

Abstract
Human Immunodeficiency Virus type 1 (HIV-1) Vpr is known to dysregulate host cellular functions through its interaction with cellular proteins. Using a protein array we assessed Vpr-mediated differential regulation of host cellular proteins expression. Results demonstrated that Vpr differentially regulated host factors that are involved in functions such as cell proliferation, differentiation and apoptosis. One of the most highly downregulated proteins attained was the sodium hydrogen exchanger, isoform 1 (NHE1), which showed a significant (60%) decrease in HIV-1 vpr(+) virus infected cells as compared to HIV-1 vpr(−) virus infected control. NHE1 downregulation further led to acidification of cells and was directly correlated with loss of ezrin, radixin and moesin (ERM) protein complex and decreased AKT phosphorylation. Vpr-mediated NHE1 dyregulation is in part through GR pathway as GR antagonist, mifepristone reversed Vpr-induced NHE1 downregulation.
Introduction
HIV-1 infects and destroys several target cell types including cells of the immune and nervous systems leading to overt diseases (Levy, 2006). It has been hypothesized that the cytopathic effects resulting from viral infection may be partly due to the interaction between virally encoded proteins and host cell proteins through direct and/or indirect mechanisms (Alimonti et al., 2003; Amendola et al., 1996). The cytopathic effects noted with HIV-1 infection have been linked to several viral proteins including Env (gp120), Tat, Nef and Vpr (Azad, 2000; Gibellini et al., 2005; Moon and Yang, 2006; Perfettini et al., 2005; Rasola et al., 2001). HIV-1 vpr gene encodes a protein of 96 amino acids with a predicted molecular weight of 14 kDa, and is conserved in HIV and Simian Immunodeficiency Virus (SIV) (Cohen et al., 1990). One of the characteristic features of Vpr is its association with virus particles through the interaction with p6 domain of HIV-1 Gag (Paxton et al., 1993) and present in multiple forms (cell-associated, virion-associated and free Vpr) within the infected milieu (Tungaturthi et al., 2003).
Vpr is a pleiotropic protein with diverse functions including cell cycle arrest at the G2/M phase, apoptosis, nuclear import of the preintegration complex, transcriptional activation, and interactions with viral and several cellular proteins (Bukrinsky and Adzhubei, 1999; Kino and Pavlakis, 2004; Mahalingam et al., 1997). Vpr is known to induce apoptosis through regulation of cell cycle arrest (Azuma et al., 2006; Stewart et al., 1997), mitochondrial dysfunction (Arunagiri et al., 1997; Roumier et al., 2002), Bcl-2 family members (Jacotot et al., 2001), DNA repair enzymes (Andersen et al., 2005), and ion channels (Piller et al., 1996). However, it is not clear whether Vpr acts at these multiple levels independently or these molecules are regulated sequentially via a common pathway. To gain a better understanding of the host cellular pathways involved in Vpr-induced apoptosis, we have employed an antibody protein microarray analysis of PBMCs infected with HIV-1 either with or without the expression of Vpr. Results indicate that Vpr targets several apoptotic regulatory proteins that are in the nuclear, cytoplasmic and cell membrane compartment. Many of the identified proteins in the intracellular compartments (cytoplasmic and nuclear) are previously shown to have a role in apoptosis, whereas NHE1, a membrane bound protein, was a new candidate. Though Vpr exposure of cells to Vpr leads to cell shrinkage followed by cell death, pathways involved in membrane associated proteins and Vpr mediated apoptosis is unknown, thus the present study focuses on the effect of Vpr on NHE1 and its subsequent role in cell death.
NHE1 is a member of sodium hydrogen exchanger family (Fliegel, 2005). Sodium hydrogen exchangers’ function at the cell membrane to exchange intracellular hydrogen ions (H+) generated during cellular metabolic processes for extracellular sodium ions (Na+). In addition to maintain the balance of these two ions, NHE1 also maintains both the intracellular pH and cell volume at homeostatic levels. Reduced capacity of NHE1 to perform either of these functions has been shown to induce cellular apoptosis. Fluctuations in intracellular pH mediated by NHE1 activity have also been linked to cell cycle control, especially at the G2 phase (Putney and Barber, 2003). Recently, (Wu et al., 2004) has discovered a role of NHE1 in maintaining cell survival, which is separate from its Na+/H+ exchange capacity. Thus, given the dual role of NHE1 as an anti-apoptotic protein, and a cell cycle regulator, a reduction of NHE1, might be expected to lead to induction of host cell apoptosis. The goal of this study is to confirm whether the downregulation of NHE1 is at the transcriptional level or at the translational level, and to determine if NHE1 downregulation is associated with loss of anti-apoptotic properties of NHE1. Results indicate that Vpr specifically downregulated NHE1, and this correlates with altered intracellular pH, ERM complex and Akt phosphorylation. Together, these results present one of the potential signaling pathway(s) contributing to the induction of apoptosis by HIV-1 Vpr.
