

Abstract
Motivation
Rational design of kinase inhibitors remains a challenge partly because there is no clear delineation of the molecular features that direct the pharmacological impact towards clinically relevant targets. Standard factors governing ligand affinity, such as potential for intermolecular hydrophobic interactions or for intermolecular hydrogen bonding do not provide good markers to assess cross reactivity. Thus, a core question in the informatics of drug design is what type of molecular similarity among targets promotes promiscuity and what type of molecular difference governs specificity. This work answers the question for a sizable screened sample of the human pharmacokinome including targets with unreported structure.
Results
We show that drug design aimed at promoting pairwise interactions between ligand and kinase target actually fosters promiscuity because of the high conservation of the partner groups on or around the ATP-binding site of the kinase. Alternatively, we focus on a structural marker that may be reliably determined from sequence and measures dehydration propensities mostly localized on the loopy regions of kinases. Based on this marker, we construct a sequence-based kinase classifier that enables the accurate prediction of pharmacological differences. Our indicator is a micro-environmental descriptor that quantifies the propensity for water exclusion around preformed polar pairs. The results suggest that targeting polar dehydration patterns heralds a new generation of drugs that enable a tighter control of specificity than designs aimed at promoting ligand–kinase pairwise interactions.
Availability
The predictor of polar hot spots for dehydration propensity, or solvent-accessible hydrogen bonds in soluble proteins, named YAPView, may be freely downloaded from the University of Chicago website http://protlib.uchicago.edu/dloads.html
1 INTRODUCTION
Kinase targeting is a central theme in drug discovery and molecular cancer therapy (Bain et al., 2003; Druker, 2004; Huse and Kuriyan, 2002; Hopkins et al., 2006; Knight and Shokat, 2005; Vieth et al., 2004), but a structural basis for rational design appears to be inconclusive (Bain et al., 2003; Hopkins et al., 2006; Huse and Kuriyan, 2002; Vieth et al., 2004). In practice, most ligands or drug leads are actually discovered through screening techniques. While the paradigm of target specificity (Bain et al., 2003; Huse and Kuriyan, 2002) may be shifting to controlled multi-target impact (Hopkins et al., 2006), the structural factors determining these possibilities are not yet fully understood, in spite of notable progress. For instance, the accessible nonpolar surface, frequently invoked to assess protein associations (Chothia, 1974; Whittle and Blundell, 1994), actually fosters promiscuity (Feng et al., 2005; Hopkins et al., 2006), as shown in this work. Progress in controlling specificity is also hampered by the lack of structural information, and this is precisely the problem dealt with in this article.
We focus on identifying the dominant molecular feature that directs nonpromiscuous drug targeting. Promiscuity is operationally defined based on extensive drug screening (Fabian et al., 2005) as significant cross reactivity (dissociation constant Kd<100 nM) extended over >30% of the sampled kinome. Conversely, specificity implies <5% cross reactivity. The problem is complicated due to the scarcity of affinity-profiled kinases with reported structure: To date, only ~20% of the human kinome (103 out of the ~520 discovered protein kinases) is reported in the PDB. Furthermore, kinase homology models by themselves are not useful to make sequence-based inferences since the level of pairwise sequence identity across the kinase superfamily is typically low (<30%, Manning et al., 2002). Instead, we take advantage of the high degree of conservation of kinase folds, arising from their common ancestry. Thus, reliable sequence-based attributes such as disorder propensity (Braken et al., 2004) are here mapped onto structurally threaded models (Fernández and Berry, 2004) to make inferences about drug specificity/promiscuity.
We search for a sequence-based attribute that enables a classification of kinase space that accurately reproduces similarities/differences in affinity profiling against a drug background (Fabian et al., 2005). The classification introduces a partition into disjoint sets of kinases within which pharmacological differences are accurately predicted from molecular differences. Our methodology could be implemented because the relevant classification of kinases was performed by comparing the regions deprived of adequate packing or intramolecular dehydration, i.e. the loopy regions, which are precisely the markers differentiating kinases at the structural level (Huse and Kuriyan, 2002).
The article is organized as follows: First, we determine the type of molecular similarity that promotes promiscuity whenever targeted. Specifically, we demonstrate that solvent-exposed nonpolar regions engaged in ligand association foster promiscuity. To draw statistics at a kinomic scale, we formally define a region, the nonpolar hull, enabling comparison of targeted exposed nonpolar regions across different kinases. Then, we examine other targeted features in molecular design aimed at inducing pair wise interactions between ligand and kinase and show that the high degree of conservation of the partner groups on the protein surface does not enable a cogent control of specificity. Subsequently, we introduce a means of calculating dehydration propensities on polar-paired regions on the protein surface and formally define a region, the environmental hull, enabling a comparison of polar dehydration propensities across targets. We demonstrate that this molecular attribute is responsible for controlling specificity. To carry out the analysis at a sequence level, a technique named environmental alignment is implemented. This technique enables the identification of residues whose microenvironments are likely to be perturbed by ligand association. Such residues are identified by aligning the kinase sequence against a background of sequences of homologous kinase–ligand complexes reported in the PDB.
