hrs post-injury. In turn, cytoskeletal degradation preceded the peak of neurodegeneration which occurred at 48 hrs post-injury. These studies have led us to the hypothesis that PN-mediated oxidative damage is an early event that contributes to a compromise of Ca++ homeostatic mechanisms which causes a massive Ca++ overload and calpain activation which is a final common pathway that results in post-traumatic neurodegeneration.
Keywords: peroxynitrite, oxidative damage, calpain, cytoskeletal degradation, neurodegeneration, controlled cortical impact (CCI), TBI
INTRODUCTION
There is compelling evidence supporting the role of oxidative damage in the delayed secondary neuronal cell death which is initiated by the primary traumatic brain injury (TBI). The first work in this regard, conducted by Kontos and his colleagues, demonstrated the formation of superoxide radicals in cerebral microvasculature in a cat fluid percussion TBI models (Kontos and Povlishock, 1986; Kontos and Wei, 1986). Using the salicylate trapping method, Hall and coworkers detected a rapid, but transient rise in brain hydroxyl radical (•OH) in experimental head injury models (Hall et al., 1993; Hall et al., 1994); (Smith et al., 1994). This was elegantly confirmed by others using brain microdialysis techniques to monitor salicylate-trapped •OH over time in the same animals (Globus et al., 1995). The increase of •OH is followed by an increase in lipid peroxidation products (Smith et al., 1994). The strongest support for a role of oxidative damage in the acute pathophysiology of TBI is derived from the fact that several antioxidant compounds have been shown to be neuroprotective in TBI models (Hall et al., 1994) (Awasthi et al., 1997); (Mori et al., 1998); (Marklund et al., 2001).
The reactive oxygen species (ROS) peroxynitrite (PN), which can produce highly reactive and cytotoxic free radicals, has been suggested to be a key player in post-traumatic secondary brain oxidative damage (Hall et al., 2004; Hall et al., 1999). Peroxynitrite is formed by the chemical reaction of nitric oxide (•NO), produced by nitric oxide synthase (NOS), with superoxide radical (O2• −) whenever the two are produced in close proximity (Saran et al., 1990). All three NOS isoforms (eNOS, iNOS, and nNOS), as a key element in production of PN, have been shown to be up-regulated in brain tissue following TBI (Cobbs et al., 1997); (Wada et al., 1998) (Gahm et al., 2000); (Orihara et al., 2001). In addition, a novel isoform of Ca++-sensitive NOS (mtNOS), discovered within mitochondria (Bates et al., 1995); (Lopez-Figueroa et al., 2000);(Giulivi, 2003), has been shown to contribute to mitochondrial production of •NO and PN (Radi et al., 2002). The PN-derived free radicals (•NO2, •OH, •CO3−) can induce extensive oxidative damage to cellular membranes, proteins and DNA (Beckman, 1996) (Murphy et al., 1998); (Radi, 1998). Each of the PN-derived free radicals can initiate lipid peroxidation (LP), or cause protein carbonylation by reaction with susceptible amino acids (e.g. lysine, cysteine, arginine). Moreover, aldehydic LP products (e.g. 4-hydroxynonenal) can bind to cellular proteins compromising their structural and functional integrity (Neely et al., 1999). Additionally, •NO2 can nitrate 3 position of tyrosine residues in proteins. As a result, 3-nitrotyrosine (3NT) is used as a specific footprint of PN-induced cellular damage (Hall et al., 1997).
In recent work we investigated the role of PN in a mouse model of diffuse closed head injury which demonstrated the spatial and temporal coincidence of PN-induced protein nitration and lipid peroxidation and that this oxidative damage precedes, and therefore may have a causal role in post-traumatic neurodegeneration (Hall et al., 2004). However, TBI is a complex disorder that is impossible to fully reproduce in a single model. For example, we have found significant differences between the time courses of neurodegeneration in mouse models of diffuse (Kupina et al., 2003) and focal (Hall et al., 2005a; Hall et al., 2005b) TBI. Hence, the magnitude and timing of post-traumatic secondary injury mechanisms probably varies across head injury models. Most importantly, if we are to accurately test the efficacy of novel neuroprotective pharmacological treatments, it is important to first understand the time course of the relevant secondary injury mechanisms in models of focal as well as diffuse TBI.
Thus, the present study was conducted to examine the temporal and spatial characteristics of PN-induced cortical oxidative damage in a model of severe controlled cortical impact (CCI) focal TBI, and its relationship to calpain-mediated cytoskeletal degradation and neurodegeneration. Markers for PN-induced protein nitration (3-nitrotyrosine, 3NT), and lipid peroxidation (4-hydroxynonenal, 4HNE) were measured using immuno-slotblotting and immuno-histochemistry. Secondly, we employed western-blotting methods to look at the time course of calpain-mediated cytoskeletal degradation in order to assess the temporal relationship of oxidative damage to Ca++−mediated proteolytic degradation. Finally, we used de Olmos silver staining to measure the evolution of post-traumatic neurodegeneration. The results demonstrate that the PN-mediated oxidative damage is an early event that probably contributes to an exacerbation of neuronal intracellular Ca++ overload, massive calpain activation and cytoskeletal degradation and neurodegeneration.
MATERIALS AND METHODS
All the surgical, injury and animal care protocols described below have been approved by the University of Kentucky Institutional Animal Care and Use Committee, and are consistent with the animal care procedures set forth in the guidelines of the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Mouse Model of Controlled Cortical Impact (Focal) Traumatic Brain Injury
Young adult male CF-1 mice (Charles River, Portage, MI) weighing 29–31g were used in this study. The mice were anesthetized with isoflurane (3.0%), shaved, and then placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA). Throughout the surgery, the mice were provided with constant isoflurane (SurgiVet, 100 Series) and oxygen (SurgiVet, O2 flowmeter, 0–4Lpm). The head was positioned in the horizontal plane of a stereotaxic frame with the nose bar set at zero. We firstly produced a 4mm craniotomy lateral to the sagittal suture, and centered between lambda and bregma. A cortical contusion was produced on the exposed cortex (the anterior-posterior coordinate for the epicenter of the injury was Bregma −2.0mm) using a pneumatically-controlled impactor device (Precision System Instruments TBI-0200 Impactor, Lexington, KY) similar to that previously described (Sullivan et al., 1999b) (Hall et al., 2005b; Sullivan et al., 1999a) except that the current device utilizes a unique contact sensor mechanism that ensures accurate and reliable determination of cortical surface prior to initiating the injury sequence. This results in increased accuracy and reproducibility in regards to the CCI injury compared to that produced by earlier CCI impactors. In the present studies, the impactor containing a 3mm diameter rod tip compressed the cortex at 3.5m/sec to a depth of 1mm with a dwell time of 50msec to produce a severe injury. After surgery and injury, a 4mm disk made from dental cement (Dentsply Trubyte) was placed over the craniotomy site and adhered to the skull using cyanoacrylate. In order to prevent immediate post-traumatic hypothermia, following the suturing of the skin, mice were placed in a Hova-Bator incubator (37°C, model 1583, Randall Burkey Co) until them regained consciousness (determined by the regain of the righting reflex and increased mobility). The injured mice were allowed to survive from 30 min to 7 days depending on their experimental group. For the neurochemical studies (measurement of oxidative damage markers and cytoskeletal degradation, see below), the N for each timepoint group was 8 animals based upon our experience with the variability seen with these measurements in previous studies (Hall et al., 2004; Hall et al., 2005b; Kupina et al., 2003). For each timepoint, two sham animals were included which were mice that received craniotomy but not cortical contusion. The two sham animals in each group were killed at each of the post-traumatic times along with their brain-injured counterparts. Eight sham mice were randomly selected for comparison with the injured groups.
Tissue Extraction and Protein Assay
At the selected times for the time course analysis (30 min, 1 hr, 3 hrs, 6 hrs, 12 hrs, 24 hrs, 48 hrs, 72 hrs and 7 days), the sham or CCI-injured mice were deeply overdosed with sodium pentobarbital (200mg/kg i.p.). Following decapitation, the ipsilateral cortex were rapidly dissected on an ice-chilled stage and immediately transferred into pre-cooled Triton lysis buffer (1% triton, 20mM tris HCL, 150mM NaCl, 5mM EGTA, 10mM EDTA, 10% glycerol) with protease inhibitors (Complete Mini™ Protease Inhibitor Cocktail tablet). Samples were then briefly sonicated and vortexed at 14,000rpm for 30 minutes at 4°C, and the supernatants were collected for protein assay. Protein concentration was determined by Bio-Rad DC Protein Assay, with sample solutions diluted to contain 1mg/ml of protein for immunoblotting.