Materials and Methods
Cells and Transfection
Blood from HIV-1-negative, healthy donors was used to isolate peripheral blood mononuclear cells (PBMCs) by Ficoll-Hypaque (Pharmacia) gradient centrifugation. Purified PBMCs were resuspended in RPMI 1640 supplemented with 10% FCS, stimulated with phytohemoagglutinin (PHA) (5μg/ml) for 3 days, and cultured in IL-2 (5U/ml) containing medium. HEK293 cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS), 1% L-glutamine, and 1% Penicillin/Streptomycin. CEMx174 cells were maintained in RPMI-1640 supplemented with 10% fetal bovine serum (FBS), 1% L-glutamine, and 1% Penicillin/Streptomycin. Monocyte-derived macrophages were isolated from PBMCs by adhesion as described (Collman et al., 1989). Briefly, PBMCs were resuspended in DMEM supplemented with 1% Penicillin-Streptomycin and 1% L-Glutamine without serum and were plated for two hours at 37°C, to allow the monocytes to adhere to the plates. Two-hours postincubation, non-adherent cells were removed and the adherent cells were washed twice with phosphate buffered saline (PBS). DMEM supplemented with 10% FBS, 10% human sera, 1% Penicillin-Streptomycin, 1% L-Glutamine, 500U/mL GM-CSF (granulocyte-macrophage colony stimulating factor) (Berlex Labs, Richmond, California), and 15ng/mL M-CSF (macrophage colony stimulating factor) (Amgen, Thousand Oaks, California) were added to the cells and cultured for 7 days. Half the media was replaced with an equal quantity of fresh media containing cytokines every three days.
Plasmids and Virus infection
HIV-1 proviral DNAs 89.6wt (wild type with an intact vpr gene), and 89.6Δvpr (containing a frame shift mutation in the Vpr coding sequence to eliminate its expression) were constructed as described (Balliet et al., 1994). HEK-293T cells (2×106 per plate) were cotransfected with 15μg of HIV-1 proviral constructs by the calcium phosphate precipitation method (Majumder et al., 2005). Cell free supernatant was collected 72 hours posttransfection and assayed for virus production by p24 ELISA. Number of infectious particles was determined by titration in TZM cell line as described (Li et al., 2005). PHA-stimulated normal human PBMCs (10 ×107) were infected with viruses containing 100ng p24 antigen equivalent of 89.6wt or 89.6Δvpr virus stocks as described (Balliet et al., 1994). Cells (as indicated per experiment) were infected at an MOI of 0.5. Infectivity (%) was determined by intracellular p24 staining (Beckman Coulter, p24 Clone KI57) and analyzed by flow cytometry. Noninfectious virus particles were generated by treatment with AT-2 as described (Rossio et al., 1998). Inactivation was confirmed by lack of infectivity on the TZM indicator cell line as described above.
Antibody microarray and data analysis
In an effort to detect changes induced by Vpr in host cellular protein expression, we performed antibody array using Ab Microarray 380 slides purchased from BD Biosciences, USA, following the manufacturer’s protocol. Briefly, normal human PBMCs were infected with 89.6wt and 89.6vpr- viruses as described above. Cell lysate preparation, protein labeling with Cy5 and Cy3, hybridization, and scanning were performed according to the manufacturer’s protocol. Labeled proteins were added to the slides and hybridization was carried out for 30 minutes at room temperature (25–27°C). Data analysis was done using GenePixPro software and expression ratios were calculated using the Microsoft Excel template provided by the manufacturer. The Internally Normalized Ratio (INR) corrects for differences in the reverse color labeling and is calculated as follows: INR =√ (Ratio 1/Ratio 2), where Ratio 1 is sample A Cy5/sample B Cy3 and Ratio 2 is sample B Cy5/sample A Cy3. An INR >1.3 or <0.7 is considered by the manufacturer to be significant. Protein that are significantly regulated were subjected to multiple pathway builder software (Pathway Assist, ariadnegenomics; Ingenuity Pathways Knowledge Base, Ingenuity Systems) that are licensed by the University of Pittsburgh and/or demo versions that are available through their web sites.
