

Abstract
Sulfotransferases (SULTs) are enzymes that catalyze the sulfation of hydroxyl-containing compounds. Sulfation regulates hormone activities and detoxifies xenobiotics. Human estrogen sulfotransferase (hSULT1E1) catalyzes the sulfation of estrogens and regulates estrogen bioactivities. Oxidative regulation provides a biological mechanism for regulating enzyme activities in vivo. The oxidative regulation of human SULTs has not been reported. In this study, we used amino acid modification, manipulation of intracellular redox state, and site-directed mutagenesis to study the redox regulation of human SULTs and specifically the mechanism of hSULT1E1 inhibitory regulation by oxidized glutathione (GSSG). Of the four major human SULTs, hSULT1A1, hSULT1A3, and hSULT2A1 do not undergo redox regulation; hSULT1E1, on the other hand, can be redox regulated. GSSG inactivated hSULT1E1 activity in an efficient, time- and concentration-dependant manner. The co-enzyme adenosine 3′-phosphate 5′-phosphosulfate protected hSULT1E1 from GSSG-associated inactivation. A reduced glutathione (GSH) inducer (N-acetyl cysteine) significantly increased while a GSH depletor (buthionine sulfoxamine) significantly decreased hSULT1E1 activity, but both failed to affect the amount of hSULT1E1 protein in human hepatocyte carcinoma Hep G2 cells. Crystal structure suggested that no Cys residues exist near the active sites of hSULT1A1, hSULT1A3, and hSULT2A1, but Cys residues do exist within the active site of hSULT1E1. Site-directed mutagenesis demonstrated that Cys83 is critical for the redox regulation of hSULT1E1. This first report on the redox regulation of human SULTs suggests that the redox regulation of hSULT1E1 may interrupt the regulation and function of estrogens under various physiological and pathological conditions.
Keywords: sulfotransferase, SULT1E1, estrogen sulfotransferase, Redox Regulation, Estrogen
1. INTRODUCTION
Sulfotransferases (SULTs) are enzymes that catalyze the sulfation (sulfonation) of hydroxyl-containing compounds [1, 2]. The universal sulfuryl group donor (co-substrate) for SULT-catalyzed sulfation is adenosine 3′-phosphate 5′-phosphosulfate (PAPS). The reaction products are a sulfated product and adenosine 3′, 5′-diphosphate (PAP). One of the main biological functions of SULTs is the regulation of various hormones [3]. Many of the endogenous hormones are substrates for different SULT isoforms [4]. Sulfation usually leads to the inactivation of hormones, as the sulfated forms of hormones are usually unable to bind to receptors [5]. Sulfated hormones could also be used for transport or storage of bioactive hormones [3].
SULTs also catalyze the sulfation of many structurally diverse drugs, carcinogens, and other xenobiotics. The substrate specificities of many SULTs are very broad [6]. Most hydroxyl groups in phenols, alcohols, and N-substituted hydroxylamines function as substrates for given cytosolic SULT isoforms. Sulfation of drugs and xenobiotics is primarily associated with detoxification by which a relatively hydrophobic xenobiotic is biotransformed into a more water-soluble sulfuric ester, which in turn is readily excreted. However, there are numerous important exceptions wherein the formation of chemically reactive sulfuric esters is an essential step in metabolic pathways leading to toxic or carcinogenic bioactivation [6]. Detoxification or bioactivation depends on the chemical properties of the sulfated product.
Human SULT1E1 (hSULT1E1) catalyzes the sulfation of estrone and estradiol with extremely high efficiency. Sulfation is believed to be involved in the inactivation of estrogens in target tissues [7]. Sulfation of active 17β-estradiol (E2) forms inactive estradiol sulfate, which can be reactivated following desulfation by estrogen sulfatase [8].
The cytoplasm is a highly reducing environment (containing millimolar levels of GSH) in which protein cysteine residues are maintained primarily in their thiol state [9]. Redox modification of Cys residues of an enzyme provides a mechanism for regulating enzyme activity [10]. Proteins can be S-glutathionylated [11] or S-nitrosylated [12], especially during oxidative stress. Oxidative stress is involved in the pathogenesis of various degenerative diseases, including cancer [13]. Oxidative stress may be involved in breast cancer, and high levels of GSH is associated with a favorable outcome and good prognosis, whereas low levels of GSH is associated with more aggressive or more advanced disease [14].
Many factors, including clinical oxygen treatment, chemical (toxicants) stress, physical stress, aging, virus infection, and different pathological conditions, can cause oxidative stress. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) can modify thiol bonds that affect a protein’s function [15]. Oxidative stress is a well-known cause of changes in GSSG/GSH ratios and levels in vivo [10]. S-glutathionylation regulates the activity of various enzymes [16, 17], although no data has been reported regarding in vivo SULT regulation. Only in vitro redox regulation using E. coli-expressed rat aryl sulfotransferase IV (AST-IV or rSULT1A1) has been reported [18]. Our recent work [19, 20] demonstrated that hyperoxia, physical stress, and chemical (parathion) stress regulate rSULT1A1 enzyme activity in vivo. Our in vitro redox regulation mechanism studies suggest that rSULT1A1 oxidative regulation occurs through Cys thiol modification. Oxidative stress that occurs in various pathological conditions may significantly alter SULT enzymatic activity, leading to changes in hormone regulation and xenobiotic drug detoxification/metabolism. Thus, it is important to understand the oxidative regulation of human SULTs and its mechanisms.