Slot-Immunoblotting Analysis of Oxidative Damage (3NT and 4HNE)
To measure 3-nitrotyrosine (3-NT) or 4-hydroxynonenal (4-HNE), an aliquot of each ipsilateral cortical sample (2μg) was diluted with 200μl of tris-buffered saline (TBS), and transferred to a Protran (0.2μm) nitrocellulose membrane (Schleicher & Schuell, Dassel, Germany) by a Minifold II vacuum slot blot apparatus (Schleicher & Schuell). After the samples were loaded into the slots, they were allowed to filter through the membrane by gravity (no vacuum). Each slot was then washed with 200μl TBS which was allowed to filter through the membrane again. The membranes were then disassembled from the apparatus, and incubated in a TBS blocking solution with 5% milk for 1 hr at room temperature. For the detection of 3NT, a rabbit polyclonal anti-nitrotyrosine antibody (Upstate Biotechnology, MA, USA) was used at a dilution of 1:2000 in TBST blocking solution with 5% milk for overnight at 4°C. For the detection of HNE, a rabbit polyclonal anti-HNE antibody (Alpha Diagnostics International) was used at a dilution of 1:5000 in TBST blocking solution with 5% milk for overnight at 4°C. A goat anti-rabbit secondary antibody conjugated to an infrared dye (1:5000, IRDye800CW, Rockland) was applied to the membrane for 1 hr at room temperature. Dry membranes were then imaged and quantified using the Li-Cor Odyssey Infrared Imaging System (Li-Cor® Biosciences). Preliminary experiments established protein concentration curves in order to ensure that quantified blots were in the linear range. From the results of these, we selected the 2 μg for use based upon that amount giving blots that were well within the linear range. A standardized protein loading control was included on each blot to normalize the band densities so that comparisons could be made across multiple blots. All the samples were run in duplicate which were then averaged. For both the 4HNE and 3NT analyses, no primary antibody controls were run to verify that the oxidative damage staining was specific. In addition, we ran positive controls using both bovine serum albumin and normal brain tissue which were exposed to PN and negative controls in which we pre-absorbed the primary antibodies with either 4HNE or 3NT which resulted in inhibition of the 4HNE and 3NT staining.
Immunohistochemical Analysis of Oxidative Damage (3NT and 4HNE)
A second set of animals was employed for immunohistochemical analysis of 3NT and 4HNE. At the appointed times for the posttraumatic time course analysis (30 min, 1 hr, 3 hrs, 6 hrs, 12 hrs), groups of 5 mice each were overdosed with sodium pentobarbital (200mg/kg i.p.). Two sham, non-injured mice that had craniotomies, but no injury, were included for each timepoint. The mice were then perfused transcardially with 0.9% sodium chloride (pH7.4) until the venous effluent (sectioned superior vena cava) was cleared of blood, followed by a fixative solution made with 4% paraformaldehyde in 0.2M PBS (pH7.4). The heads were decapitated after fixation and stored in the same fixative solution overnight at 4°C. The brains were removed and equilibrated in the same fixative solution with 15% sucrose overnight at 4°C. The equilibrated brains were sectioned at a thickness of 35μm into the same fixative solution with 15% sucrose at −20°C using a microtome with a freezing stage.
For the 4HNE detection, the free-floating sections were pre-treated with 0.1M NaBH4 in 0.1M MOPS (pH8.0) for 10 min and rinsed off with 0.2M PBS for three times. The sections were then incubated in 0.3% H2O2 for 30 min and rinsed three times with 0.2M phosphate-buffered saline (PBS). Following that, the sections were blocked with a blocking solution (5% goat serum, 0.25% Triton X-100, 1% dry milk, 0.2M PBS) for 2 hrs at room temperature, and then incubated in the primary anti-HNE antibody (rabbit anti-HNE Michael Adducts, Calbiochem) diluted 1:5500 in the same blocking solution overnight at room temperature. The sections were then rinsed with 0.2M PBS six times, and then incubated in secondary goat anti-rabbit antibody (Vectastain ABC-AP kit) for 2 hrs at room temperature. The sections were then rinsed with 0.1M Tris-HCl (pH8.2) and incubated in Vector blue (Vector blue alkaline phosphatase substrate kit III). They were then rinsed with tap water four times, and mounted onto gelatinized slides. For the 3NT detection, the sections were not pre-treated with 0.1M NaBH4 in 0.1M MOPS (pH8.0), but were treated the same for the rest of the procedure, except that they were incubated in the primary anti-3NT antibody (rabbit anti-nitrotyrosine, Upstate) diluted 1:1200 in the blocking solution. For both the 4HNE and 3NT analyses, control slides were run without the primary antibodies to verify that the oxidative damage staining was specific. In addition, we ran additional controls in which we pre-absorbed the primary antibodies with either 4HNE or 3NT which resulted in inhibition of the 4HNE and 3NT immunostaining. After the immunohistochemical procedure, all slides mounted with brain sections were counterstained with nuclear fast red (Vector Lot# Q1214), and then cover slipped. The brain sections were photographed on an Olympus Provis A70 microscope at 1.25x, 20x and 40x magnification using an Olympus Magnafire digital camera.
Western-blotting Analysis of Calpain-mediated Cytoskeletal Degradation
To measure calpain-mediated α-spectrin proteolysis, aliquots of each cortical sample (5μg) were run on a SDS/PAGE Precast gel (3–8% Tris-Acetate Criterion™ XT Precast gel, Bio-Rad) and then transferred to a nitrocellulose membrane using a semi-dry electro-transferring unit set at 15V for 15 min. Preliminary experiments established protein concentration curves in order to ensure that quantified bands were in the linear range as measured with the Li-Cor Odyssey Infrared Imaging System. The membranes were incubated in a TBS blocking solution with 5% milk for 1 hr at room temperature. For the detection of α-spectrin and its breakdown products, a mouse monoclonal anti-α-spectrin antibody (Affiniti FG6090) was used at a dilution of 1:5000 in TBST blocking solution with 5% milk for overnight at 4°C. A goat anti-mouse secondary conjugated to an infrared dye (1:5000, IRDye800CW, Rockland) was then applied for 1 hr at room temperature. After drying, the membranes were then imaged and quantified using the Li-Cor Odyssey Infrared Imaging System. A standardized protein loading control was included on each blot to normalize the band densities so that comparisons could be made across multiple blots (Hall et al., 2005b). This was made up of pooled brain tissue protein collected from previously run TBI mice which gave strong bands corresponding to the 280kD parent α-spectrin, the 150 kD and the 145 kD breakdown products. The amount of protein in the loading control had been previously determined and shown to be within the linear range as measured with the Li-Cor Odyssey Infrared Imaging System. Following the transfer, the gels were stained with Coomassie Blue to verify even transfer. All the samples were run in duplicate and averaged.
De Olmos Silver Staining Analysis of Neurodegeneration
Neurodegeneration was examined using the de Olmos aminocupric silver histochemical technique in a third set of 27 mice as previously described (de Olmos et al., 1994; Hall et al., 2005a; Hall et al., 2005b; Switzer, 2000). At either 6, 12, 24, 48, or 72 hrs or 7 days the injured mice (N=3–4 per time point) were overdosed with sodium pentobarbital (200mg/kg i.p) and transcardially perfused with 0.9% sodium chloride, followed by a fixative solution containing 4% paraformaldehyde; a sham group of 4 mice that received craniotomy only were sacrificed 24 hrs following surgery. Following decapitation, the heads were stored in a fixative solution containing 15% sucrose for 24 hrs after which the brains were removed, placed in fresh fixative and shipped for histological processing to Neuroscience Associates Inc (Knoxville TN). The 27 brains used for this study were embedded into one gelatin block (Multiblock® Technology, Neuroscience Associates). The block was then frozen and thirteen 35μm coronal sections were taken 420μm apart between 1.1mm anterior and 4.4mm posterior to bregma, were de Olmos silver-stained to reveal degenerating neurons and neuronal processes, and then counterstained with Nuclear Fast Red. The brain sections were photographed on an Olympus Provis A70 microscope at 1.25x magnification using an Olympus Magnafire digital camera and the image was analyzed by Image-Pro Plus (4.0). The percentage area of silver staining for each brain section was calculated by dividing the area of silver staining in each section by the area of the total hemispheric section and multiplying by 100. The volume of silver staining in the hemisphere as a percentage of the overall hemispheric volume was estimated by the by the equation % V = t × Σ % a(s), where % V is percent silver stain volume, t = the distance between sections analyzed (420μm) and Σ % a (s) is the sum of percent area of silver staining in all sections examined (13 for each brain) (Hall et al., 2005b).
Statistical Analysis
For all of the time course analyses, we used Statview 5.0 to perform a one-way analysis of variance (ANOVA), followed by Fisher’s PLSD post-hoc analysis to determine the significance of differences between individual time points and the non-injured Sham group. For the ANOVA, a p<0.05 was required to establish a statistically significant difference across the groups. However, for the post-hoc Fisher’s analysis, the program determined significance based upon a correction for multiple comparisons comparing sham to each of the 9 post-injury time points. Differences between pairs of post-injury timepoints (e.g. 30 min. vs. 1 hr) were also analyzed.
RESULTS
Quantitative Post-Traumatic Time Course of Protein Nitration and Lipid Peroxidation
Figure 1 displays the complete quantitative time course study for 3NT and 4HNE in the ipsilateral cortical samples taken from sham or CCI-injured mice. Figure 1A indicates schematically the dorsal and coronal view of contusion site and peri-contusional cortical tissue that was collectively sampled for the current study. Figure 1B shows the time course of changes in 3NT, a selective marker of PN-mediated damage. As noted in the MATERIALS AND METHODS, two non-injured sham animals corresponding to each post-traumatic timepoint were run. For the nine timepoints, this resulted in a total of 18 shams. A comparison of the bands obtained from different pairs of shams, showed little evidence that those animals killed at the later timepoints had any more 3NT or 4HNE than those sampled at the earlier time points. Therefore, we randomly selected 8 of the 18 shams to use as the sham group for comparison with the different injured groups. The ANOVA showed that there was a highly significant overall difference across the collective sham and post-traumatic groups in regards to 3NT levels [F(9,70)=2.744; p<0.0001]. Post-hoc analysis revealed a significant increase in 3NT at 30 min after injury compared to the sham group. At 1 hr after injury, the 3NT level reached its peak and was approximately twice as much as that seen in sham, non-injured mice. The 3NT level maintained significantly high until 12 hrs after injury, and at 24 hrs, it returned to baseline (sham level). However, a small, but statistically significant secondary increase in 3NT was also observed at 48, but not at 72 hrs.