Western blot
Infected PBMCs were lysed in lysis buffer containing; 1M Tris pH 7.5, 5M NaCl, 10% Triton, 100mM Na3VO4, 1M NaF, 50mM PMSF, Aprotanin, Pepstatin, Leupeptin, and Chymostatin. Equivalent amount of protein was electrophoresed on a 12% sodium dodecyl sulfate- polyacrylamide gel (SDS-PAGE), transferred and immunoblotted with the following primary antibodies as per manufacturer’s suggested protocol: mouse monoclonal anti-NHE1 (1:250, BD Transduction Laboratories, California), rabbit polyclonal anti-Vpr (1:500, kind gift of John Kappes, University of Alabama), anti-Gag (1:500, NIH-RRRP), anti-alpha tubulin (1:500, NeoMarkers), anti-Akt-P (serine 473, 1:250, Cell Signaling), and anti-Akt (1:250, Cell Signaling). After washing three times in Tris-buffered saline-Tween-20 (TBS-T), membranes were incubated 1 hour. at room temperature in appropriate goat anti-mouse or anti-rabbit HRP-conjugated secondary antibodies (1:5000, Caltag). Following 5 washes in TBS-T, antibody bound proteins were detected by autoradiography using ECL system.
Immunofluorescence
HeLa-CD4 cells were infected with HIV-1 vpr(+)/EGFP or HIV-1 vpr(−)/EGFP reporter virus at an MOI of 0.1. Twenty-four hours post infection cells were fixed with 3.7% methanol-free paraformaldehyde for 15 minutes at room temperature. After washing 3 times with PBS, cells were incubated with anti-ERM antibody (1:100 dilution; Cell signaling) at 4°C in a humidified chamber for 16 hours. Primary antibody was washed three times with PBS and then incubated with anti-rabbit RRX (Jackson Immunochemicals) to detect ERM. Following several washes with PBS, cells were mounted with Vectashield containing DAPI (Vector Laboratories, CA). Confocal microscopy was performed using a Leica TCS NT confocal tri-laser scanning inverted microscope (Wetzlar, Germany) at the center for biological imaging facility at University of Pittsburgh.
Quantitative RT-PCR analysis
To confirm the expression ratios seen with antibody array, the mRNA levels were quantitated by real-time RT-PCR. RNA was extracted from infected PBMCs using Qiagen RNeasy mini kit as per manufacturer’s instructions, with optional on-column DNase digestion. RNA integrity and concentration were assessed by agarose gel electrophoresis and spectrophotometry. RNA (1μg per triplicate reaction) was reverse transcribed to cDNA using the Taqman Gold Reverse Transcription kit (Applied Biosystems). Real-time RT-PCR was carried out using 2X Taqman Universal Master Mix without UNG, and NHE1 specific primer and probe mix. Data were collected on an ABI PRISM 7000 and analyzed via Sequence Detector v1.1 software. All values were normalized to the endogenous control RPLPO to control for variation. Assays were performed in triplicate and average threshold cycle (CT) values were used to determine relative concentration differences based on the ΔΔCT method of relative quantitation described in the manufacturer’s protocol.
HIV-1 p24 staining
To determine the percent of infection, cells were washed and fixed in 2% paraformaldehyde for 1 hour at 4°C. Following fixation, cells were permeabilized for intracellular staining through incubation in a buffer containing 0.1% saponin (Sigma) in PBS at room temperature for 10 minutes. Intracellular staining for p24 antigen was carried out by the addition of 2.5μl of anti-p24-FITC. Two hours post staining, cells were washed twice in permeabilization buffer, resuspended in PBS containing 5% FBS, and analyzed on a Beckman Coulter XL-MCL flow cytometer and data were analyzed using CellQuest software.