To the best of our knowledge, oxidative regulation of human SULTs has not been reported. In the present investigation, we used human liver cytosol and purified recombinant human SULTs to study the effect of GSSG on human SULT activity. We applied either a GSH inducer (N-acetyl cysteine [NAC]) or a GSH depletor (buthionine sulfoxamine [BSO]) to human hepatocyte carcinoma (Hep G2 cells) to test how varying intracellular GSH levels affect hSULT1E1 enzymatic activity. These results, as well as site-directed mutagenesis studies we report here, clearly reveal that hSULT1E1 activity can be regulated by redox modification of its cysteine residues.
2. METHODS AND MATERIALS
2.1 Materials
Ampicillin,17β-estradiol (E2), 3′-phosphoadenosine-5′-phosphosulfate (PAPS), dithiothreitol, buthionine sulfoxamine (BSO), N-acetylcysteine (NAC), dithiobis-2-nitrobenzoic acid (DTNB), reduced glutathione (GSH), and oxidized glutathione (GSSG) were purchased from Sigma. SDS-polyacrylamide gel electrophoresis reagents were purchased from Bio-Rad (Hercules, CA). Western blot chemiluminescence reagent kits (Super Signal West Pico Stable Peroxide and Super Signal West Pico Luminol/Enhancer solutions) were purchased from Pierce Chemical (Rockford, IL). Nitrocellulose membranes (Immobilon-P; Millipore Corporation, Bedford, MA) were purchased from Fisher Scientific Co. (Fair Lawn, NJ). Protein assay reagent was purchased from Bio-Rad. All other reagents and chemicals were of the highest analytical grade available.
2.2 hSULT1E1 Enzyme Activity Assay
hSULT1E1 activities in human liver cytosol, Hep G2 cell cytosol, bacterial cytosol (50 μg protein from each) and in purified enzyme (3 μg protein) were determined by the radioactive assay method [21–24]. [3H]E2 (0.9 Ci/mmol; 0.15 μM final concentration) was used as substrate for the reaction. For all assays, 20 μM PAPS was used in a total of 250 μl reaction mixture containing 50 mM Tris buffer (pH 6.2). After a 30-min incubation in a shaking water bath (37°C), the reaction was stopped by adding 250 μl of 0.25 M Tris (pH 8.7). Extraction was performed twice by adding 0.5 ml of water-saturated chloroform each time. After the final extraction, 100 μl of aqueous phase was used for scintillation counting. The data collected from the enzymatic assay from each protein source were the average of results obtained from three independent experiments.
2.3 hSULT1E1 Inactivation by GSSG
Samples containing either human liver cytosol (final concentration, 1.0 mg/ml) or purified hSULT1E1 (final concentration, 0.1 mg/ml) were incubated in Tris buffer (pH 6.2) at room temperature with various concentrations of GSSG for different durations, as indicated in the figures. Aliquots (50 μl) of the mixture were used to determine E2 sulfation (hSULT1E1) activity as described above. The proper control experiments were performed by adding an equal volume of water.
2.4 Western Blot Analysis of hSULT1E1 Protein
Cytosol protein from Hep G2 cells (10 μg) and SULT1E1 expressed in bacterial cytosol (1 or 5 μg) were subjected to electrophoresis on 12% polyacrylamide gels (Novex, San Diego, CA) [23] at 200 V for about 45 minutes. Separated proteins were electro-transferred onto nitrocellulose membranes at a voltage of 30 V overnight. Membranes were blocked in TBST (50 mM Tris [pH 7.5], 150 mM NaCl, 0.05% Tween-20) containing 5% dried milk for 1 h on a shaker at room temperature. After blocking, membranes were incubated with rabbit anti-hSULT1E1 (1:2000) (PanVera, Madison, WI) in TBST containing 5% dried milk for 2 h on a shaker at room temperature. After incubation, membranes were washed with TBST containing 5% dried milk for 4 × 15 min and incubated in secondary antibody (horseradish peroxidase-conjugated Immuno-Pure goat anti-rabbit IgG; H+L) at a dilution of 1:5000 in the same buffer for 2 h. The membranes were washed with the same buffer for 4 × 15 min and then with TBS for 3 × 5 min. Fluorescent bands were developed with 1 ml of substrate containing the same volume of each Super Signal West Pico Luminol Enhancer solution and Super Signal West Pico Stable Peroxidase solution at room temperature for 5 min. The X-ray films were exposed to the membrane and then developed. Films were scanned and densitometric analyses were performed with a Gel Documentation and Analysis System from Advanced American Biotechnology and with AAB software (Fullerton, CA).
2.5 Effects of NAC and BSO on hSULT1E1 Catalytic Activity and GSH Level in Hep G2 Cells
Hep G2 cells were obtained from American Type Culture Collection (Manassas, VA). Cells were grown and maintained in Dulbecco’s Modified Eagles’s Medium Nutrient Mixture F-12 Ham (Sigma) supplemented with L-glutamine and 15 mM HEPES, and 10% fetal bovine serum. On day 0, cells were seeded in 100-cm2 dishes at a density of 1.0×105 cells per dish. The cultures were incubated at 37°C in a humidified incubator containing 5% CO2 and 95% air. On day 1, various concentrations of NAC (0, 1, 3, or 10 mM final concentrations in normal medium) or various concentrations of BSO (0, 0.05, 0.15, or 0.5 mM final concentrations in normal medium) were added to the properly marked dishes. On day 6, cells were harvested using 0.25% trypsin-EDTA solution (Sigma), washed with phosphate buffered saline (PBS), and then homogenized in 1 ml lysis buffer (0.1 mM PMSF, 50 mM Tris, 250 mM sucrose, 0.1 mM EDTA [pH 7.5]). Lysis buffer was deoxygenated by bubbling the buffer with excess nitrogen gas. The homogenate was centrifuged at 12,000x g for 30 min and the supernatant was used for the hSULT1E1 activity assays and Western blotting. Our protocols for cell culture and cytosol preparation have been described earlier [21].