2 METHODS
2.1 Construction of the nonpolar hull
In order to assess differences in the exposed nonpolar regions of kinase targets that interact with different ligands we define a common region, named the nonpolar hull. A residue a is defined as making contact with a ligand L within a PDB-reported complex if a side chain heavy atom (H excluded) is found to be within 3.6 Å (upper bound for any bond length) of a heavy atom in the ligand. The nonpolar hull for protein chain i, Hnp(i ), is dependent on a ‘structural background set’ of chains, S(i), which includes all homolog chains that align with chain i (Higgins et al., 1996) for which there are protein–ligand complexes with reported structure. Thus, the nonpolar hull is defined as Hnp(i) = Uj∈S(i)Φi(Rnp(j)), where Φi(a), with a ∈ chain j, is the residue in chain i that aligns with residue a in chain j and Rnp(j) is the set of nonpolar residues in chain j in contact with its respective ligand Lj. For any pair i, j, the following property holds: Φi (Hnp(j)) = Hnp(i), enabling a comparison of kinases by examining differences in nonpolar hulls.
2.2 Identification of localized dehydration propensities
A preformed hydrogen bond microenvironment may be determined from the atomic coordinates of the protein by calculating the extent of intramolecular dehydration, ρ, quantified as the number of the side chain carbonaceous nonpolar groups within a dehydration domain. This domain consists of two intersecting balls of radius 6.0 Å (~width of three solvation layers (Fernández and Berry, 2004)) centered at the α-carbons of the hydrogen-bond paired residues. In soluble protein domains, at least two-thirds of the backbone hydrogen bonds lie in the range ρ = 26.6 ± 7.5. The extent of intramolecular dehydration of a solvent-accessible hydrogen bond (SAHB) lies in the tails of the distribution, i.e. with 19 or fewer nonpolar groups in its microenvironment. That is, its ρ-value is below the mean, minus one Gaussian dispersion. Such bonds constitute dehydration-propensity hot spots as demonstrated before (Fernández, 2004; Fernández and Berry, 2004). The SAHBs may be determined directly from a PDB file using the program YAPView (University of Chicago). This program is inspired in earlier desolvation calculations (Fernández et al., 2002).
YAPView identifies SAHBs, the hot spots for polar dehydration propensity in soluble proteins. The SAHBs may be identified by loading the PDB file, choosing a structure display/representation and enabling a desolvation calculation. The latter is needed to determine the extent of intramolecular dehydration of hydrogen bonds. This operation requires the selection: Configuration → General Options → Desolvation. Thereafter, one needs to enable the desolvation calculator and select the appropriate parameters, especially desolvation radius and desolvation threshold, according to the indications given above.
Thus, YAPView displays the dehydration-propensity hot spots directly on the protein surface: The hydrogen bonds that are poorly dehydrated intramolecularly, that is, below the pre-selected threshold are shown in green in the structure display and promote the exclusion of surrounding water.
2.3 Validation of active structure prediction
Our analysis is not constrained to PDB-reported kinases [32 out of the assayed 119 (Fernández and Maddipati, 2006)]. Robetta/Rosetta predictions of active structures (Bonneau et al., 2002; Chivian et al., 2005) become reliable given the extent of PDB representation (~27%) of paralogs within the kinase superfamily and since SAHBs may be directly inferred from sequence (Fernández and Berry, 2004), and contrasted with the structure predictions for mutual validation (Supplementary Material). Such SAHB inferences make use of a strong correlation between the extent of dehydration of the preformed hydrogen bonds and the disorder score (Braken et al., 2004), an accurate sequence-based attribute that indicates the propensity of a chain window to be structurally disordered. The correlation is maintained irrespectively of whether the structure is a Rosetta prediction or PDB reported (Supplementary Material) and implies that native disorder arises essentially from the impossibility to dehydrate intramolecular hydrogen bonds. The choice of disorder score to benchmark Rosetta predictions is due to their lower accuracy on loopy regions, as quantified in Supplementary Material.
The disorder propensity is given by a score determined by the program PONDR®, a neural-network predictor of native disorder. Only 0.4% of more than 900 nonhomologous PDB proteins give false-positive predictions in regions with 40 or more consecutive sites of predicted disorder. Even this 0.4% of false positives is an overestimation, as many disordered regions in monomeric chains become ordered upon ligand binding or in crystal contacts (Braken et al., 2004). The false negatives error rate (~11% for regions of 40 or more consecutive predicted ordered residues) is also compelling with regard to the predictor quality.
The correlation between solvent exposure of hydrogen bonds and disorder propensity implies that it is possible to predict SAHBs directly from sequence (Fernández and Berry, 2004): It suffices to determine the PONDR-generated pattern associated with the desired feature. The correlation implies that the propensity to adopt a natively disordered state becomes pronounced for proteins which, due to a chain composition reflecting high hydrophilicity, cannot protect even minimally (see Supplementary Material) the backbone hydrogen bonds. Thus, we can infer the existence of SAHBs from the PONDR score (λd) with 92% accuracy in regions with λd > 0.35 provided such regions are flanked by well-protected regions (λd < 0.35), to ensure the existence of structure. The accuracy of this sequence-based SAHB predictor was established by inferring the location of SAHBs in proteins with reported structure, for which the microenvironment of each hydrogen bond can be determined unambiguously (Fernández and Berry, 2004). The false negatives constitute 368 of the 8215 SAHBs in a PDB database of 1466 proteins free from structural redundancy and <25% sequence identity in pairwise alignment. The false positives correspond to 2721 of the 133 623 backbone hydrogen bonds examined. Similar statistics were found when contrasting PONDR and Rosetta predictions (Supplementary Material).