Intracellular pH staining by flow cytometry
The pH sensitive dye 5-(and-6)-carboxy SNARF-1, acetoxymethyl ester acetate was used to determine intracellular pH. SNARF-1 is excited at one wavelength and emits at two wavelengths, with the ratio of fluorescent intensity between the two wavelengths, varying with pH. Stock solution of SNARF-1 was made at 10mM in cell culture grade DMSO, and diluted to 10μM in PBS. Cells to be stained (1×106) were washed twice in PBS and resuspended in SNARF-1. Dye loading was carried out at room temperature for 30 minutes in the dark. Cells were washed twice, resuspended in 1ml PBS and immediately analyzed on a Beckman-Coulter XL-MCL using System II. Dye was excited at 488nm and fluorescence was measured using 575BP and 645DLP filters. In order to generate calibration curves for each cell type used, uninfected PBMCs, CEMx174 or Jurkat cells were stained as described above. Following staining, cells were incubated in high K+ buffers of known pH (range 6.0–9.0) as suggested by Bond and Varley (2005). Nigericin (2μg/ml) was added to the buffers in order to equilibrate the intracellular pH to the extracellular buffer pH. Flow cytometry was performed as described above. A graph of input pH versus fluorescence ratio was generated and a third order polynomial best-fit curve was generated. The equation of the best-fit line was used to compute changes in pH from change in mean fluorescent intensity. Values reported are the mean of four independent experiments.
Statistical analysis
Results were expressed as mean ± SEM. Data were analyzed using student t test for normally distributed data with equal variances and p <0.02 was considered as significant.
Results
Antibody array reveals changes at the protein level induced by Vpr
Previous work from our laboratory indicated that HIV-1 Vpr induced changes in host cellular transcription (Janket et al., 2004). Based on these findings, we rationalized that the changes at the transcriptional level could directly alter the protein expression of the same genes and/or indirectly alter the expression of other proteins that are involved in either the downstream or upstream cellular events. In order to determine the cellular pathways that are potentially affected by Vpr, we performed an antibody array, which consists of antibodies corresponding to 378 human cellular proteins spotted in duplicate. Cell lysates prepared from PBMCs infected with HIV-1 vpr(+) or HIV-1 vpr(−) virus was subjected to antibody microarray according to the manufacturer’s suggested instructions and the results presented in Table 1. Average R/R and average internally normalized ratio (INR) were calculated using internal normalization controls to improve the stringency and quality of the data. Average R/R represents the raw ratio of the wt versus the vpr- virus, averaged over both sets of data (each sample was labeled and run twice). INR represents corrected for differences in the way proteins bound the dyes, Cy3 and Cy5. In order to generate the INR, samples were labeled twice, once with Cy3 and once with Cy5. The Cy3/Cy5 ratio and the Cy5/Cy3 ratio of each sample were used to correct the sample ratio. In this report we considered only INR values >2.00 and <0.6 for upregulated and downregulated protein expression, respectively, to increase the confidence level. Results indicate that of the 378 proteins tested, 16 proteins showed upregulation at a ratio greater than 2 fold (data not shown). Conversely, only six proteins of the 378 tested showed downregulation at a ratio <0.6 (Table 1), representing diverse signaling families. In order to further determine the relevance of these proteins in cellular signaling, the data obtained from this protein array was used as input in several pathway prediction software models.
Table 1.
Host cellular proteins downregulated by HIV-1 Vpr:
| Ab-Ag | Average R/R | Average INR | Accession # |
|---|---|---|---|
| PARP | 0.1428 | 0.3779 | P09874 |
| NHE1 | 0.1718 | 0.4145 | P19634 |
| Dynamin 1 | 0.3359 | 0.5795 | Q05193 |
| P63 | 0.3384 | 0.5817 | Q9UBV9 |
| WT1 | 0.3834 | 0.6192 | P19544 |
| CDC34 | 0.3849 | 0.6204 | P49427 |
| IL-13 | 0.3921 | 0.6262 | P35225 |
Normal human PBMCs infected with HIV-1 vpr(+) or HIV-1 vpr(−) virus were lysed, and host cellular protein expression level was analyzed using the Ab Microarray as described in methods. R/R represents the raw ratio of protein expression in the HIV-1 vpr(+) versus HIV-1 vpr(−) virus infected samples. INR is the ratio of expression corrected for differences in the ability of individual proteins to bind the Cy3 and Cy5 dyes.