2.6 Determination of Intracellular GSH
The intracellular GSH levels from NAC- or BSO-treated Hep G2 cells were determined using reported method [25] with slight modification. Briefly, treated Hep G2 cells were harvested and washed with ice-cold PBS and re-suspended in 100 μl of 10 mM HCl. The cells were subjected to rapid freeze thaw over liquid nitrogen several times and vortexed for 1 min. Trichloroacetic acid (100 μl, 10% w/v) was added to the lysate to precipitate the protein. The mixture was centrifuged at 10,000x g for 10 min to obtain protein-free supernatant. The following fluids were then mixed to make a color reaction: 0.7 ml of 0.5 M potassium phosphate buffer (pH 8.0), 0.05 ml protein-free supernatant, and 0.1 ml of 0.04% (w/v) DTNB. After 5 min, the absorbance at 412 nm was measured using a spectrophotometer, and GSH concentrations were calculated using a GSH standard curve.
2.7 Site-directed Mutagenesis of hSULT1E1
The cDNA encoding hSULT1E1 in the pKK233-3 vector was from Dr. Charles Falany [26]. All mutant cDNAs were created with the QuikChange mutagenesis kit (Stratagene, La Jolla, CA) as outlined previously [27] according to the manufacturer’s protocol. Primers were designed by using Gene Fisher primer designing and Multialignment software. All primers used for mutagenesis were obtained from Integrated DNA Technologies Inc. (Coralville, IA). The primers used for the mutations are as follows:
Cys69SerF, 5′GGGTGATGTGGAAAAGAGCAAAGAAGATG3′;
Cys69SerR, 5′CCCAAAATGAGGCAGGAAGAAGTTCAGG3′;
Cys83SerF, 5′CGAATACCTTTCCTGGAAAGCAGAAAAG3′;
Cys83SerR, 5′GCCACATCCTTTGCATTCCGGCAAAGATAG3′;
Cys122SerF, 5′GGGAAAAGGATAGTAAGATAATCTATC3′;
Cys122SerR, 5′CCCACCAAGATTTTACATGTTTATACCAGG3′;
Cys128SerF, 5′GATAATCTATCTTAGCCGGAATGCAAAGG3′;
Cys128SerR, 5′CCCACCAAGATTTTACATGTTTATACCAGG3′.
Mutant cDNAs were generated and selected through a series of three steps consisting of primer extension/thermo-cycling, digestion of parental DNA, and transformation. Transformed colonies were grown in sterile medium using standard protocols. Plasmids were isolated from cells with the QIAprep Spin Miniprep Kit (Qiagen Inc., Valencia, CA). The presence of the desired mutations was confirmed by DNA sequencing, which was performed at The Recombinant DNA/Protein Resource Facility, Oklahoma State University. Recombinant bacteria carrying desired mutations were grown on a large scale in proper medium, and bacterial cytosol was prepared by using standard protocol.
All data presented in the figures represent means ± SEM of the data collected separately from three individual experiments.
3. RESULTS
3.1 Redox Regulation of Human SULTs
To investigate the potential oxidative regulation of human SULTs, the effects of GSSG on human SULTs in human liver cytosol (Figure 1) and on purified human SULTs (data not shown) was investigated. Our results suggest that hSULT1E1 is sensitive to GSSG treatment, whereas hSULT2A1, hSULT1A1, and hSULT1A3 are not. These results on Cys modification agree crystal structures (not shown) of the four human SULTs. The structures indicated that Cys70 and Cys287 of hSULT1A1 and hSULT1A3, and Cys55, Cys154, and Cys199 of hSULT2A1 are located on the surface of the molecules, remote from the enzymes’ active sites. GSSG modification of these Cys residues may not significantly affect the enzymatic activity of these SULTs. For hSULT1E1, Cys83 directly contacts the substrate (6Å), whereas Cys128 is located near the active site of the sulfuryl-group transfer (PAPS side, 9Å). Chemical modification of Cys83 and Cys128 may inactivate hSULT1E1.
Figure 1. GSSG inactivation of human SULTs in human liver cytosol.
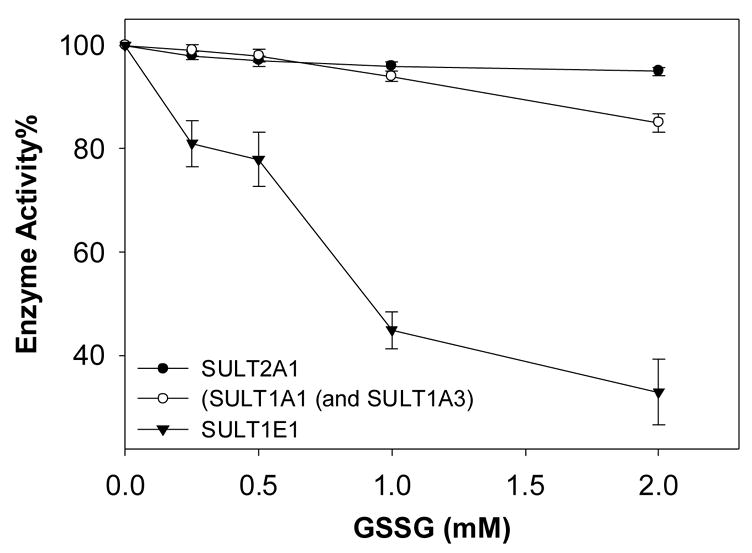
Human liver cytosol (from a 15-year-old male) was treated with the indicated concentrations of GSSG for 20 min at room temperature. Then enzyme activities against [14C]2-naphthol (hSULT1A1 and hSULT1A3), [3H]estradiol (hSULT1E1), and [3H]DHEA (hSULT2A1) were determined [21, 22]. Relative (%) activities to controls are plotted as a function of GSSG concentration.