2.4 The environmental alignment technique
To assess differences in the dehydration propensities of polar regions for purported kinase targets, we introduce a common region named environmental hull. First, the set Renv(j) is defined for protein chain j as the set of residues paired by SAHBs in chain j within a protein–ligand complex with reported structure subject to the following condition: ligand Lj contributes to the dehydration of the SAHB, that is, it has some carbonaceous nonpolar group within the dehydration domain of the SAHB. Then, the set Eenv(j) is defined as the set of residues from chain j that contribute to the dehydration microenvironment of a SAHB contained in Renv(j), that is they are either paired by the SAHB or they contain a side chain nonpolar group within the microenvironment of the SAHB. Then, the environmental hull for chain j, Henv(j), is defined as the union of the residues in chain j that align with residues framing the environments of SAHBs that in turn are environmentally affected by ligands in PDB-reported complexes: Henv(j) = Ui∈S( j )Φj (Eenv(i)) (notation has been followed consistently, the structural background S(j) is defined above). As with nonpolar hulls, for any pair i, j, the following property also holds: Φi (Henv(j)) = Henv(i). This property is needed to actually compare environments of different proteins. The dependence of denv on the structural background S defining the region for comparison is illustrated in Supplementary Material.
3 RESULTS
The elucidation of the molecular factors governing promiscuity and specificity in molecularly targeted drug therapy requires that we attempt to correlate different molecular attributes with available screening data for a sizable set of kinase targets. The large assayed set adopted (Fabian et al., 2005) is highly underreported in the PDB and consequently, a reliable sequence-based predictor of the relevant molecular attributes had to be implemented. The predictor is based on alignments against sequences of PDB-reported kinases that are used to define the windows for comparison (Section 2, Supplementary Material).
We first define a pharmacological distance, dphar, that quantifies differences in the affinity profiling of kinases against a background of available drugs (Fabian et al., 2005). This metric is the Euclidean distance between affinity vectors with entries given in negative logarithmic or ΔG/RT units (ΔG = free energy change for protein–ligand association, R = universal gas constant, T = absolute temperature). A positive cutoff value ΔG/RT = ln10 ~ 2.3 is adopted for affinities reported as ‘no hit’ in the screening (Kd > 10 μM, Fabian et al., 2005). Figure 1a displays the matrix Dphar = [dphar(i, j )] for all pairs (i, j ) from the 119 assayed kinases. The affinity profiling adopted included 19 of the 20 drugs originally screened (Fabian et al., 2005): only the promiscuous staurosporine was initially excluded since it does not belong to the pharmacology realm.
Fig. 1.


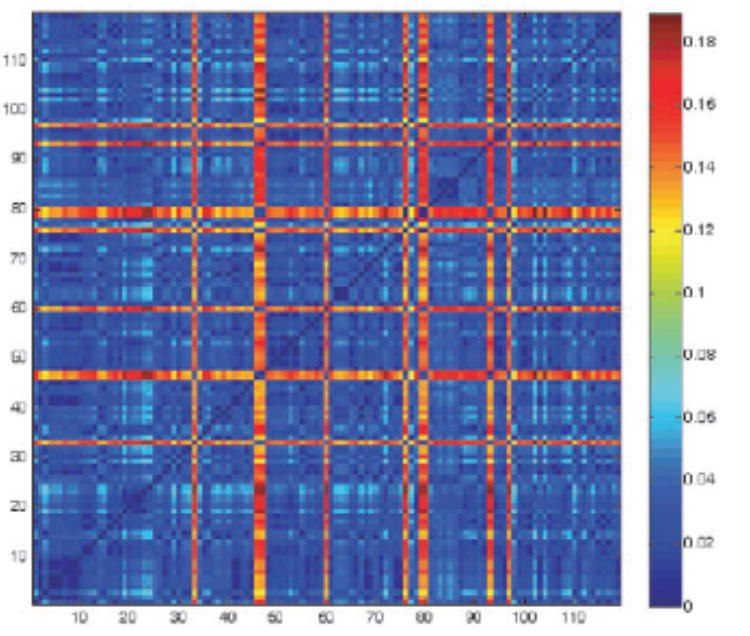

1a. Pharmacological distance matrix Dphar = [dphar(i, j)] for all pairs (i, j) from the 119 kinases assayed through affinity profiling against a background of 19 drugs (Fabian et al., 2005): SB202190; SB203580; sp600125; imatinib (Gleevec); VX-745; BIRB 796; BAY-43-9006; GW-2016; gefitinib; erlotinib; CI-1033; EKB-569; ZD-6474; Vatalanib; SU11248; MLN-518; LY-333531; roscovitine/CYC202 and flavopiridol. The distance is given by dphar(i, j) = [Σm∈ inhibitors (K(i, m) – K(j, m))2]1/2, where K(i, m), K(j, m) represent respectively the negative logarithm of equilibrium constants for complexation of kinase i and kinase j with drug inhibitor m.