Figure 1.
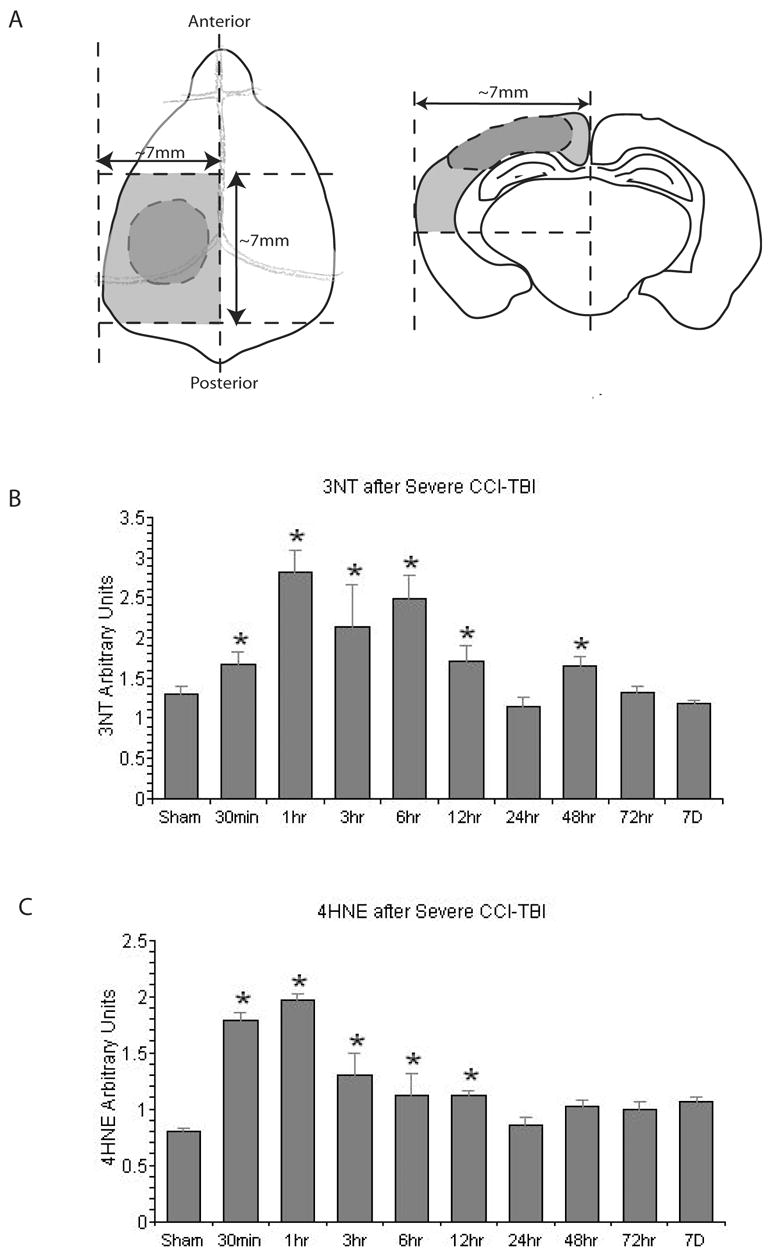
Slot-blotting studies in ipsilateral cortical traumatic brain injury tissues showing the temporal changes in protein nitration (3-nitrotyrosine; 3NT) and lipid peroxidation. A. Schematic showing peri-contusional cortical tissue samples; B. time course of changes in 3-NT; C. Time course of LP end product 4HNE. N = 8 animals per timepoint; values = mean ± standard error; one-way ANOVA and Fisher’s PLSD post hoc test: *P < 0.0001 vs. Sham.
Figure 1C displays the time course of post-traumatic changes in 4HNE, a marker for lipid peroxidation, at different timepoints after injury. As with the 3NT analysis, there was a highly significant overall difference across the collective sham and post-traumatic groups in regards to 4HNE content [F(9,70)=15.211; p<0.0001]. Post-hoc analysis of between group differences showed that there was a significant increase in 4HNE at 30 min after injury, the 4HNE level reached its peak at 1 hr after injury and it stayed significantly higher than the sham level until 12hr before returning to the baseline levels seen in the Sham animals.
Spatial and Temporal Distribution of Peroxynitrite-Induced Protein Nitration and Lipid Peroxidation
Figure 2 demonstrates the temporal and spatial characteristics of the immunohistochemical staining related to post-traumatic oxidative damage in the ipsilateral hemisphere of mice after CCI injury. Two adjacent brain sections in the epicenter (Bregma-2.0mm) of the contusion were selected for 3NT staining and 4HNE staining. In Figure 2, the staining for 3NT and 4HNE markers were concentrated within and around the contusion area in the ipsilateral cortex, and the 3NT and 4HNE staining was largely overlapping. Staining for both markers was observed as early as 30 min. Staining remained intense up to 6 hrs after injury. At 12 hrs, the staining showed signs of decrease no doubt due to proteolytic degradation of the oxidatively modified proteins. This time course is in good agreement with the quantitative time course for 3NT and 4HNE shown in Figure 1.
Figure 2.
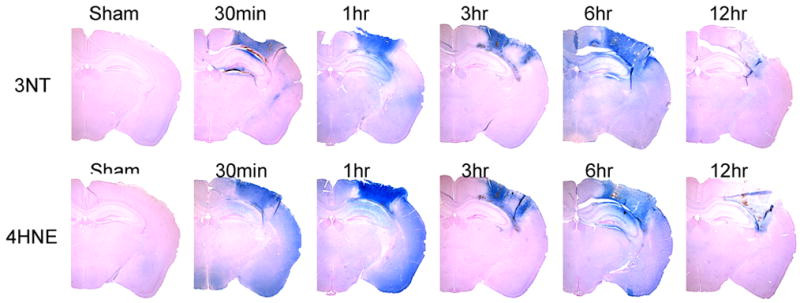
Immunohistochemical (IHC) staining studies showing the time course and spatial extent of PN-induced 3NT and 4HNE at the epicenter (Bregma-2.0mm) of the injury site during the first 12 hrs compared to Sham. The IHC staining indicated prominent increase of both markers after injury and similar distribution between the two.
Figure 3 shows high power photomicrographs of 3NT and 4HNE immunostaining within the cortical contusion site in a sham, non-injured brain compared to staining at the contusion site at 1 hr after injury. At 20x and 40x magnification, intense staining for both oxidative damage markers is seen throughout the neuropil. In addition, microvascular staining for both 3NT and 4HNE is seen to clearly outline the microvessels deep within the injured cortex. Although the sham brain shows some light staining in the neuropil, there is no evidence of staining of non-injured microvessels. The pattern of staining in both the parenchyma and vasculature has also been seen previously after a diffuse brain injury (Hall et al., 2004).
Figure 3.
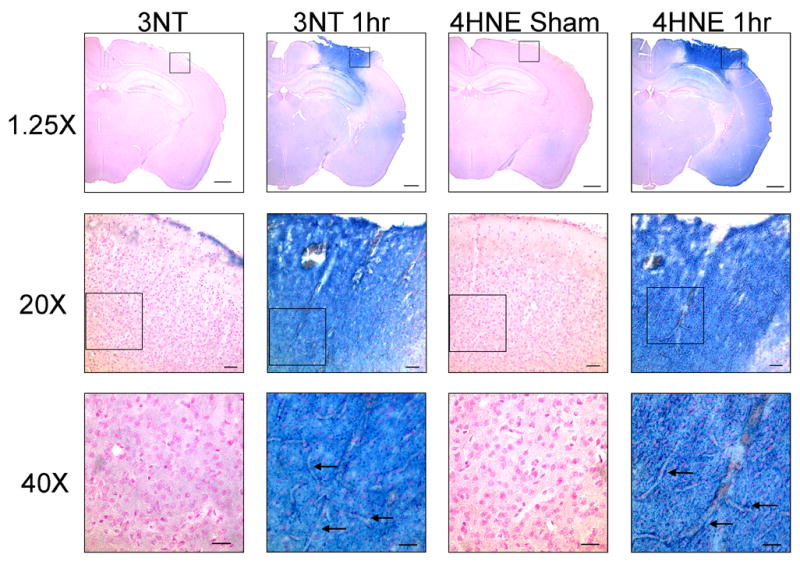
High power photomicrographs of 3NT and 4HNE immunostaining in the contusion site in a sham, non-injured brain compared to staining at the contusion site at 1 hr after injury. At 20x and 40x magnification, intense staining for both oxidative damage markers is seen throughout the neuropil. In addition, microvascular staining for both 3NT and 4HNE is seen to clearly outline the microvessels (arrows) deep within the injured cortex. Although the Sham brain shows some light staining in the neuropil, there is no evidence of staining of non-injured microvessels. Please note that the 20x and 40x focus was adjusted to emphasize the microvascular staining, and as a result the background tissue appears slightly out of focus. The calibration bar for the 1.25x photomicrographs is 2.0mm; for 20x the calibration bar is 100μm; for 40x the calibration bar is 50μm.