3.2 GSSG-mediated Inactivation of 17β-Estradiol Sulfation Catalyzed by Purified hSULT1E1 and by SULTs in Human Liver Cytosol
GSSG inactivated E2 sulfation by purified hSULT1E1 (Figures 3A) and by human liver cytosol (Figures 3B) in a time- and concentration-dependent manner. According to the equation d[E]/dt = kapp [E] or log[E] = 2.3kappt (where E represents active hSULT1E1; t is time; kapp is apparent first-order rate constant), kapp can be calculated from the slopes in Figure 2A or 2B. According to the equation, kapp = k[GSSG]n or log kapp = log k + n log[GSSG] (where k is the rate constant; n is the reaction order for GSSG), plotting of Log(Kapp) versus log[GSSG] gave a straight line for both purified hSULT1E1 (Figure 2C) and human liver cytosol (Figure 2D).
Figure 3. PAPS protects purified hSULT1E1 from being inactivated by GSSG.
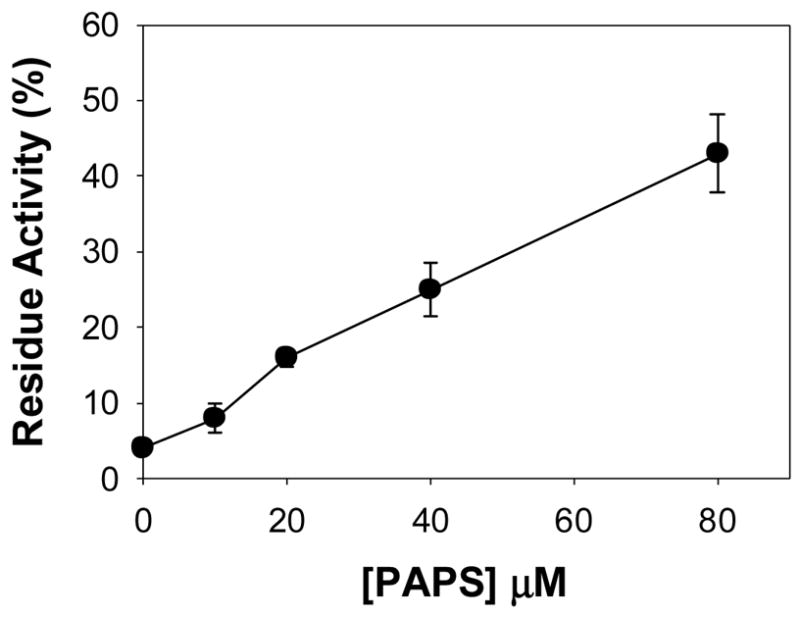
Purified hSULT1E1 was protected from inactivation by 1 mM GSSG by adding various concentrations of PAPS in the pre-incubation mixture.
Figure 2. GSSG-mediated time- and concentration-dependent inactivation of 17β-estradiol sulfation catalyzed by purified hSULT1E1 and by SULTs in human liver cytosol.
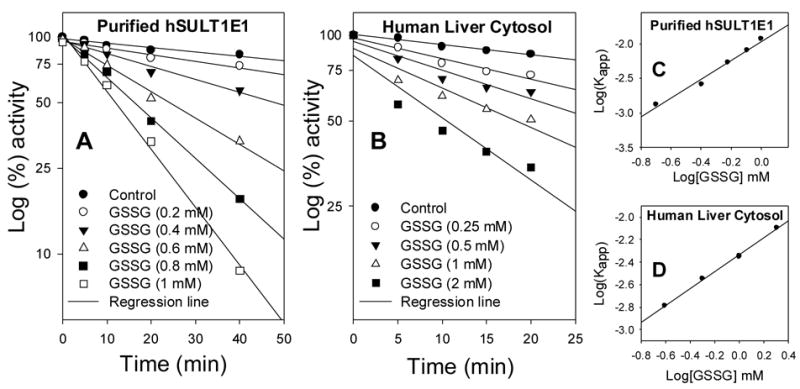
Human liver cytosol (final concentration, 1.0 mg/ml) or purified hSULT1E1 (final concentration, 0.1 mg/ml) were incubated in Tris buffer (pH 6.2) at room temperature with various GSSG concentrations and durations, as indicated. [3H]E2 sulfation (hSULT1E1) activity was determined using the radioactive assay method [21, 22]. The apparent first-order rate constant (Kapp) in Figure 2C and 2D was calculated using data shown in Figure 2A and 2B, respectively.
These results support the hypothesis that GSSG inactivation of hSULT1E1 is caused by active site Cys modification. The calculated reaction order relative to GSSG was 1.3 for the inactivation of purified hSULT1E1-catalyzed E2 sulfation and 0.8 for the inactivation of human liver cytosol-catalyzed E2 sulfation. The calculated second-order rate constant for hSULT1E1 inactivation was 0.011 mM−1min−1 and that for human liver cytosol was 0.0047 mM−1min−1. Based on the GSSG inactivation reaction order, modification of one Cys residue caused the inactivation of hSULT1E1.
3.3 PAPS prevented hSULT1E1 Inactivation by GSSG
PAPS or substrate protection of hSULT1E1 from GSSG-mediated inactivation can demonstrate whether Cys modification occurs in the active site. If PAPS or substrate prevents hSULT1E1 from being inactivated by GSSG, this would suggest that GSSG modifies Cys residue(s) in the active site. Results shown in Figure 3 demonstrate that PAPS protected hSULT1E1 from being inactivated by GSSG in a concentration-dependent manner. This suggests that Cys residue(s) exist in the hSULT1E1 active site and that the modification of these Cys residue(s) can inactivate hSULT1E1.