1b Aligned backbones (Hogue, 1997) (RMSD 3 0.33 Å) for paralog kinases PDK1 (blue) and CHK1 (lilac) in their active folds. The structures were reported in complex with ligands BIM8 (PDB.1UVR) and 3A3 (PDB.2GCU), respectively. The nonpolar hulls are depicted in yellow (see Supplementary Material for details), and were computed taking into account only the two PDB complexes.
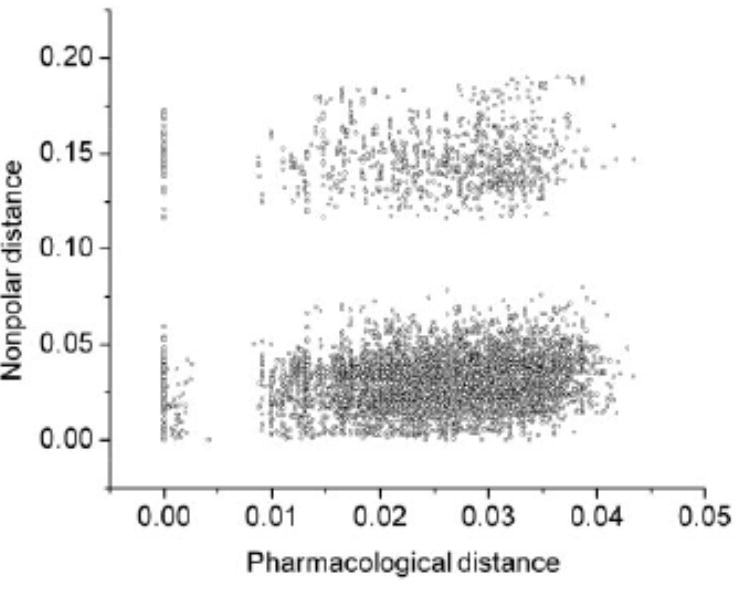
1c. Nonpolar distance matrix Dnp = [dnp(i, j)] over the 119 assayed kinases. The numerals in rows and columns follow Figure 1a. The plot dnp versus dphar for all (119 × 118)/2 kinase pairs (i, j) shows no correlation between the two metrics (Fig. 1d). However, when the highly promiscuous affinity-dominant staurosporine is incorporated to the affinity profile (Fabian et al., 2005) and the affinity-based distance matrix is recalculated (dphar→ dps = pseudopharmacological distance), a good correlation (R2 = 0.875) between dps and dnp is obtained (Fig. 1e). This correlation reveals that promiscuity, the dominant affinity trait when staurosporine is incorporated, is fostered by targeting accessible nonpolar moieties, in turn, a highly conserved feature of protein interfaces (Ma et al., 2003). The strong correlation shown in Figure 1e implies that staurosporine should bind mainly through hydrophobic contacts, as it is indeed the case in its PDB complexes (Fernández and Maddipati, 2006).
1d. Plot of nonpolar distance versus pharmacological distance. Each circle represents a kinase pair. No correlation is observed, while there is some bimodality in each dimension.
1e. Correlation between pseudopharmacological distance (including staurosporine in the drug screening background) and nonpolar distance between kinases. The sole outliers are pairs involving the EGFR kinase, the kinase whose affinity vector is only dominated by staurosporine (cf. Fabian et al., 2005, Fig. 5).
To determine whether pharmacological differences are dictated by differences in nonpolar accessible surfaces of the targets within ligand-binding sites, a nonpolar distance, dnp(i, j), between the affinity-assayed kinases i, j is introduced. The dnp(i, j) is determined by differences in accessible nonpolar surface areas of the respective nonpolar hulls, Hnp(i), Hnp(j). The nonpolar hull of a kinase (Section 2) is comprised of the nonpolar residues of the kinase in contact with its drug ligands, whenever the complexes are reported in PDB, and residues in the kinase (not necessarily nonpolar) that align with nonpolar residues in contact with ligands in homolog PDB-reported ligand–kinase complexes (Fig. 1b). The latter constitute the structural background for the comparison. Introducing the hull becomes necessary to compare kinases at this level. Thus, the nonpolar distance is expressed as dnp(i, j) = [A(Hnp(i))– 1 Σa∈Hnp(i)|A(a) – A(Φj(a))|, with A = nonpolar accessible area (Fraczkiewicz and Braun, 1998; Ooi et al., 1987); a = generic residue in the nonpolar hull of chain i; A(a) = 0 if a is polar; Φj(a) = residue in chain j that aligns with a.