Quantitative Post-traumatic Time Course of Calpain-Mediated Cytoskeletal Degradation
Figure 4 displays the quantitative post-traumatic time course analysis of calpain-mediated cytoskeletal degradation in the ipsilateral cortical tissue in terms of the levels of α-spectrin breakdown products SBDP145 and 150 measured by western-blotting. The spectrin breakdown product 145 (kD) and 150 (kD) in each time group were compared to the sham SBDP145 and the sham SBDP150 respectively. The SBDP150 band can be produced by either calpain or caspase 3, whereas the SBDP145 band is calpain specific. ANOVA showed that there was a significant overall post-traumatic increase in SBDP145 [F(9,70)=9.444; p<0.0001] and SBDP150 [F(9,70)=8.857; p<0.0001]. Accordingly, post-hoc analysis of the significance of increases seen at individual post-traumatic time points was compared to sham, non-injured animals. At 30 min after injury, the mean SBDP150 level was significantly increased. The levels of this mixed calpain/caspase 3 marker remained significantly higher than sham levels at all subsequent timepoints out to, and including, 48 hrs post-injury. The maximum increase in SBDP150 was observed at 24 hrs post-injury. The calpain-specific SBDP145 showed a significant increase vs. sham beginning at 1 hr post-injury, and increased slightly more at 3, 6 and 12 hrs.
Figure 4.
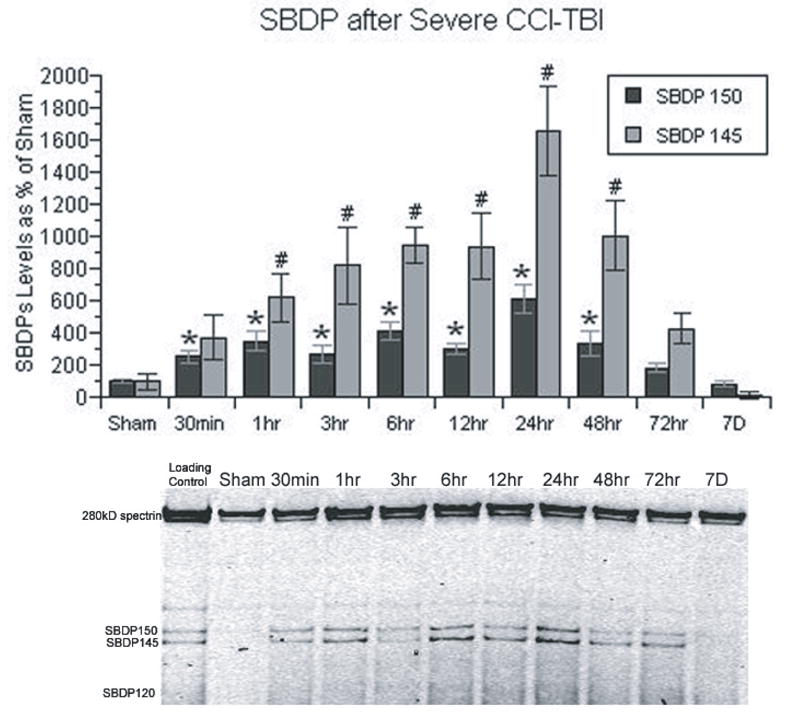
Time course of the post-traumatic increase in calpain-mediated α-spectrin breakdown products in CCI cortical tissues. Spectrin breakdown product (SBDP) 145 is specific to calpain activity, whereas SBDP 150 is produced by both calpain and caspase 3. Both SBDPs showed similar patterns, except that calpain-mediated SBDP 145 showed a more prominent increase. Both SBDPs have an immediate increase following injury, and did not reach their peak until 24 hrs. N = 8 animals per timepoint; values = mean + standard error; one-way ANOVA and Fisher’s PLSD post hoc test: *P < 0.0001 vs. Sham SBDP150; one-way ANOVA and Fisher’s PLSD post hoc test: #P < 0.0001 vs. Sham SBDP145.
Between 12 and 24hrs, there was a statistically significant (12 hr vs. 24 hr; p<0.0004) 75% jump in SBDP145 levels with the 24 hrs time point manifesting the maximum post-traumatic increase. In parallel, there was an equally significant (12 vs. 24 hr; p<0.0001) increase in the SBDP150 levels SBDP150. The magnitude of the increase in SBDP145 was greater (compared to the sham group) (18-fold) in comparison to the SBDP150 increase (6-fold). The level of SBDP145 remained significantly elevated at 48 hrs before returning to levels at 72 hrs after TBI that were no longer significantly higher than those seen in sham animals. The overall time course patterns were similar for both SBDP150 and SBDP145.
It should be noted that the current post-traumatic time course of α-spectrin degradation, although similar in several respects to a time course study we published earlier (Hall et al., 2005b) using the CCI model, shows some differences that are worthy of explanation. In the previous study, the earliest post-traumatic timepoint examined was 6 hrs at which time we observed a statistically significant increase in SBDP145 and 150 compared to sham, just as in the present study. However, the elevation in SBDPs did not increase further at 24 or 48 hrs, but rather appeared to plateau before returning to the sham baseline level at 1 week. This is in contrast to the significantly higher peak in SBDP 145 and 150 observed in the current study. There are two likely reasons for the discrepancy between the current and the previously published time course (Hall et al., 2005b). First of all, in the current study we employed a more sensitive infrared imaging method for performing the densitometric analysis of the blots which has a broader linear range compared to the less sensitive enhanced chemiluminescence (ECL) method that we previously employed which is only semi-quantitative. Consequently, the previous plateau between 6 and 72 hrs post-injury (Hall et al., 2005b), in contrast to the current results is probably due to the less accurate nature of ECL vs. infrared imaging. Secondly, the current study was performed with a newer CCI device than the one used in the previous study. The newer device (PSI TBI 0200 Impactor) utilizes a unique contact sensor mechanism that insures accurate and reliable determination of cortical surface prior to initiating the injury sequence. This results in increased accuracy and reproducibility in regards to the CCI injury. Thus, for these two reasons, we have more confidence in the accuracy of the current results in regards to the time course of post-traumatic cytoskeletal degradation. Furthermore, the current time course of α-spectrin degradation in the CCI-injured cortex is similar in pattern to the time course of α-spectrin degradation we have seen in the injured hippocampus using the same injury device, injury severity and infrared imaging of the blots (Thompson et al., 2006)
Quantitative Post-Traumatic Time Course of Neurodegeneration
Figure 5 shows the quantitative de Olmos aminocupric silver staining analysis of neurodegeneration in the ipsilateral hemisphere of mice subjected to CCI along with histological examples of the spatial and temporal characteristics of silver staining between 6 hrs and 7 days after injury. At 6 hrs after injury, there was a significant increase of silver staining volume compared to minimal level seen in sham, non-injured mouse brains. The silver staining spread over time and peaked at 48 hrs after injury. After 48 hrs, the volume of silver staining slowly waned, but remained significantly higher than the sham level even at 7 days. As shown in the selected brain sections, at 6 hrs, the silver staining was mainly concentrated in all layers in the ipsilateral cortical contusion. A cavitation lesion began to develop at 12 hrs and became evident at 24 hrs. At 24 hrs, the silver staining had spread into and throughout the ipsilateral hippocampus and dorsolateral aspect of the thalamus. At 48 hrs, the silver staining volume reached its greatest extent. Silver staining also extended to the contralateral hippocampus, indicative of the degeneration of CA3 commissural fiber projections to the contralateral dentate gyrus and CA1 region as previously shown (Hall et al., 2005b).
Figure 5.
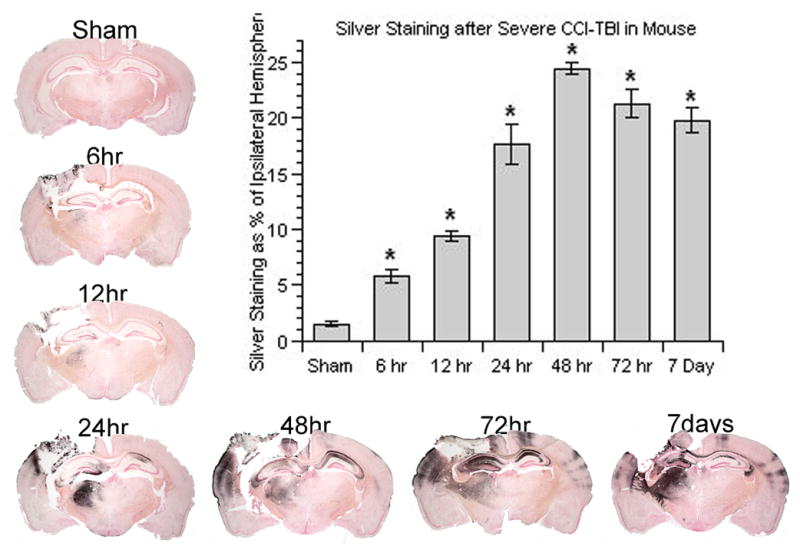
Time course of post-traumatic neurodegeneration in CCI model as revealed by the de Olmos silver staining technique. The bar chart provides the quantification of the lesion volume abut measurement of the silver staining. An increase in silver staining volume determined by image analysis (see Materials and Methods) occurred as early as 6 hrs and reached its peak at 48 hrs post-injury. N = 3–4 animals per time point; values = mean ± standard error; one-way ANOVA and Fisher’s PLSD post hoc test: *P < 0.0001 vs. Sham.