The wide availability of array data has produced the need for powerful prediction software packages to generate relevant models from the lists generated by arrays. Using such software, disjointed lists of unrelated genes may be conjugated into useful models for future studies. Following such rationale, we utilized several pathway prediction models (Pathway Assist, Ingenuity Pathway Knowledge Base software, etc) to assess the potential signaling pathways mediated by HIV-1 Vpr. Pathway analysis results indicate that protein upregulated by HIV-1 Vpr modulated several cell cycle, apoptosis, cancer-related molecules, and transcription factors (data not shown). However, more interestingly, all the downregulated proteins indicated a role for Vpr in apoptosis with distinct subcellular distribution pattern (Fig. 1). Here, we chose to focus on the effect of Vpr on the NHE1 due to its unique localization pattern and its role in apoptosis (Putney et al., 2002; Schneider et al., 2004).
Figure 1.
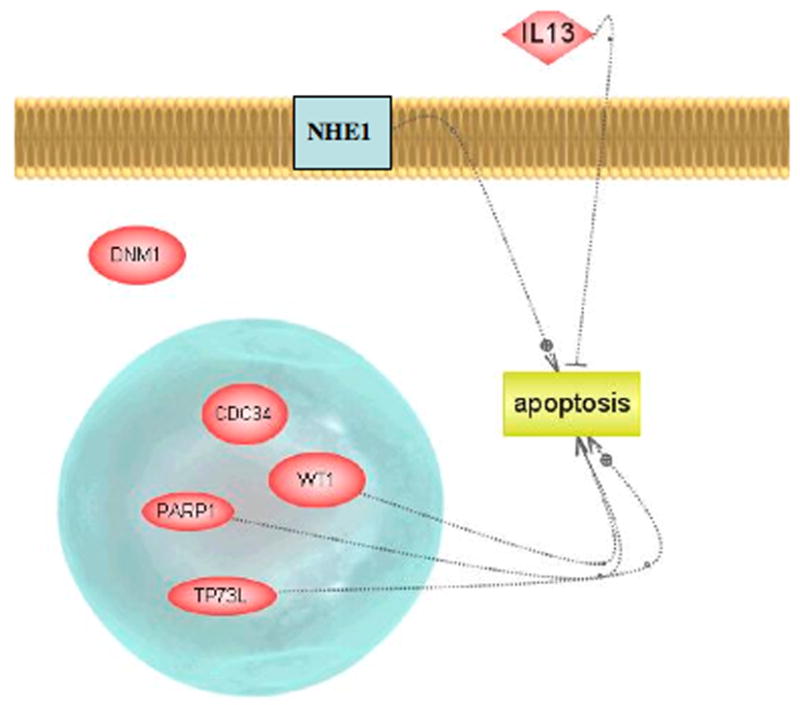
Pathway analysis depicting cellular proteins down regulated by HIV-1 Vpr: Role in apoptosis.
NHE1 is specifically downregulated by HIV-1 Vpr
NHE1 was found to be downregulated by a ratio of <0.41 using antibody array analysis. Results from the antibody array suggested that downregulation of NHE1 was specific, as NHE3 (isoform 3) was not affected by Vpr (ratio 1.01). In order to confirm the specific downregulation of NHE1 by Vpr, we employed western blot using an NHE1-specific antibody. PBMCs isolated from healthy donors were stimulated for three days followed by infection with HIV-1 vpr(+) or HIV-1 vpr(−) virus. Seventy-two hours post infection cells were assessed for percent infection by intracellular staining for p24 antigen and flow cytometry. A similar percentage of infection was observed in both HIV-1 vpr(+) and HIV-1 vpr(−) virus infected cells (Fig. 2A). Cells were lysed, and subjected to SDS-PAGE followed by western blot with the specific antibodies noted in Figure 2B. Results indicate that NHE1 is specifically downregulated by Vpr in the context of infection. To further confirm this finding, multiple donor PBMCs were infected with 0.5 and 1.0 MOI of HIV-1 vpr(+) or HIV-1 vpr(−) virus and assessed for NHE1 expression. Results indicate that NHE1 expression was downregulated by Vpr in a dose dependent manner without altering the total cellular protein level (alpha-Tubulin control) in multiple donors (N=6), suggesting that Vpr-mediated down regulation of NHE1 is specific. Given the ability of HIV-1 to infect different cell types within the immune system, we next determined whether Vpr-mediated NHE1 downregulation is cell type specific. Similar analyses were performed in infected macrophages and the cell line CEMx174. NHE1 was found to be downregulated in each of these cells types, in multiple experiments, indicating that Vpr-mediated NHE1 downregulation is not cell type specific.