3.4 Site-directed Mutagenesis of Cys Residues in hSULT1E1
To investigate the mechanism underlying the oxidative regulation of hSULT1E1, four hSULT1E1 Cys mutations—Cys69Ser, Cys83Ser, Cys122Ser, and Cys128Ser—were generated. Vector DNA sequencing results confirmed the mutations. The mutated proteins were expressed in E. coli. Figure 4 shows the Western blot of the cytosols from the mutants. Enzyme assay results suggest that changing Cys to Ser did not significantly alter the enzymatic activity of the four mutants. This implies that they are not critical residues.
Figure 4. Western blot of hSULT1E1 mutant proteins.
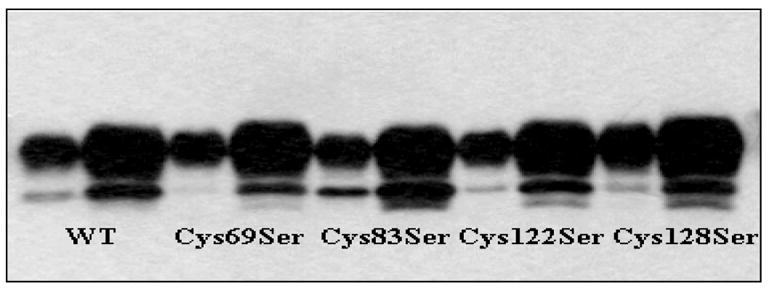
For each pair of lanes containing each mutant protein, one lane contains 1 μg of E. coli cytosol protein and the other contains 5 μg of E. coli cytosol protein.
Time- (Figure 5A) and concentration- (Figure 5B) dependent GSSG inactivation results demonstrated that the Cys69Ser, Cys122Ser, and Cys128Ser mutants were similarly sensitive to GSSG inactivation as was wild-type hSULT1E1. By contrast, Cys83Ser was much less sensitive to GSSG treatment. Taken together, these findings suggested that residues Cys69, Cys122, and Cys128 were not involved in GSSG inactivation of the enzyme, and that Cys83 was mainly responsible for the GSSG inactivation of wild-type hSULT1E1. These results agreed with crystal structure predictions of the redox regulation of hSULT1E1.
Figure 5. Time- and concentration-dependent inactivation of hSULT1E1 mutants by GSSG.
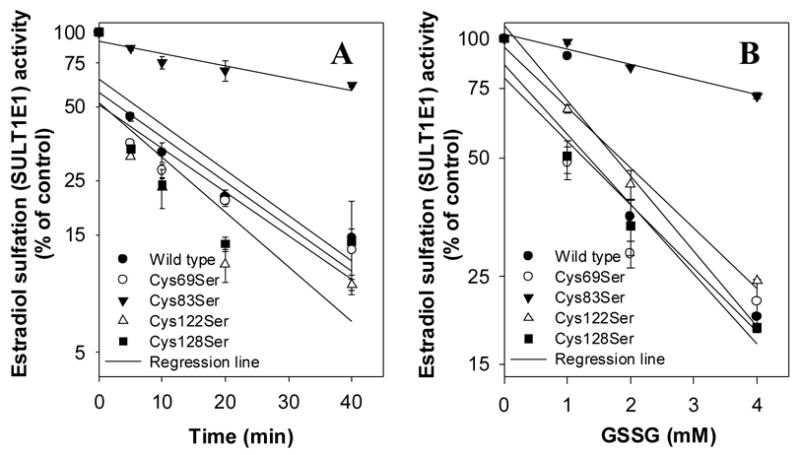
hSULT1E1 mutant cytosols (0.2 mg/ml) were inactivated with 1 mM GSSG at the indicated times or after 20 minutes at the indicated GSSG concentrations as described in the legend for Figure 2. Enzyme activity was plotted using relative activity to control (%) at time zero (Figure 5A) or at a GSSG concentration of zero (Figure 5B).
3.5 Effects of BSO and NAC on E2 Sulfation Activity and GSH Levels in Hep G2 Cells
It is well known that BSO depletes GSH and that NAC increases GSH in cell cultures [28]. Results shown in Figure 6 indicate that BSO treatment decreased and NAC treatment increased E2 sulfation activity (hSULT1E1) in Hep G2 cells (Figure 6A). These changes in enzyme activity (Figure 6A) corresponded with the changes in GSH levels (Figure 6B).
Figure 6. Effects of BSO and NAC on 17β-estradiol sulfation activity (A) and GSH levels (B) in Hep G2 cells.
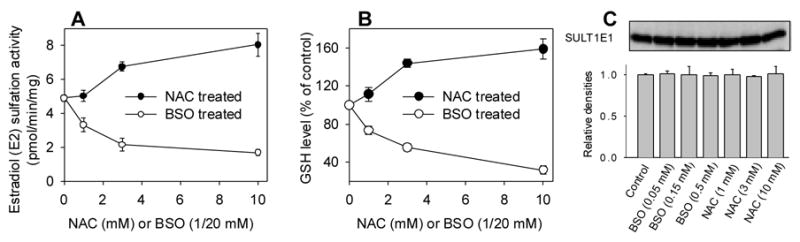
Hep G2 cells were treated with various concentrations of BSO or NAC for 5 days. Treated cells were collected and used for hSULT1E1 enzyme activity assays or GSH level assays as described in the Materials and Methods.