DISCUSSION
The present study in the context of the focal controlled cortical impact (CCI) mouse model has uniquely defined in a parallel fashion the time courses of oxidative damage, calpain-mediated cytoskeletal degradation and neurodegeneration. A careful analysis of these three secondary injury parameters has revealed clues concerning their mechanistic interrelationships. The following discussion lays out the hypothesis that 1) the potent ROS PN is a key mediator of post-traumatic oxidative damage; 2) a major source of PN is Ca++−overloaded mitochondria; 3) a major consequence of PN-mediated oxidative damage is exacerbation of intracellular Ca++ overload by impairment of Ca++ homeostatic mechanisms and 4) this leads to an enhancement of calpain-mediated cytoskeletal degradation which is the immediate precursor of post-traumatic neuronal degeneration.
Temporal and Spatial Characteristics of Oxidative Damage and the Role of Peroxynitrite
The two oxidative markers: tyrosine nitration (3NT) and LP-derived 4HNE modified proteins (see Halliwell and Gutteridge, 1999), increased in unison. Their increase occurred as early as 30 min after injury and peaked at 1hr. The coincidence of the early time course of 3NT and 4HNE suggests that they share a common ROS as their initiating source. The most likely candidate ROS in this regard is PN. The nitration of the 3 position of tyrosine residues in proteins is a result of PN activity and is used as a selective marker for PN-induced oxidative damage. Therefore, it is generally accepted as evidence of a role of PN in tissue injury models where 3NT elevations are seen (Beckman, 1996). Moreover, the indictment of PN as a source of LP (4HNE) is strongly implied by the fact that 3NT and 4HNE share the same early post-traumatic onset (30 min), time to peak (1 hr) and duration (12 hrs). Furthermore, spatial overlap of 3NT and 4HNE seen in the immunohistochemical examples also indicates that nitration and LP are both initiated by PN. Previous work from our laboratory using a mouse model of diffuse TBI, has also shown the spatial and temporal coincidence of 3NT and 4HNE immunostaining leading to a similar conclusion that PN was key player in the post-traumatic oxidative damage (Hall et al., 2004). However, in the case of the previously studied diffuse TBI model, the duration of the increase in 3NT and 4HNE was much longer (at least 96 hrs) in contrast to the much shorter expression of oxidative damage in the presently employed focal paradigm. Particularly important is the fact that PN-mediated oxidative damage was observed in both the cortical microvessels and in the neural parenchyma consistent with our earlier studies in the mouse diffuse TBI model (Hall et al., 2004; Hall et al., 1999)
Sources of Peroxynitrite
Regarding the sources of PN in the injured brain, •NO is produced by multiple NOS isoforms, and is a mediator of physiological (Garthwaite and Boulton, 1995) (Bicker, 2001) and neuroprotective (Mohanakumar et al., 1998) (Chiueh, 1999) actions as well as being an effecter of PN formation and oxidative damage (Dawson and Dawson, 1996); (Vicente et al., 2006). An earlier immunocytochemical study revealed the induction of endothelial NOS (eNOS) isoform in microvessels surrounding the cortical contusion post-injury, which may contribute to blood-brain barrier (BBB) breakdown and hyperemia followed by TBI (Cobbs et al., 1997). Other isoforms of NOS, neuronal NOS (nNOS) and inducible NOS (iNOS), have also been shown to be upregulated in TBI models. In a focal brain contusion model, all three isoforms of NOS (nNOS, eNOS, iNOS) have been found to be increased. However, each is expressed in different compartments and cells, and is differentially regulated (Gahm et al., 2000). In addition, a possibly novel isoform of NOS (mtNOS) is constitutively present in mitochondria, is Ca++-dependent (Giulivi, 2003) and appears to be involved in mitochondrial respiratory regulation (Elfering et al., 2002). Under hypoxic conditions, mtNOS activity is induced in comparison to that seen in non-stressed mitochondria (Lacza et al., 2001). In rat liver mitochondria, mtNOS has been shown to stimulated by Ca++ influx (Ghafourifar and Richter, 1997) and leads to intramitochondrial PN formation (Ghafourifar et al., 1999). Furthermore, there is evidence that mitochondria may be the primary target for oxidative damage by PN under a variety of pathological conditions (Radi et al., 1994). Consistent with this view, recent experiments from our laboratory, using the same mouse CCI model as in the present study, have shown that the early post-traumatic impairment of mitochondrial ultrastructure and bioenergetics are associated with PN-mediated protein nitration and 4HNE conjugation together with a severe attenuation of Ca++ buffering capacity (Singh et al., 2006). Furthermore, our in vitro studies with isolated brain mitochondria have demonstrated that application of compounds such as penicillamine, which scavenges PN anion (ONOO−) and peroxynitrous acid (ONOOH) (Hall et al., 1999), or tempol, which catalytically scavenge PN-derived oxygen radicals (e.g. •NO2, •CO3) (Carroll et al., 2000), can protect against PN-mediated mitochondrial respiratory dysfunction and oxidative damage (Singh et al, 2006b). Both penicillamine (Hall et al., 1999) and tempol (Beit-Yannai et al., 1996) have been reported to improve neurological recovery in rodent TBI models. Moreover, administration of the NOS inhibitors nitro-arginine methyl ester and 7-nitroindazole have been shown to improve neurological recovery in head-injured mice (Mesenge et al., 1996) together with a reduction in brain 3NT levels (Mesenge et al., 1998).
Temporal Characteristics of Calpain-Mediated Cytoskeletal Damage
Several experimental TBI studies from different laboratories have documented the significant contribution of calpain activity to the post-traumatic damage and neurodegeneration (Arrigoni and Cohadon, 1991) (Posmantur et al., 1996; Saatman et al., 1996). Pathological calpain activation is known to be triggered by excessive intracellular Ca++ accumulation (Bartus et al., 1995) (Kampfl et al., 1997), which is associated with the glutamate release and sustained NMDA activation, as well as depolarization-induced opening of voltage-dependent Ca++ channels immediately after CNS trauma. Preferred substrates for calpain include cytoskeletal proteins, membrane-associated proteins, signaling transduction proteins and transcription factors (Carafoli and Molinari, 1998); Wang, 2000a). As noted earlier, calpain-mediated proteolysis of the major membrane cytoskeletal protein, α-spectrin, results in the appearance of two highly stable breakdown products, calpain-specific SBDP145 and the non-specific calpain/caspase 3-generated SBDP150. Our results showed that there is a rapid accumulation of both SBDPs by 1 hr after injury which further increases at 3 hrs before reaching a plateau at 6 and 12 hrs. Nevertheless, at 24 hrs, both SBDPs showed a significant increase above the level seen at 12 hrs spiked to their peak with a particularly prominent SBDP145 increase. Although a role of caspase 3 cannot be ruled out in regards to the increase in SBDP150, the predominant elevation in the calpain-specific SBDP145 at 24 hrs indicates that the major contribution of calpain to cytoskeletal damage. In addition, the caspase 3 specific SBDP120 (Wang, 2000b) showed no increase over time which suggests only a very minor role of caspase 3 in post-traumatic spectrin proteolysis. After 24 hrs, the SBDP 145 and 150 levels progressively subsided, and both returned to the baseline level (sham) by 72 hrs after injury.
Interaction of Peroxynitrite-Induced Oxidative and Calpain-Mediated Proteolytic Damage Mechanisms
Multiple lines of evidence have linked ROS-initiated oxidative damage to the loss of intracellular Ca++ homeostasis and calpain activation. It has been commonly indicated that ROS cause a rapid increase in cytoplasmic Ca++ concentration in diverse cell types (Roveri et al., 1992) (Chakraborti et al., 1999) (Okabe et al., 2000), which can contribute to the early transient calpain activation. However, the current study indicated that the full activity of calpain did not occur until a later timepoint that follows the early wave of oxidative damage and the peak of mitochondrial dysfunction (Singh et al., 2006). At the point at which mitochondrial functional failure occurs, this results in a loss of mitochondrial Ca++ buffering and release of accumulated Ca++ into the cytoplasm which would be expected to exacerbate calpain activation and spectrin breakdown. It has been shown that mitochondria-derived PN plays a role in mitochondrial Ca++ overload, and can actually promote Ca++ release from intact mitochondria (Bringold et al., 2000). Moreover, we have previously demonstrated that the mitochondrial failure is coincident with oxidative damage to mitochondrial proteins and loss of mitochondrial Ca++ buffering capacity (Singh et al., 2006). Moreover, in an in vitro study, nitric oxide-elicited neuronal apoptosis through excitotoxicity and receptor-mediated intracellular Ca++ overload mechanisms was blocked by calpain inhibitors (Volbracht et al., 2005). Another in vitro study has also indicated that PN-initiated cell death in human articular chondrocytes is mediated by mitochondrial dysfunction and calpain activity (Whiteman et al., 2004).