Only 32 of the 119 assayed kinases are reported in PDB complexes (Fernández and Maddipati, 2006), yet, structural inferences can be made with confidence (Bonneau et al., 2002;Chivian et al., 2005;Fernández and Berry, 2004) given the homology (Manning et al., 2002) and high structural alignment (RMSD < 1 Å) across reported structures (Fernández and Maddipati, 2006). The prediction accuracy decreases somewhat but in a quantifiable manner (~12%) on loopy regions (Section 2 and Supplementary Material). The matrix Dnp = [dnp(i, j)] (Fig. 1c) reveals remarkable nonpolar similarity across kinases: 〈dnp〉 /maximum(dnp)~11% (〈 〉 = average over kinase pairs);〈[dnp – 〈dnp〉]2〉1/2/〈dnp〉~ 16%.This similarity implies a high conservation of accessible nonpolar surface, an indication that ligands whose affinity is dominated by hydrophobic interactions should be highly promiscuous.
With typical ligand-dissociation constants in the nanomolar range, the coordinate for staurosporine dominates the affinity vector for each kinase to such an extent that most affinity profiles look similar when this compound is included in the drug background. This similarity is reflected in the low rank (~7, Supplementary Material) of the pseudopharmacological distance matrix. The nonpolar matrix possess a comparable rank (~6) because the basis for comparison, i.e. exposed nonpolar residues, is highly conserved (Ma et al., 2003), leading to high levels of target similarity.
Significantly, the most promiscuous drug target, the pregnane X nuclear receptor (PXR, PDB accession code 1ILH), believed to bind to over 50% of human drugs (Hopkins et al., 2006) contains the most extensive nonpolar hull per 1000 Å2 of ligand surface of all complexes reported in PDB (Supplementary Material ), and the highest density of nonpolar accessible surface: 660 Å2 per 1000 Å2 of accessible surface within the nonpolar hull.
On the other hand, sequence alignment including the 32 assayed kinases with PDB-reported complexes (Fernández and Maddipati, 2006) reveals that residues in ATP-binding sites engaged in hydrogen bonding with ligands are highly conserved, with 0 ≤σ(n) ≤ 0.21 (n = chain position for hydrogen-bonding residue; σ = information entropy reflecting aminoacid variability after sequence alignment; average σ in Hnp = 0.87; maximum (σ = ln 20 ~ 4.2) (Higgins et al., 1996; Shenkin et al., 1991). As expected, differences in hydrogen-bonding capabilities do not appreciably correlate with dps (R2 ~ 0.19), and there is no correlation with dphar.
These observations lead to the question: What feature may be targeted to promote specificity? We need to identify a feature with sufficient variability across homologs and capable of influencing the affinity for the ligands by modulating the local propensity for water exclusion. Thus, we focus on ‘environmental residues’, i.e. those framing the microenviron-ment of intramolecular SAHBs (Fernández and Berry, 2004). These bonds may become intermolecularly dehydrated upon ligand association (Section 2). They promote such associations because the enhancement and stabilization of electrostatic interactions overcomes the thermodynamic cost associated with removing the surrounding water molecules that hydrate amide or carbonyl groups (Fernández and Berry, 2004). Environmental residues include the hydrogen-bonded residues themselves. To compare environments of different kinases, we define the environmental hull of a kinase, Henv, as the reunion of all environmental residues in the chain and residues aligning with environmental residues from other chains (Fig. 2a and b).
Fig. 2.






2a. Environmental hull (light blue) for CHK1 (obtained from alignment with PDK1). Solvent-accessible hydrogen bonds (SAHBs) are indicated as green segments joining the α-carbons of the paired residues. The virtual bonds are shown as blue segments. The three SAHBs perturbed by the ligand (named 3A3) are C87-G90; G90-L138; G16-V23.
2b. Aligned backbones for PDK1 (blue) in complex with BIM8 (PDB.1UVR) and CHK1 (lilac) in complex with 3A3 (PDB.2GCU), with the environmental hulls depicted in light blue.
2c. Environmental distance matrix Denv = [denv(i, j)] for the 119 kinases assayed (Fabian et al., 2005).
2d. Correlation of environmental versus pharmacological distance. The line indicates the optimal linear fit. The red diamonds correspond to the six pairs including ABL1, the primary target for imatinib, and each of its six mutants, listed in Figure. 1a, that confer different degrees of drug resistance.
2e. Relation between packing and environmental distance as function of the size, #S, of the structural background set used to define the environmental hull. The 103 structurally reported kinases were used for the analysis and their environmental distances were computed as if the structures were unknown. For a reduced background (#S<5), the packing metric is well approximated by denv, although with significant dispersion (~25%, error bars). As more structural background is included (#S>4), packing distance becomes an overestimation.
2f. Renormalized difference matrix Ddif = (Dphar/0.045) ‒ (Denv/0.042).