BSO treatment of Hep G2 cells significantly suppressed hSULT1E1 activity at 0.05 mM (34%, p<0.05); 0.15 mM (58%, p<0.01); and 0.5 mM (68%, p<0.01). NAC treatment significantly increased hSULT1E1 activity in Hep G2 cells at 3 mM (36%, p<0.001) and 10 mM (60%, p<0.01). BSO treatment significantly decreased GSH levels in Hep G2 cells at 0.05 mM (28%, p<0.01); 0.15 mM (44%, p<0.01); and 0.5 mM (60%, p<0.01). NAC treatment significantly increased GSH levels in Hep G2 cells at 1 mM (11%, p<0.05); 3 mM (48 %, p<0.01); and 10 mM (59%, p<0.01). Western blot results (Figure 6C) demonstrated that neither BSO nor NAC treatment affected hSULT1E1 protein levels, suggesting that the hSULT1E1 enzyme activity changes we observed were not caused by protein level changes.
4. DISCUSSION
Protein cysteine residues in the cytoplasm are maintained primarily in their thiol state [9). Redox modification of Cys residues of an enzyme provides a mechanism for regulating enzyme activity, especially during oxidative stress {Ghezzi, 2005 #33602]. S-glutathionylation has been reported to regulate the activity of various enzymes [16, 17], although no data has been reported regarding the oxidative regulation of human SULTs. Our recent work [19, 20] demonstrated that hyperoxia, physical stress, and chemical (parathion) stress regulate rat SULT1A1 enzyme activity in vivo. In the present study, we investigated the potential oxidative regulation of human SULTs and its mechanisms.
Our results demonstrated that hSULT1A1, hSULT1A3, and hSULT2A1 are not sensitive to GSSG treatment. One earlier report also suggested that the conserved cysteine residue (Cys70) in human SULT1A1 does not play a role in enzyme’s catalytic activity, but this residue does play a role in the enzyme’s thermostability [29]. In the present investigation, we showed that hSULT1E1, both in purified form and in human liver cytosol, can be redox regulated. Moreover, manipulation of GSH levels in Hep G2 cells significantly altered hSULT1E1 enzyme activity without changing hSULT1E1 protein level.
Our results suggest that active site Cys residue modification caused the inactivation of hSULT1E1. The inactivation reaction order suggested that one Cys residue in the active site of hSULT1E1 is crucial for redox regulation of its enzyme activity. These results are in agreement with crystal structures of human SULTs. Crystal structures suggest that no Cys residues exist near the active sites of hSULT1A1, hSULT1A3, and hSULT2A1. For hSULT1E1, Cys83 and Cys128 are located near the active site. Our site-directed mutagenesis results demonstrated that the presence of Cys83 is crucial for the GSSG inactivation of hSULT1E1.
To the best of our knowledge, oxidative regulation of human SULTs has not been reported. Our results suggest a potential oxidative regulation mechanism for hSULT1E1 through Cys83 redox modification. Cys83 is located in the active site and is in direct contact (6Å) with the substrate E2. Moreover the –SH group of Cys83 is directed toward the E2 molecule based on its crystal structure. Cys83 modification by a bulky molecule like -SG was sufficient to inactivate hSULT1E1, probably by inhibiting substrate binding or product release. Mutant Cys83Ser remained enzymatically active, suggesting that Cys83 is not a critical residue for hSULT1E1 catalytic activity. Ser residue is structurally similar to Cys residue. The Cys/Ser mutation only results a replacement of the –SH group with the –OH group. The mutation should not cause other structural or chemical property changes of hSULT1E1 except the redox regulation property. When the –SH group is replaced with –OH group, although the catalytic activity of hSULT1E1 is not significantly changed, its redox regulation property is significantly changed. This strongly supports the hypothesis that Cys83 is responsible for the redox regulation of hSULT1E1. This also suggests the potential oxidative regulation mechanism for hSULT1E1.
Other E2 binding proteins also are regulated by Cys residue modification. For example, modification of Cys residues inactivates mouse 17β-hydroxysteroid dehydrogenase1 (m17β-HSD1) [30]. Another report showed that modification of Cys residues in human 17β-hydroxysteroid dehydrogenase1 (h17β-HSD1) abolishes the catalytic activity of this enzyme [31]. Also, S-nitrosylation of specific Cys residues in the DNA-binding zinc finger domain of human estrogen receptor α (hERα) is responsible for nitric oxide-mediated structural modifications of hERα, leading to impaired transcriptional activity [32]. Nine Cys residues are present in the DNA-binding domain and four Cys residues in the ligand (E2)-binding domain of hERα [33]. A homology analysis of the atomic structures of the E2-binding region of these proteins (part of a large family of proteins responsible for estrogen metabolism) reveals an evolutionary relationship among them [34]. Our present results on the redox modification of important Cys residues that lead to the regulation of hSULT1E1 are consistent with all of these findings. Together with results discussed from the literature, our results suggest that oxidative stress may importantly affect E2 function and regulation.
Estrogens act on the growth, differentiation, and functioning of many target tissues. These targets include tissues of the reproductive system, such as the mammary gland and uterus, cells in the hypothalamus and pituitary, bone, and liver, as well as the cardiovascular system, where estrogens exert cardioprotective effects [35]. In addition to stimulating normal mammary gland growth and duct development, pathological levels of estrogens increase proliferation and metastatic activity of breast cancer cells and stimulate proliferation of uterine cells [36, 37]. While much is known about the physiology of estrogens, little has been documented about the oxidative regulation of SULTs. Our results on the redox regulation of hSULT1E1, along with possible physiological or pathological consequences of this regulation via altered E2 metabolism, contribute to our understanding of the catalytic mechanism of SULTs and of the metabolic role of SULTs in disease processes during oxidative stress.