In addition to the apparent role of ROS in the post-traumatic impairment of Ca++ homeostasis, ROS also are involved in directly regulating calpain proteolytic activity. As indicated in Figure 4, following an immediate and rapid increase of SBDPs at 1 hr after injury, there is essentially a plateau in regards to the SBDP levels between 3 hrs and 12 hrs. Then, between 12 hrs and 24 hrs, both SBDPs exhibited a greater than 75% increase which was highly significant (p<0.0004 for SBDP145 and p<0.0001 for SBDP150). A similar biphasic pattern of calpain-mediated α-spectrin degradation has also been observed by our laboratory in the context of a rat spinal cord injury model (Xiong et al., 2006) and by others in a transient ischemic brain injury model (Neumar et al., 2001). In a recent hippocampal organotypic slice study, stretch-induced injury also showed a biphasic activation of calpain (DeRidder et al., 2006). This repeatedly demonstrated biphasic time course of calpain-mediated proteolysis can potentially be explained by an initial partial ROS-mediated inhibition of calpain activity in the early post-traumatic hrs during which PN generation and evidence of oxidative damage are at their highest level. Consistent with this hypothesis, it has been shown that ROS can inhibit calpain activity via oxidation of the sulfhydryl groups of cysteine residues at the active site of the enzyme (Benuck et al., 1992) (Guttmann et al., 1997) (Guttmann and Johnson, 1998). Recent work by another group has shown that another cysteine protease caspase-3 in the traumatized brain is similarly inhibited by PN specifically, and that this is prevented by application of the sulfhydryl reducing agent dithiothreitol (Lau et al., 2006). The latter is also relevant since the SBDP150 fragment which also showed a biphasic time course is partially generated by caspase 3 as well as calpain (Wang, 2000b). Therefore, it is reasonable to speculate that even though intracellular Ca++ overload is triggered immediately after injury (Zhou et al., 2001), the peak of calpain- and caspase-mediated cytoskeletal degradation may not be achieved until as much as 24 hrs post-injury after which an initial ROS-mediated calpain and caspase inhibition has subsided. In other words, as the intensity of the PN generation decreases, the oxidative damage-mediated impairment of various Ca++ homeostatic mechanisms and the neurodegenerative consequences of post-traumatic intracellular Ca++ overload and peak calpain activity would have the opportunity to be fully seen. This peak in calpain-mediated damage at 24 hrs closely precedes the progressive increase in post-traumatic neurodegeneration (silver staining) which peaks at 48 hrs.
We have observed the same close association between calpain-mediated cytoskeletal degradation and neurodegeneration in the mouse diffuse TBI model except that the peak of the former is seen at 72 hrs and that of the latter is seen between 72 and 96 hrs (Hall et al., 2004). The different time courses of PN-mediated oxidative damage and calpain-mediated cytoskeletal degradation seen in focal and diffuse (Kupina et al., 2003) TBI models suggests that the therapeutic window and optimum treatment durations for either PN-directed antioxidant agents or calpain inhibitor treatment may differ between the two types of TBI.
Summary
Our working hypothesis, based upon the findings in this study, is illustrated in Figure 6. Initial mechanical trauma to the brain causes membrane depolarization, resulting in the opening the voltage-dependent Na+, K+ and Ca++ channels, and the release of glutamate into the extracellular spaces, which will lead to the activation of NMDA receptor. Both mechanisms directly or indirectly elevate intracellular Ca++ and rapidly initiate the activation of intracellular calpains. However, mitochondria take up the excessive Ca++ in the cell, which subsequently activates mitochondrial NOS and results in overproduction of •NO radical. Elevated •NO out-competes superoxide dismutase (SOD) for superoxide radical (•O2−), leading to the formation of PN. Then, PN-derived free radicals could be expected to initially produce a partial inhibition of calpain and caspase-3 activity. Mitochondria, however, as a major intracellular ROS source not only become a primary location for PN production, but also are susceptible to PN-induced oxidative damage due to their enriched thiol and iron-rich structures within the electron transport chain (ETC). In addition, the biochemical properties of PN enable it to diffuse out of the mitochondria to induce further oxidative damage to the cell membrane and cellular proteins. This increase in oxidative stress during the first 12 hrs, induces cellular oxidative damage by LP, protein oxidation and protein nitration. The calpain-mediated proteolysis displays a biphasic activity probably as a result of an initial partial PN-mediated inhibition of calpain. However, as this subsides, the extensive compromise of Ca++ homeostasis in the mitochondrion and the rest of the cell becomes manifest and calpain activity is free to reach its peak. In the end, calpain-mediated cytoskeletal degradation becomes a final common pathway leading to neuronal cell death.
Figure 6.
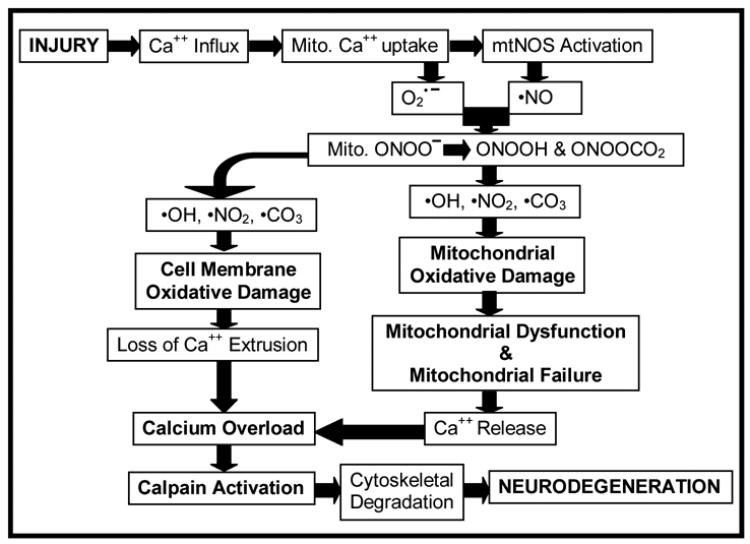
Hypothetical interrelationship between PN-induced oxidative damage in neuronal mitochondria and the rest of the neuron, compromise of Ca++ homeostasis, calpain-mediated proteolysis and neurodegeneration. Our results suggest that PN-induced oxidative damage plays a key role in the post-traumatic secondary injury, which leads to exacerbation of Ca++ overload, calpain proteolysis of cytoskeletal and other cellular proteins and neurodegeneration. See Discussion for a further description.
The findings of this study strongly support the concept of an important pathophysiological role of PN-mediated oxidative damage following TBI. Peroxynitrite-induced oxidative damage and calpain activation interact and are probably responsible for much of the secondary injury and neurodegeneration that occurs. If so, then PN-targeted antioxidants should be neuroprotective. However, the combination of a PN-targeted antioxidant and a calpain inhibitor might have a better neuroprotective action than either approach alone. Future studies will explore these possibilities.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arrigoni E, Cohadon F. Calcium-activated neutral protease activities in brain trauma. Neurochem Res. 1991;16:483–487. doi: 10.1007/BF00965570. [DOI] [PubMed] [Google Scholar]
- Awasthi D, Church DF, Torbati D, Carey ME, Pryor WA. Oxidative stress following traumatic brain injury in rats. Surg Neurol. 1997;47:575–581. doi: 10.1016/s0090-3019(96)00461-2. discussion 581–572. [DOI] [PubMed] [Google Scholar]
- Bartus RT, Elliott PJ, Hayward NJ, Dean RL, Harbeson S, Straub JA, Li Z, Powers JC. Calpain as a novel target for treating acute neurodegenerative disorders. Neurol Res. 1995;17:249–258. doi: 10.1080/01616412.1995.11740322. [DOI] [PubMed] [Google Scholar]
- Bates TE, Loesch A, Burnstock G, Clark JB. Immunocytochemical evidence for a mitochondrially located nitric oxide synthase in brain and liver. Biochem Biophys Res Commun. 1995;213:896–900. doi: 10.1006/bbrc.1995.2213. [DOI] [PubMed] [Google Scholar]
- Beckman JS. Oxidative damage and tyrosine nitration from peroxynitrite. Chem Res Toxicol. 1996;9:836–844. doi: 10.1021/tx9501445. [DOI] [PubMed] [Google Scholar]
- Beit-Yannai E, Zhang R, Trembovler V, Samuni A, Shohami E. Cerebroprotective effect of stable nitroxide radicals in closed head injury in the rat. Brain Res. 1996;717:22–28. doi: 10.1016/0006-8993(95)01492-6. [DOI] [PubMed] [Google Scholar]
- Benuck M, Banay-Schwartz M, Ramacci MT, Lajtha A. Peroxidative stress effects on calpain activity in brain of young and adult rats. Brain Res. 1992;596:296–298. doi: 10.1016/0006-8993(92)91560-2. [DOI] [PubMed] [Google Scholar]