Thus, an environmental distance denv(i, j) between kinases i and j is obtained by comparing the aligned hydrogen-bond microenvironments within Henv(i) and Henv(j): denv(i, j) = M(i, j)−1[Σn=1,. . .,M(i, j) Δn(i, j )], where M(i, j) = number of residue pairs in Henv(i) corresponding to SAHBs in kinase i or to hydrogen bonds or nonbonded residue pairs that align with SAHBs in Henv(j), n = dummy index denoting residue pair and Δn(i, j) = 0 if residue pair n corresponds to a SAHB in Henv(i) that aligns with a SAHB in Henv(j) and Δn(i, j) = 1, otherwise. Thus defined, the environmental distance compares local dehydration propensities associated with SAHB patterns in kinases. The validity of the relation: Φi(Henv(j)) = Henv(i) enables a rigorous comparison between environments of two different proteins, in contrast with earlier attempts (Fernández and Maddipati, 2006).
The matrix Denv = [denv(i, j)] for the 119 kinases assayed (Fabian et al., 2005) (Fig. 2c) is obtained after inference of the SAHBs for the 87 kinases unreported in PDB from direct structure prediction (Bonneau et al., 2002; Chivian et al., 2005). The predictions are validated through a correlation with an independent and accurate sequence-based prediction of another structural attribute: the disorder propensity (Braken et al., 2004; Fernández and Berry, 2004) (Section 2). This attribute was chosen because loopy regions, in the twilight between order and disorder, compromise somewhat the accuracy of a structure prediction. The validation is based on the fact that the extent of intramolecular hydration of a hydrogen bond correlates strongly with the disorder propensity: disorder arises from an impossibility to sufficiently hinder hydration of amides and carbonyls (Fernández and Berry, 2004). The strong correlation between denvand dphar(R2~0.917) (Fig. 2d) reveals that the impact of drugs on the human kinome is dictated by differences in hydration microenvironments across the ligand-binding regions of the kinases. To the best of our knowledge, the hydration differences across kinases, quantified through the metric denv, were not considered in the development of the drugs screened in (Fabian et al., 2005). The diversity in hydration microenvironments needed to yield specificity across paralog kinases results from the variability (〈σ〉 ~1.38) of environmental residues, while 〈σ〉 ~0.21 when the average is restricted to residues paired by hydrogen bonds that are environmentally affected by the ligands.
The ranks and Shannon entropies of Dphar, Denv, Dnp and Dps were determined at different coarse-grained resolutions (Supplementary Material) to compare the respective levels of information complexity of the matrices. Again, the comparable levels of complexity point to the appropriateness of denv as a metric to assess specificity, while dnp adequately maps promiscuity.
The metric denv is defined at the sequence level by identifying the comparison window through environmental alignment (Section 2). This technique required aligning residues with those in homolog kinases with reported structure whose microenvironments are known to be affected by ligand association. Thus, in contrast with the structure-based packing distance that compares SAHBs wrapped by ligands in the structures of two complexes (cf. Fernández and Maddipati, 2006), a background of homologs, S (Section 2), is needed to compare sequence pairs using denv. If S is limited (cardinal <5), denv is well approximated by dpack but for a more extended and reliable window of comparison (determined by aligning the test sequence to more PDB homologs), denv becomes a distinctive metric (Fig. 2e). This extended comparison is required to make reliable sequence-based inferences and incorporates for a given protein the alternative binding regions found in homologs, as illustrated in Supplementary Material.
We also identified sets of kinases—cliques—within which inhibitory impact may be inferred from environmental distances with confidence higher than 98%. This required computing the matrix of differences between pharmacological and environmental distances, both appropriately rescaled to a single [0, 1] range (Fig. 2f), followed by a block diagonalization of this matrix, leading to four sets of kinases (blocks I–IV, Table 1).
Table 1.
Kinase groups from block diagonalization of matrix Ddif
| I | AAK1, Aurora2, Aurora3, BIKE, CAMK1, CAMK1D, CAMK1G, CAMKK2, CDK2, CLK2, CLK4, CSNK1E, CSNK1G1, DAPK2, DAPK3, FGFR2, GAK, JNK1, MARK2, PCTK1, PHKG2, PIM2, RPS6KA2, RPS6KA5,STK16, STK17A, STK18, STK3_m, STK38L, STK4, ULK3_m |
| II | ABL1, ABL1(M351T), ABL1(Q252H), ABL1(T315I), ABL1(Y253F), CDK5, EPHA2, EPHA4, EPHA5, EPHA6, EPHA7, EPHA8, EPHB1, FLT3, FLT4, FRK, LCK, NTRK1, p38-alpha, p38-beta, PDGFRB, STK10, TEK VEGFR2 |
| III | ABL1(E255K), ABL1(H396P), ABL2, ACK1, BMX, BTK, CAMK2A, CAMK2B, CAMK2D, CAMK2G, CAMKK1, CLK1, CLK3, CSK, CSNK1G2, EGFR, EPHB4, ERBB2, FER, FGFR1, FGFR3, FGR, FYN, HCK, INSR, JAK1, JAK2, KIT, LIMK1, LYN, MAP3K4, MAP3K5, MAP4K5, MYLK2, NEK2, NEK6, NEK9, PAK1, PAK3, PAK4, PAK6, PAK7/PAK5, PHKG, PIM1, PKMYT1, PRKAA1, PRKACA, PTK2, PTK6, RIPK2, RPS6KA3, SRC, STK17B, STK36, SYK, YES |
| IV | EPHA3, JNK2, JNK3, MKNK2, p38-gamma, SLK, TNIK, TTK |
Four cliques (I–IV) of kinases obtained from block diagonalization of matrix Ddif. Pairs of kinases within each clique have optimal correlation between pharmacological and environmental distance.