Redox regulation of hSULT1E1 activity is important in relation to E2 metabolism, function, and regulation in both normal and pathological conditions. Our findings strongly suggest a correlation between increased cellular GSH levels and increased hSULT1E1 activity. Redox regulation of hSULT1E1 changes bioactive E2 levels in vivo. This in turn is related to E2-dependent tumorigenesis in the breast, endometrium, and prostate [36, 37].
Abbreviations
- SULT
sulfotransferase
- SULT1E1
estrogen sulfotransferase
- NAC
N-acetyl cysteine
- BSO
buthionine sulfoxamine
- PAPS
adenosine 3′-phosphate 5′-phosphosulfate
- E2
17β-estradiol
- ER
estrogen receptor
- ROS
reactive oxygen species
- and RNS
reactive nitrogen species
Footnotes
This work was supported in part by NIH Grant GM59873 (G.C.) and the Oklahoma Center for the Advancement of Science and Technology (OCAST) grant HR05-015 (G.C.)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Coughtrie MW. Sulfation through the looking glass--recent advances in sulfotransferase research for the curious. Pharmacogenomics J. 2002;2:297–308. doi: 10.1038/sj.tpj.6500117. [DOI] [PubMed] [Google Scholar]
- 2.Duffel MW, Marshal AD, McPhie P, Sharma V, Jakoby WB. Enzymatic aspects of the phenol (aryl) sulfotransferases. Drug Metab Rev. 2001;33:369–95. doi: 10.1081/dmr-120001394. [DOI] [PubMed] [Google Scholar]
- 3.Falany CN. Enzymology of human cytosolic sulfotransferases. FASEB Journal. 1997;11:206–16. doi: 10.1096/fasebj.11.4.9068609. [DOI] [PubMed] [Google Scholar]
- 4.Dooley TP. Cloning of the human phenol sulfotransferase gene family: three genes implicated in the metabolism of catecholamines, thyroid hormones and drugs. Chemico-Biological Interactions. 1998;109:29–41. doi: 10.1016/s0009-2797(97)00118-x. [DOI] [PubMed] [Google Scholar]
- 5.Roy AK, Lavrovsky Y, Song CS, Chen S, Jung MH, Velu NK, et al. Regulation of androgen action. Vitamins & Hormones. 1999;55:309–52. doi: 10.1016/s0083-6729(08)60938-3. [DOI] [PubMed] [Google Scholar]
- 6.Glatt H. Sulfotransferases in the bioactivation of xenobiotics. Chem Biol Interact. 2000;129:141–70. doi: 10.1016/s0009-2797(00)00202-7. [DOI] [PubMed] [Google Scholar]
- 7.Duanmu Z, Weckle A, Koukouritaki SB, Hines RN, Falany JL, Falany CN, et al. Developmental expression of aryl, estrogen, and hydroxysteroid sulfotransferases in pre- and postnatal human liver. J Pharmacol Exp Ther. 2006;316:1310–7. doi: 10.1124/jpet.105.093633. [DOI] [PubMed] [Google Scholar]
- 8.Iwamori M. Estrogen sulfatase. Methods Enzymol. 2005;400:293–302. doi: 10.1016/S0076-6879(05)00017-0. [DOI] [PubMed] [Google Scholar]
- 9.Rahman I. Regulation of glutathione in inflammation and chronic lung diseases. Mutat Res. 2005;579:58–80. doi: 10.1016/j.mrfmmm.2005.02.025. [DOI] [PubMed] [Google Scholar]
- 10.Ghezzi P. Regulation of protein function by glutathionylation. Free Radic Res. 2005;39:573–80. doi: 10.1080/10715760500072172. [DOI] [PubMed] [Google Scholar]
- 11.Chen FC, Ogut O. Decline of contractility during ischemia-reperfusion injury: actin glutathionylation and its effect on allosteric interaction with tropomyosin. Am J Physiol Cell Physiol. 2006;290:C719–27. doi: 10.1152/ajpcell.00419.2005. [DOI] [PubMed] [Google Scholar]
- 12.Aracena P, Tang W, Hamilton SL, Hidalgo C. Effects of S-glutathionylation and S-nitrosylation on calmodulin binding to triads and FKBP12 binding to type 1 calcium release channels. Antioxid Redox Signal. 2005;7:870–81. doi: 10.1089/ars.2005.7.870. [DOI] [PubMed] [Google Scholar]
- 13.Niedowicz DM, Daleke DL. The role of oxidative stress in diabetic complications. Cell Biochem Biophys. 2005;43:289–330. doi: 10.1385/CBB:43:2:289. [DOI] [PubMed] [Google Scholar]
- 14.Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J Nutr. 2004;134:489–92. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- 15.Cross JV, Templeton DJ. Oxidative stress inhibits MEKK1 by site-specific glutathionylation in the ATP-binding domain. Biochem J. 2004;381:675–83. doi: 10.1042/BJ20040591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Di Stefano A, Frosali S, Leonini A, Ettorre A, Priora R, Di Simplicio FC, et al. GSH depletion, protein S-glutathionylation and mitochondrial transmembrane potential hyperpolarization are early events in initiation of cell death induced by a mixture of isothiazolinones in HL60 cells. Biochim Biophys Acta. 2006;1763:214–25. doi: 10.1016/j.bbamcr.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 17.Dixon DP, Fordham-Skelton AP, Edwards R. Redox regulation of a soybean tyrosine-specific protein phosphatase. Biochemistry. 2005;44:7696–703. doi: 10.1021/bi047324a. [DOI] [PubMed] [Google Scholar]