Figure 2. Confirmation of NHE1 downregulation by western blot.
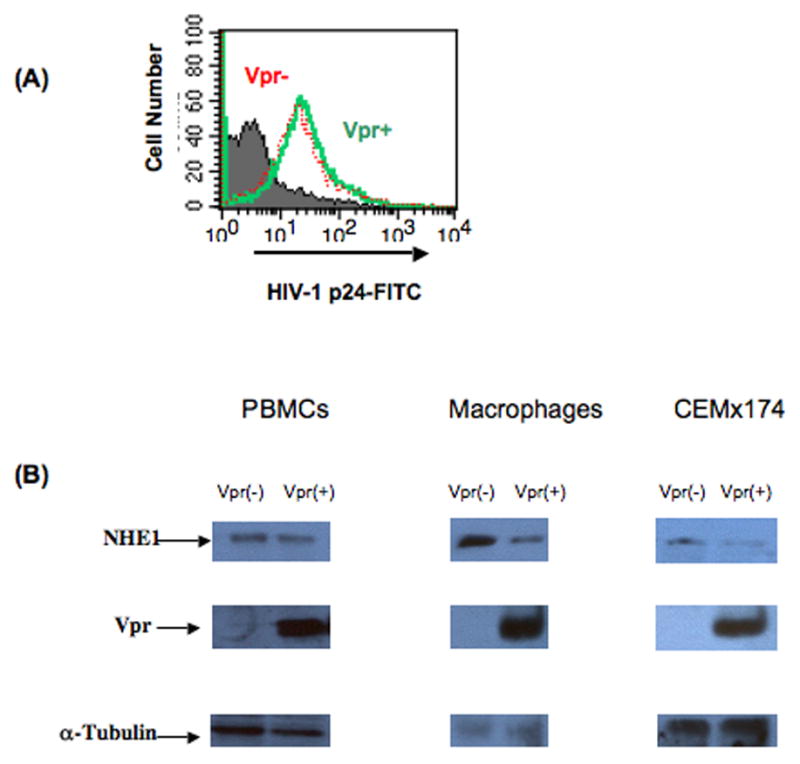
Total PBMCs and macrophages from healthy donors or the cell line, CEM x174, were infected with HIV-1 vpr(+) or HIV-1 vpr(−) virus. (A) Percentage of cells infected by HIV-1 vpr(+) and HIV-1 vpr(−) virus was determined by flow cytometry: PBMCs infected as above were fixed, permeabilized, and stained for intracellular p24 antigen expression using FITC conjugated anti-p24 antibody. Histogram represents the HIV-1 vpr(+) virus infected (green) and HIV-1 vpr(−) virus infected cells (red). Histogram (gray) represents the isotype control (IgG-FITC) in infected cells. (B) Immunoblot analysis of NHE1 in HIV-1 vpr(+) or HIV-1 vpr(−) virus infected cell: Postinfection cells were lysed and subjected to immunoblot with the indicated antibodies. Anti-Vpr and anti-tubulin are shown as controls for Vpr expression and protein loading, respectively. Results are representative of six independent experiments.
Vpr mediated NHE1 downregulation is at the transcriptional level
Previously we have shown that HIV-1 Vpr differentially regulated expression of host cellular genes at the level of transcription (Janket et al., 2004). To assess whether the downregulation of NHE1 is at the transcriptional level, RNA obtained from PBMCs infected with HIV-1 vpr(+) or HIV-1 vpr(−) virus was subjected to real time RT-PCR using NHE1 specific primers and Taqman probes. All values were normalized to RPLPO and calibrated to the HIV-1 vpr(−) virus infected sample using the ΔΔCT method of relative gene quantitation. Keeping HIV-1 vpr(−) virus infected sample as 1, NHE1 was found to be downregulated by a ratio of approximately 0.5 in several donors (N=5). The average and standard deviation results from five donors are shown, indicating that NHE1 was downregulated by Vpr in a statistically significant manner (P<0.005). Similar results were observed in both macrophages and lymphocytes, indicating that downregulation was not cell type specific (data not shown). Taken together, the results of the immunoblot and real time RT-PCR indicate that NHE1 is downregulated by Vpr is at the transcriptional level.