Local hydration differences determining specificity of drug inhibitors may arise in kinase pairs with high degree of structural alignment (Hogue, 1997) (RMSD ~ 0.3 Å), such as SRC and LCK. The latter is a known target for Gleevec (imatinib), which may thus act as immunosuppressant (Dietz et al., 2004), while SRC shows no affinity for the drug (Fabian et al., 2005). Strikingly, two SAHBs in LCK, G254-G257 and R397-A400 that promote their own dehydration through protein–imatinib association (Fig. 3), are absent from SRC: regions in the twilight zone between order and disorder in LCK become fully disordered in SRC, as shown by the absence of electron density (Fig. 3). Thus, while structurally similar, the environmental distance between SRC and LCK is sufficient to account for the selective affinity of imatinib.
Fig. 3.

Environmental differences between the highly alignable native folds of LCK (blue) and SRC (lilac). The two SAHBs G254-G257 and R397-A400 are present only in LCK, a target for imatinib, while SRC has no affinity for the ligand.
Drug-resistant mutations in another imatinib target, Bcr-ABL (ABL1), produce environmental differences that correlate approximately with the pharmacological distances between mutant and wild type (Fig. 2d). Upon close examination (Fig. 4), the most effective mutations (Fabian et al., 2005) T315I and E255K are precisely the ones that have the most dramatic effect in increasing the dehydration (by adding nonpolar groups to the microenvironment) of preformed SAHBs Q300-E316, and G251-G254, respectively. These SAHBs are part of the environmental hull of wild-type Bcr-ABL. All reported ABL1 mutations actually perturb the dehydration propensity of SAHBs (Fig. 4). The poorest correlation between environmental and pharmacological distance (Fig. 2d) arises for mutations E255K, H396P, likely to perturb affinity through other mechanisms that supersede environmental change. Overall, the drug-resistant mutations significantly decrease the dehydration propensity of the target surface and accordingly decrease the inhibitor affinity.
Fig. 4.

Environmental impact of the drug-resistant mutations of ABL, a primary target for imatinib (PDB.1IEP, ligand shown in complex). Only the side chains of the mutating residues are indicated, together with the SAHBs (green) whose microenvironments they affect. Hydrogen bonds not accessible to solvent are shown as thin segments in light grey. The mutations with the SAHBs affected (in brackets) are: T315I (Q300-E316); E255K (G251-G254); Q252H (L248-G251; G249-Q252; G251-G254); Y253F (L248-G251; G249-Q252; G251-G254); M351T (E352-K356); H396P (H396-A399).
4 DISCUSSION
In this work, we identified the molecular code for promiscuity and specificity in targeting drugs that impact the human kinome. Thus, significant progress in the informatics of drug design has been achieved by determining the type of similarity among targets that promotes promiscuity and the type of difference that controls specificity. The former corresponds to highly conserved exposed nonpolar moieties in alignable regions of protein–ligand interfaces, while the latter corresponds to nonconserved hot spots of high dehydration propensity around amide-carbonyl pairs.
Kinase structures may be tightly aligned except in the regions of highest conformational plasticity, precisely the regions of catalytic and regulatory relevance (Huse and Kuriyan, 2002). These loopy regions are obviously rich in packing defects, as their backbone hydrogen bonds are not fully buried and thus contain hot spots of dehydration propensity. Thus, a way to achieve a classification of kinases is to assess differences in the nonconserved microenvironments of their loopy regions. As shown in this work, such microenvironmental differences reflect the differences in binding affinity against a representative set of drugs.
The analysis was carried out at a genomic scale, since the environmental assessment of local dehydration propensities can be reliably determined from sequence (cf. Fernández and Berry, 2004). The sequence-based predictor became operational because of the high level of structural similarity reported for the kinase superfamily, combined with relatively low sequence homology levels that enable target discrimination. These properties paved the way to the mapping of a reliable sequence-based attribute, the disorder propensity, onto a molecular feature, the dehydration propensity, shown to be effectively sculpted in drug ligands to modulate specificity.
In future work, our methodology will be adapted to model specificity control in drug-targeted nuclear hormone receptors. The moderate structure conservation of the ligand-binding domain (Escriva et al., 2000) and the significant PDB representation needed to construct a reliable structural background instills confidence in the success of our environmental predictor.
Our results lead us to the difficult inverse problem of determining affinity profiles of individual kinases from their predicted pharmacological distances. A troubling aspect of this problem is the uniqueness of the solution. We would need to specify n + 1 affinity profiles for a background of n drugs to be able to determine uniquely the remaining profiles solely from their distances.
Supplementary Material
Supplementary data are available at Bioinformaticsonline.
Acknowledgments
The research of A.F. was supported by NIH grant R01-GM072614, by the Rice Computational Research Cluster (NSF Grant CNS-0421109), and by a grant from the John and Ann Doerr Fund for Computational Biomedicine (Program GC4R 2005). The authors thank Drs Chen Su and Harry Harlow (Eli Lilly) and Ridgway Scott (University of Chicago) for their valuable insights.