- Bicker G. Nitric oxide: an unconventional messenger in the nervous system of an orthopteroid insect. Arch Insect Biochem Physiol. 2001;48:100–110. doi: 10.1002/arch.1062. [DOI] [PubMed] [Google Scholar]
- Bringold U, Ghafourifar P, Richter C. Peroxynitrite formed by mitochondrial NO synthase promotes mitochondrial Ca2+ release. Free Radic Biol Med. 2000;29:343–348. doi: 10.1016/s0891-5849(00)00318-x. [DOI] [PubMed] [Google Scholar]
- Carafoli E, Molinari M. Calpain: a protease in search of a function? Biochem Biophys Res Commun. 1998;247:193–203. doi: 10.1006/bbrc.1998.8378. [DOI] [PubMed] [Google Scholar]
- Carroll RT, Galatsis P, Borosky S, Kopec KK, Kumar V, Althaus JS, Hall ED. 4-Hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl (Tempol) inhibits peroxynitrite-mediated phenol nitration. Chem Res Toxicol. 2000;13:294–300. doi: 10.1021/tx990159t. [DOI] [PubMed] [Google Scholar]
- Chakraborti T, Das S, Mondal M, Roychoudhury S, Chakraborti S. Oxidant, mitochondria and calcium: an overview. Cell Signal. 1999;11:77–85. doi: 10.1016/s0898-6568(98)00025-4. [DOI] [PubMed] [Google Scholar]
- Chiueh CC. Neuroprotective properties of nitric oxide. Ann N Y Acad Sci. 1999;890:301–311. doi: 10.1111/j.1749-6632.1999.tb08007.x. [DOI] [PubMed] [Google Scholar]
- Cobbs CS, Fenoy A, Bredt DS, Noble LJ. Expression of nitric oxide synthase in the cerebral microvasculature after traumatic brain injury in the rat. Brain Res. 1997;751:336–338. doi: 10.1016/s0006-8993(96)01429-1. [DOI] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM. Nitric oxide in neuronal degeneration. Proc Soc Exp Biol Med. 1996;211:33–40. doi: 10.3181/00379727-211-43950e. [DOI] [PubMed] [Google Scholar]
- de Olmos JS, Beltramino CA, de Olmos de Lorenzo S. Use of an amino-cupric-silver technique for the detection of early and semiacute neuronal degeneration caused by neurotoxicants, hypoxia, and physical trauma. Neurotoxicol Teratol. 1994;16:545–561. doi: 10.1016/0892-0362(94)90033-7. [DOI] [PubMed] [Google Scholar]
- DeRidder MN, Simon MJ, Siman R, Auberson YP, Raghupathi R, Meaney DF. Traumatic mechanical injury to the hippocampus in vitro causes regional caspase-3 and calpain activation that is influenced by NMDA receptor subunit composition. Neurobiol Dis. 2006;22:165–176. doi: 10.1016/j.nbd.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Elfering SL, Sarkela TM, Giulivi C. Biochemistry of mitochondrial nitric-oxide synthase. J Biol Chem. 2002;277:38079–38086. doi: 10.1074/jbc.M205256200. [DOI] [PubMed] [Google Scholar]
- Gahm C, Holmin S, Mathiesen T. Temporal profiles and cellular sources of three nitric oxide synthase isoforms in the brain after experimental contusion. Neurosurgery. 2000;46:169–177. [PubMed] [Google Scholar]
- Garthwaite J, Boulton CL. Nitric oxide signaling in the central nervous system. Annu Rev Physiol. 1995;57:683–706. doi: 10.1146/annurev.ph.57.030195.003343. [DOI] [PubMed] [Google Scholar]
- Ghafourifar P, Richter C. Nitric oxide synthase activity in mitochondria. FEBS Lett. 1997;418:291–296. doi: 10.1016/s0014-5793(97)01397-5. [DOI] [PubMed] [Google Scholar]
- Ghafourifar P, Schenk U, Klein SD, Richter C. Mitochondrial nitric-oxide synthase stimulation causes cytochrome c release from isolated mitochondria. Evidence for intramitochondrial peroxynitrite formation. J Biol Chem. 1999;274:31185–31188. doi: 10.1074/jbc.274.44.31185. [DOI] [PubMed] [Google Scholar]
- Giulivi C. Characterization and function of mitochondrial nitric-oxide synthase. Free Radic Biol Med. 2003;34:397–408. doi: 10.1016/s0891-5849(02)01298-4. [DOI] [PubMed] [Google Scholar]
- Globus MY, Alonso O, Dietrich WD, Busto R, Ginsberg MD. Glutamate release and free radical production following brain injury: effects of posttraumatic hypothermia. J Neurochem. 1995;65:1704–1711. doi: 10.1046/j.1471-4159.1995.65041704.x. [DOI] [PubMed] [Google Scholar]
- Guttmann RP, Elce JS, Bell PD, Isbell JC, Johnson GV. Oxidation inhibits substrate proteolysis by calpain I but not autolysis. J Biol Chem. 1997;272:2005–2012. doi: 10.1074/jbc.272.3.2005. [DOI] [PubMed] [Google Scholar]
- Guttmann RP, Johnson GV. Oxidative stress inhibits calpain activity in situ. J Biol Chem. 1998;273:13331–13338. doi: 10.1074/jbc.273.21.13331. [DOI] [PubMed] [Google Scholar]
- Hall ED, Andrus PK, Yonkers PA. Brain hydroxyl radical generation in acute experimental head injury. J Neurochem. 1993;60:588–594. doi: 10.1111/j.1471-4159.1993.tb03189.x. [DOI] [PubMed] [Google Scholar]
- Hall ED, Andrus PK, Yonkers PA, Smith SL, Zhang JR, Taylor BM, Sun FF. Generation and detection of hydroxyl radical following experimental head injury. Ann N Y Acad Sci. 1994;738:15–24. doi: 10.1111/j.1749-6632.1994.tb21785.x. [DOI] [PubMed] [Google Scholar]
- Hall ED, Detloff MR, Johnson K, Kupina NC. Peroxynitrite-mediated protein nitration and lipid peroxidation in a mouse model of traumatic brain injury. J Neurotrauma. 2004;21:9–20. doi: 10.1089/089771504772695904. [DOI] [PubMed] [Google Scholar]
- Hall ED, Gibson TR, Pavel KM. Lack of a gender difference in post-traumatic neurodegeneration in the mouse controlled cortical impact injury model. J Neurotrauma. 2005a;22:669–679. doi: 10.1089/neu.2005.22.669. [DOI] [PubMed] [Google Scholar]
- Hall ED, Kupina NC, Althaus JS. Peroxynitrite scavengers for the acute treatment of traumatic brain injury. Ann N Y Acad Sci. 1999;890:462–468. doi: 10.1111/j.1749-6632.1999.tb08025.x. [DOI] [PubMed] [Google Scholar]
- Hall ED, Oostveen JA, Andrus PK, Anderson DK, Thomas CE. Immunocytochemical method for investigating in vivo neuronal oxygen radical-induced lipid peroxidation. J Neurosci Methods. 1997;76:115–122. doi: 10.1016/s0165-0270(97)00089-7. [DOI] [PubMed] [Google Scholar]
- Hall ED, Sullivan PG, Gibson TR, Pavel KM, Thompson BM, Scheff SW. Spatial and temporal characteristics of neurodegeneration after controlled cortical impact in mice: more than a focal brain injury. J Neurotrauma. 2005b;22:252–265. doi: 10.1089/neu.2005.22.252. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JMC. Free radicals in biology and medicine. 3. Oxford Science Publications; 1999. Chapter 2.5.4; 4.9.11; 5.13.4. [Google Scholar]
- Kampfl A, Posmantur RM, Zhao X, Schmutzhard E, Clifton GL, Hayes RL. Mechanisms of calpain proteolysis following traumatic brain injury: implications for pathology and therapy: implications for pathology and therapy: a review and update. J Neurotrauma. 1997;14:121–134. doi: 10.1089/neu.1997.14.121. [DOI] [PubMed] [Google Scholar]
- Kontos HA, Povlishock JT. Oxygen radicals in brain injury. Cent Nerv Syst Trauma. 1986;3:257–263. doi: 10.1089/cns.1986.3.257. [DOI] [PubMed] [Google Scholar]
- Kontos HA, Wei EP. Superoxide production in experimental brain injury. J Neurosurg. 1986;64:803–807. doi: 10.3171/jns.1986.64.5.0803. [DOI] [PubMed] [Google Scholar]
- Kupina NC, Detloff MR, Bobrowski WF, Snyder BJ, Hall ED. Cytoskeletal protein degradation and neurodegeneration evolves differently in males and females following experimental head injury. Exp Neurol. 2003;180:55–73. doi: 10.1016/s0014-4886(02)00048-1. [DOI] [PubMed] [Google Scholar]
- Lacza Z, Puskar M, Figueroa JP, Zhang J, Rajapakse N, Busija DW. Mitochondrial nitric oxide synthase is constitutively active and is functionally upregulated in hypoxia. Free Radic Biol Med. 2001;31:1609–1615. doi: 10.1016/s0891-5849(01)00754-7. [DOI] [PubMed] [Google Scholar]
- Lau A, Arundine M, Sun HS, Jones M, Tymianski M. Inhibition of caspase-mediated apoptosis by peroxynitrite in traumatic brain injury. J Neurosci. 2006;26:11540–11553. doi: 10.1523/JNEUROSCI.3507-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Figueroa MO, Caamano C, Morano MI, Ronn LC, Akil H, Watson SJ. Direct evidence of nitric oxide presence within mitochondria. Biochem Biophys Res Commun. 2000;272:129–133. doi: 10.1006/bbrc.2000.2748. [DOI] [PubMed] [Google Scholar]