Results presented above indicate that Vpr induces downregulation of NHE1 in the infected cells. In the infected host, Vpr is present in multiple forms; virion associated, de novo expressed, and free Vpr protein. In order to assess the potential effect of various forms of Vpr on NHE1, we further explored whether virion-associated Vpr in the absence of de novo synthesis or free Vpr, could induce similar effects on NHE1. Using noninfectious HIV-1 vpr(−) and HIV-1 vpr(+) virus particles that were generated via treatment with the 2,2′-dithiodipyridine (AT-2), healthy donor PBMCs were treated with 500ng p24 equivalent of inactivated HIV-1 vpr(+) or HIV-1 vpr(−) virus. Similarly recombinant Vpr (rVpr) protein or control Gag protein (rGag) was also used to treat normal donor PBMCs. RNA was extracted from these samples and subjected to real time RT-PCR for NHE1 and results are presented in Figure 3 as a mean of five experiments. Interestingly, we observed NHE1 downregulation by Vpr delivered via AT-2 treated noninfectious particles or as rVpr in a manner similar to that seen in viral infection. These results indicate that the virion associated Vpr or free Vpr was sufficient to induce downregulation of NHE1 in the absence of de novo synthesis of Vpr.
Figure 3. Real time RT-PCR analysis of NHE1 expression.
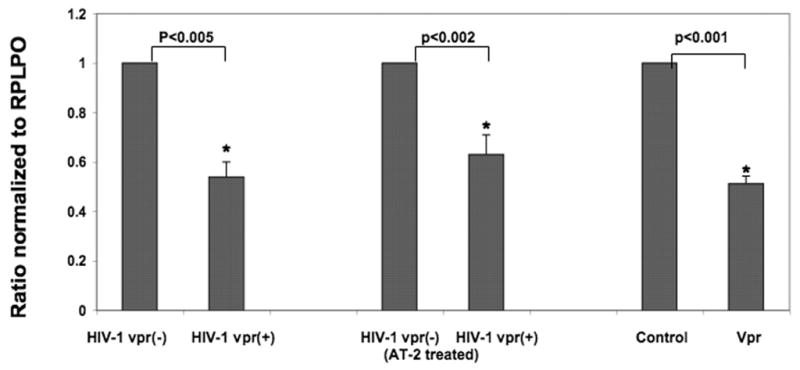
Healthy donor PBMCs infected with replication competent or AT-2 treated HIV-1 vpr(+) or HIV-1 vpr(−) virus or treated with Vpr (rVpr) or rGag (control) protein. RNA extracted from infected and control groups was reverse transcribed to cDNA and real time PCR was carried out using primers and probe specific for NHE1 or RPLPO. Ratios were generated using the ΔΔCT method of relative quantitation and values were normalized to the endogenous control RPLPO within each sample. In each experiment, the results attained in HIV-1 vpr(−) virus infected cells or rGag treated samples were considered as a ratio of 1.0. P values were based on results obtained from multiple experiments (N=5).
Vpr-mediated NHE downregulation results in change in intracellular pH
NHE1 functions at the cell surface, exchanging intracellular H+ ions generated during metabolism with extracellular Na+ ions (Baumgartner et al., 2004). NHE1 thus plays a vital role in maintaining intracellular pH, as well as maintaining the cell volume. Loss of intracellular pH within the tight range considered physiologically acceptable and has been linked to apoptosis (Putney et al., 2002; Schneider et al., 2004). To assess whether Vpr-mediated NHE1 downregulation affects intracellular pH, we performed staining with the pH sensitive dye SNARF-1. Infected PBMCs and Jurkat cells were stained with SNARF-1 as described in Methods. Following the method of Bond and Varley (2005), we performed calibration by the nigericin method of cellular calibration (Fig. 4A). Average change in intracellular pH was computed from the appropriate calibration curves (Fig. 4B). Results show that the presence of Vpr correlates with a decrease in intracellular pH. While the observed change in intracellular pH is small, these results are similar to those seen by other investigators with loss of the NHE1 ion exchange function (Bond and Varley, 2005; Wieder et al., 1993). The tight pH range within which the cell operates, makes even small changes in intracellular pH intolerable for normal cellular functioning. Thus, it was important to decipher in this experiment between change in intracellular pH prior to, and corresponding with, early apoptosis as well as with loss of pH associated with post-apoptotic/necrotic cell death. Thus, cells were stringently gated on live cells via forward and side scatter profile indicating the change in intracellular pH represents that induced by Vpr and not changes expected post-cell death.