Footnotes
Associate Editor: Dmitrij Frishman
Conflict of Interest: none declared.
References
- Bain J, et al. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonneau R, et al. De novo prediction of three-dimensional structures for major protein families. J Mol Biol. 2002;322:65–78. doi: 10.1016/s0022-2836(02)00698-8. [DOI] [PubMed] [Google Scholar]
- Braken C, et al. Combining prediction, computation and experiment for the characterization of protein disorder. Curr Opin Struct Biol. 2004;14:570–576. doi: 10.1016/j.sbi.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Chivian D, et al. Prediction of CASP6 structures using automated Robetta protocols. Proteins. 2005;61(Suppl 7):157–166. doi: 10.1002/prot.20733. Supplementary Material. [DOI] [PubMed] [Google Scholar]
- Chothia C. Hydrophobic bonding and accessible surface area in proteins. Nature. 1974;248:338–339. doi: 10.1038/248338a0. [DOI] [PubMed] [Google Scholar]
- Dietz AB, et al. Imatinib mesylate inhibits T-cell proliferation in vitro and delayed-type hypersensitivity in vivo. Blood. 2004;104:1094–1099. doi: 10.1182/blood-2003-12-4266. [DOI] [PubMed] [Google Scholar]
- Druker BJ. Molecularly targeted therapy: have the floodgates opened? Oncologist. 2004;9:357–360. doi: 10.1634/theoncologist.9-4-357. [DOI] [PubMed] [Google Scholar]
- Escriva H, et al. Ligand binding and nuclear receptor evolution. Bioessays. 2000;22:717–727. doi: 10.1002/1521-1878(200008)22:8<717::AID-BIES5>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Fabian MA, et al. A small molecule kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- Feng BY, et al. High throughput assays for promiscuous inhibitors. Nat Chem Biol. 2005;1:146–148. doi: 10.1038/nchembio718. [DOI] [PubMed] [Google Scholar]
- Fernández A. Keeping dry and crossing membranes. Nat Biotech. 2004;22:1081–1084. doi: 10.1038/nbt0904-1081. [DOI] [PubMed] [Google Scholar]
- Fernández A, Berry RS. Molecular dimension explored in evolution to promote proteomic complexity. Proc Natl Acad Sci USA. 2004;101:13460–13465. doi: 10.1073/pnas.0405585101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández A, Maddipati S. A priori inference of cross reactivity for drug-targeted kinases. J Med Chem. 2006;49:3092–3100. doi: 10.1021/jm060163j. [DOI] [PubMed] [Google Scholar]
- Fernández A, et al. Dynamics of hydrogen bond desolvation in protein folding. J Mol Biol. 2002;321:659–675. doi: 10.1016/s0022-2836(02)00679-4. [DOI] [PubMed] [Google Scholar]
- Fraczkiewicz R, Braun W. Exact and efficient analytical calculation of the accessible surface areas and their gradient for macromolecules. J Comput Chem. 1998;19:319–333. [Google Scholar]
- Higgins DG, et al. Using CLUSTAL for multiple sequence alignments. Methods Enzymol. 1996;266:383–402. doi: 10.1016/s0076-6879(96)66024-8. [DOI] [PubMed] [Google Scholar]
- Hogue CWV. Cn3D: a new generation of three-dimensional molecular structure viewer. Trends Biochem Sci. 1997;22:314–316. doi: 10.1016/s0968-0004(97)01093-1. [DOI] [PubMed] [Google Scholar]
- Hopkins AL, et al. Can we rationally design promiscuous drugs? Curr Opin Struct Biol. 2006;16:127–136. doi: 10.1016/j.sbi.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–282. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- Knight ZA, Shokat KM. Features of selective kinase inhibitors. Chem Biol. 2005;12:621–637. doi: 10.1016/j.chembiol.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Ma B, et al. Protein-protein interactions structurally conserved residues distinguish between binding sites and exposed protein surfaces. Proc Natl Acad Sci USA. 2003;100:5772–5777. doi: 10.1073/pnas.1030237100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning G, et al. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Ooi T, et al. Accessible surface area as a measure of the thermodynamic parameters of hydration of peptides. Proc Natl Acad Sci USA. 1987;84:3086–3090. doi: 10.1073/pnas.84.10.3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenkin SP, et al. Information-theoretical entropy as a measure of sequence variability. Proteins: Struct Funct Genet. 1991;11:297–313. doi: 10.1002/prot.340110408. [DOI] [PubMed] [Google Scholar]
- Vieth M, et al. Kinomics-structural biology and chemogenomics of kinase inhibitors and targets. Biochim Biophys Acta. 2004;1697:243–257. doi: 10.1016/j.bbapap.2003.11.028. [DOI] [PubMed] [Google Scholar]
- Whittle PJ, Blundell TL. Protein structure-based drug design. Annu Rev Biophys Biomol Str. 1994;23:349–375. doi: 10.1146/annurev.bb.23.060194.002025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data are available at Bioinformaticsonline.