- 18.Marshall AD, McPhie P, Jakoby WB. Redox control of aryl sulfotransferase specificity. Archives of Biochemistry & Biophysics. 2000;382:95–104. doi: 10.1006/abbi.2000.2020. [DOI] [PubMed] [Google Scholar]
- 19.Maiti S, Dutta SM, Baker SM, Zhang J, Narasaraju T, Liu L, et al. In vivo and in vitro oxidative regulation of rat aryl sulfotransferase IV (AST IV) J Biochem Mol Toxicol. 2005;19:109–18. doi: 10.1002/jbt.20064. [DOI] [PubMed] [Google Scholar]
- 20.Maiti S, Grant S, Baker SM, Karanth S, Pope CN, Chen G. Stress regulation of sulfotransferases in male rat liver. Biochem Biophys Res Commun. 2004;323:235–41. doi: 10.1016/j.bbrc.2004.08.074. [DOI] [PubMed] [Google Scholar]
- 21.Maiti S, Chen X, Chen G. All-trans retinoic acid induction of sulfotransferases. Basic Clin Pharmacol Toxicol. 2005;96:44–53. doi: 10.1111/j.1742-7843.2005.pto960107.x. [DOI] [PubMed] [Google Scholar]
- 22.Chen X, Baker SM, Chen G. Methotrexate induction of human sulfotransferases in Hep G2 and Caco-2 cells. J Appl Toxicol. 2005;28:28. doi: 10.1002/jat.1071. [DOI] [PubMed] [Google Scholar]
- 23.Maiti S, Chen G. Methotrexate is a novel inducer of rat liver and intestinal sulfotransferases. Arch Biochem Biophys. 2003;418:161–8. doi: 10.1016/j.abb.2003.08.019. [DOI] [PubMed] [Google Scholar]
- 24.Maiti S, Chen G. Tamoxifen induction of aryl sulfotransferase and hydroxysteroid sulfotransferase in male and female rat liver and intestine. Drug Metab Dispos. 2003;31:637–44. doi: 10.1124/dmd.31.5.637. [DOI] [PubMed] [Google Scholar]
- 25.Shen DX, Shi X, Fu JL, Zhang YM, Zhou ZC. The role of thiol reduction in hydroquinone-induced apoptosis in HEK293 cells. Chem Biol Interact. 2003;145:225–33. doi: 10.1016/s0009-2797(03)00003-6. [DOI] [PubMed] [Google Scholar]
- 26.Falany CN, Krasnykh V, Falany JL. Bacterial expression and characterization of a cDNA for human liver estrogen sulfotransferase. Journal of Steroid Biochemistry & Molecular Biology. 1995;52:529–39. doi: 10.1016/0960-0760(95)00015-r. [DOI] [PubMed] [Google Scholar]
- 27.Chen G, Chen X. Arginine residues in the active site of human phenol sulfotransferase (SULT1A1) J Biol Chem. 2003;16:16. doi: 10.1074/jbc.M306045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirano S, Kobayashi Y, Cui X, Kanno S, Hayakawa T, Shraim A. The accumulation and toxicity of methylated arsenicals in endothelial cells: important roles of thiol compounds. Toxicol Appl Pharmacol. 2004;198:458–67. doi: 10.1016/j.taap.2003.10.023. [DOI] [PubMed] [Google Scholar]
- 29.Falany CN, Zhuang W, Falany JL. Characterization of expressed human phenol-sulfating phenol sulfotransferase: effect of mutating cys70 on activity and thermostability. Chemico-Biological Interactions. 1994;92:57–66. doi: 10.1016/0009-2797(94)90053-1. [DOI] [PubMed] [Google Scholar]
- 30.Nakayama T, Tanabe H, Deyashiki Y, Shinoda M, Hara A, Sawada H. Chemical modification of cysteinyl, lysyl and histidyl residues of mouse liver 17 beta-hydroxysteroid dehydrogenase. Biochim Biophys Acta. 1992;1120:144–50. doi: 10.1016/0167-4838(92)90262-c. [DOI] [PubMed] [Google Scholar]
- 31.Inano H. Chemical modification of lysine residues at active-site of human placental estradiol 17 beta-dehydrogenase. Biochem Biophys Res Commun. 1988;152:789–93. doi: 10.1016/s0006-291x(88)80107-4. [DOI] [PubMed] [Google Scholar]
- 32.Garban HJ, Marquez-Garban DC, Pietras RJ, Ignarro LJ. Rapid nitric oxide-mediated S-nitrosylation of estrogen receptor: regulation of estrogen-dependent gene transcription. Proc Natl Acad Sci U S A. 2005;102:2632–6. doi: 10.1073/pnas.0409854102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–8. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 34.Nahoum V, Gangloff A, Shi R, Lin SX. How estrogen-specific proteins discriminate estrogens from androgens: a common steroid binding site architecture. Faseb J. 2003;17:1334–6. doi: 10.1096/fj.02-0524fje. [DOI] [PubMed] [Google Scholar]
- 35.Wang M, Tsai BM, Reiger KM, Brown JW, Meldrum DR. 17-beta-Estradiol decreases p38 MAPK-mediated myocardial inflammation and dysfunction following acute ischemia. J Mol Cell Cardiol. 2006;40:205–12. doi: 10.1016/j.yjmcc.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 36.Nakamura Y, Suzuki T, Fukuda T, Ito A, Endo M, Moriya T, et al. Steroid sulfatase and estrogen sulfotransferase in human prostate cancer. Prostate. 2006 doi: 10.1002/pros.20426. [DOI] [PubMed] [Google Scholar]
- 37.Suzuki T, Miki Y, Nakamura Y, Moriya T, Ito K, Ohuchi N, et al. Sex steroid-producing enzymes in human breast cancer. Endocr Relat Cancer. 2005;12:701–20. doi: 10.1677/erc.1.00834. [DOI] [PubMed] [Google Scholar]