- Marklund N, Lewander T, Clausen F, Hillered L. Effects of the nitrone radical scavengers PBN and S-PBN on in vivo trapping of reactive oxygen species after traumatic brain injury in rats. J Cereb Blood Flow Metab. 2001;21:1259–1267. doi: 10.1097/00004647-200111000-00002. [DOI] [PubMed] [Google Scholar]
- Mesenge C, Charriaut-Marlangue C, Verrecchia C, Allix M, Boulu RR, Plotkine M. Reduction of tyrosine nitration after N(omega)-nitro-L-arginine-methylester treatment of mice with traumatic brain injury. Eur J Pharmacol. 1998;353:53–57. doi: 10.1016/s0014-2999(98)00432-4. [DOI] [PubMed] [Google Scholar]
- Mesenge C, Verrecchia C, Allix M, Boulu RR, Plotkine M. Reduction of the neurological deficit in mice with traumatic brain injury by nitric oxide synthase inhibitors. J Neurotrauma. 1996;13:209–214. [PubMed] [Google Scholar]
- Mohanakumar KP, Hanbauer I, Chiueh CC. Neuroprotection by nitric oxide against hydroxyl radical-induced nigral neurotoxicity. J Chem Neuroanat. 1998;14:195–205. doi: 10.1016/s0891-0618(98)00032-5. [DOI] [PubMed] [Google Scholar]
- Mori T, Kawamata T, Katayama Y, Maeda T, Aoyama N, Kikuchi T, Uwahodo Y. Antioxidant, OPC-14117, attenuates edema formation, and subsequent tissue damage following cortical contusion in rats. Acta Neurochir Suppl. 1998;71:120–122. doi: 10.1007/978-3-7091-6475-4_36. [DOI] [PubMed] [Google Scholar]
- Murphy MP, Packer MA, Scarlett JL, Martin SW. Peroxynitrite: a biologically significant oxidant. Gen Pharmacol. 1998;31:179–186. doi: 10.1016/s0306-3623(97)00418-7. [DOI] [PubMed] [Google Scholar]
- Neely MD, Sidell KR, Graham DG, Montine TJ. The lipid peroxidation product 4-hydroxynonenal inhibits neurite outgrowth, disrupts neuronal microtubules, and modifies cellular tubulin. J Neurochem. 1999;72:2323–2333. doi: 10.1046/j.1471-4159.1999.0722323.x. [DOI] [PubMed] [Google Scholar]
- Neumar RW, Meng FH, Mills AM, Xu YA, Zhang C, Welsh FA, Siman R. Calpain activity in the rat brain after transient forebrain ischemia. Exp Neurol. 2001;170:27–35. doi: 10.1006/exnr.2001.7708. [DOI] [PubMed] [Google Scholar]
- Okabe E, Tsujimoto Y, Kobayashi Y. Calmodulin and cyclic ADP-ribose interaction in Ca2+ signaling related to cardiac sarcoplasmic reticulum: superoxide anion radical-triggered Ca2+ release. Antioxid Redox Signal. 2000;2:47–54. doi: 10.1089/ars.2000.2.1-47. [DOI] [PubMed] [Google Scholar]
- Orihara Y, Ikematsu K, Tsuda R, Nakasono I. Induction of nitric oxide synthase by traumatic brain injury. Forensic Sci Int. 2001;123:142–149. doi: 10.1016/s0379-0738(01)00537-0. [DOI] [PubMed] [Google Scholar]
- Posmantur RM, Kampfl A, Liu SJ, Heck K, Taft WC, Clifton GL, Hayes RL. Cytoskeletal derangements of cortical neuronal processes three hours after traumatic brain injury in rats: an immunofluorescence study. J Neuropathol Exp Neurol. 1996;55:68–80. doi: 10.1097/00005072-199601000-00007. [DOI] [PubMed] [Google Scholar]
- Radi R. Peroxynitrite reactions and diffusion in biology. Chem Res Toxicol. 1998;11:720–721. doi: 10.1021/tx980096z. [DOI] [PubMed] [Google Scholar]
- Radi R, Cassina A, Hodara R, Quijano C, Castro L. Peroxynitrite reactions and formation in mitochondria. Free Radic Biol Med. 2002;33:1451–1464. doi: 10.1016/s0891-5849(02)01111-5. [DOI] [PubMed] [Google Scholar]
- Radi R, Rodriguez M, Castro L, Telleri R. Inhibition of mitochondrial electron transport by peroxynitrite. Arch Biochem Biophys. 1994;308:89–95. doi: 10.1006/abbi.1994.1013. [DOI] [PubMed] [Google Scholar]
- Roveri A, Coassin M, Maiorino M, Zamburlini A, van Amsterdam FT, Ratti E, Ursini F. Effect of hydrogen peroxide on calcium homeostasis in smooth muscle cells. Arch Biochem Biophys. 1992;297:265–270. doi: 10.1016/0003-9861(92)90671-i. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Murai H, Bartus RT, Smith DH, Hayward NJ, Perri BR, McIntosh TK. Calpain inhibitor AK295 attenuates motor and cognitive deficits following experimental brain injury in the rat. Proc Natl Acad Sci U S A. 1996;93:3428–3433. doi: 10.1073/pnas.93.8.3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saran M, Michel C, Bors W. Reaction of NO with O2-implications for the action of endothelium-derived relaxing factor (EDRF) . Free Radic Res Commun. 1990;10:221–226. doi: 10.3109/10715769009149890. [DOI] [PubMed] [Google Scholar]
- Singh IN, Sullivan PG, Deng Y, Mbye LH, Hall ED. Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications for neuroprotective therapy. J Cereb Blood Flow Metab. 2006a;26:1407–18. doi: 10.1038/sj.jcbfm.9600297. [DOI] [PubMed] [Google Scholar]
- Singh IN, Carrico KM, Wang J, Sullivan PG, Hall ED. Protective effects of neuroprotective antioxidants penicillamine and tempol against peroxynitrite-induced dysfunction in isolated brain mitochondria. J Neurotrauma. 2006b;23:991. [Google Scholar]
- Smith SL, Andrus PK, Zhang JR, Hall ED. Direct measurement of hydroxyl radicals, lipid peroxidation, and blood-brain barrier disruption following unilateral cortical impact head injury in the rat. J Neurotrauma. 1994;11:393–404. doi: 10.1089/neu.1994.11.393. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Bruce-Keller AJ, Rabchevsky AG, Christakos S, Clair DK, Mattson MP, Scheff SW. Exacerbation of damage and altered NF-kappaB activation in mice lacking tumor necrosis factor receptors after traumatic brain injury. J Neurosci. 1999a;19:6248–6256. doi: 10.1523/JNEUROSCI.19-15-06248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PG, Thompson MB, Scheff SW. Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp Neurol. 1999b;160:226–234. doi: 10.1006/exnr.1999.7197. [DOI] [PubMed] [Google Scholar]
- Switzer RC., 3rd Application of silver degeneration stains for neurotoxicity testing. Toxicol Pathol. 2000;28:70–83. doi: 10.1177/019262330002800109. [DOI] [PubMed] [Google Scholar]
- Thompson SN, Gibson TR, Thompson BM, Deng Y, Hall ED. Relationship of calpain-mediated proteolysis to the expression of axonal and synaptic plasticity markers following traumatic brain injury in mice. Exp Neurol. 2006;201:253–265. doi: 10.1016/j.expneurol.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Vicente S, Perez-Rodriguez R, Olivan AM, Martinez Palacian A, Gonzalez MP, Oset-Gasque MJ. Nitric oxide and peroxynitrite induce cellular death in bovine chromaffin cells: Evidence for a mixed necrotic and apoptotic mechanism with caspases activation. J Neurosci Res. 2006;84:78–96. doi: 10.1002/jnr.20853. [DOI] [PubMed] [Google Scholar]
- Volbracht C, Chua BT, Ng CP, Bahr BA, Hong W, Li P. The critical role of calpain versus caspase activation in excitotoxic injury induced by nitric oxide. J Neurochem. 2005;93:1280–1292. doi: 10.1111/j.1471-4159.2005.03122.x. [DOI] [PubMed] [Google Scholar]
- Wada K, Chatzipanteli K, Kraydieh S, Busto R, Dietrich WD. Inducible nitric oxide synthase expression after traumatic brain injury and neuroprotection with aminoguanidine treatment in rats. Neurosurgery. 1998;43:1427–1436. doi: 10.1097/00006123-199812000-00096. [DOI] [PubMed] [Google Scholar]
- Wang KK. Calpain and caspase: can you tell the difference? Trends Neurosci. 2000a;23:20–26. doi: 10.1016/s0166-2236(99)01536-2. [DOI] [PubMed] [Google Scholar]
- Wang KK. Calpain and caspase: can you tell the difference? by kevin KW WangVol 23, pp 20–26. Trends Neurosci. 2000b;23:59. doi: 10.1016/s0166-2236(99)01536-2. [DOI] [PubMed] [Google Scholar]
- Whiteman M, Armstrong JS, Cheung NS, Siau JL, Rose P, Schantz JT, Jones DP, Halliwell B. Peroxynitrite mediates calcium-dependent mitochondrial dysfunction and cell death via activation of calpains. Faseb J. 2004;18:1395–1397. doi: 10.1096/fj.03-1096fje. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Rabchevsky AG, Hall ED. Role of peroxynitrite in secondary oxidative damage after spinal cord injury. J Neurochem. 2006 doi: 10.1111/j.1471-4159.2006.04312.x. in press. [DOI] [PubMed] [Google Scholar]
- Zhou F, Xiang Z, Feng WX, Zhen LX. Neuronal free Ca(2+)and BBB permeability and ultrastructure in head injury with secondary insult. J Clin Neurosci. 2001;8:561–563. doi: 10.1054/jocn.2001.0980. [DOI] [PubMed] [Google Scholar]