Figure 4. Intracellular pH in cells infected with HIV-1 vpr(+) or HIV-1 vpr(−) virus.
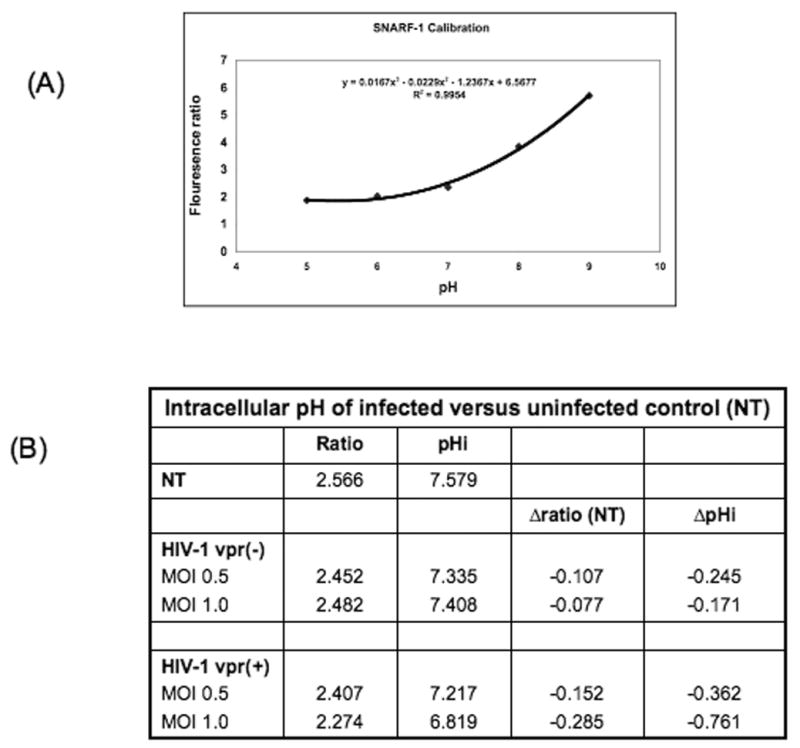
PBMCs infected with HIV-1 vpr(+) or HIV-1 vpr(−) virus were used for measuring the intracellular pH. Cells were loaded with 10μM SNARF-1 in PBS at room temperature for 30 minutes in the dark. SNARF-1 was excited at 488nm and fluorescent intensity assessed at 575 and 645nm. (A) SNARF-1 calibration curve was generated using normal cells (B) Summary of the mean change in fluorescence intensity ratio and intracellular pH in HIV-1 vpr(+) versus HIV-1 vpr(−) virus infected cells (N=4).
Vpr-mediated NHE1 downregulation results in loss of ERM complex and a decreased level of phosphorylation of the pro-survival kinase Akt
Previous studies utilizing mutation analyses have shown that in addition to its ion transport domain, the domain of NHE1 responsible for interaction with the ezrin/radixin/moesin complex (ERM) is necessary for cell survival (Baumgartner et al., 2004). Wu et al. (2004) showed that NHE1 at the cell membrane performs a scaffolding function, recruiting ERM to the cell membrane and facilitating interaction of ERM with the pro-survival kinase, Akt. This interaction resulted in increased phosphorylation and activation of Akt, leading to cell survival. Loss of NHE1 at the cell surface led to decreased Akt phosphorylation and cell death by altering the signaling events regulated by NHE1. Next, we assessed whether the decrease in cellular NHE1 mediated by Vpr affects the ERM complex and the phosphorylation of Akt. One of the pathways NHE1 uses to influence the signaling events is through the scaffolding proteins such as ERM that is complexed with NHE1 and the signaling molecules. HeLa-T4 cells infected with HIV-1 vpr(+)/EGFP or HIV-1 vpr(−)/EGFP reporter virus was immunostained with ERM antibody and assessed for ERM distribution within the infected population by confocal microscopy. Results indicate that in the uninfected and HIV-1 vpr(−) virus infected cells, ERM is localized in the cell membrane indicating distinct cell morphology, whereas in cells infected with HIV-1 vpr(+) virus there is a partial or total loss of ERM staining (Fig. 5A). The loss of ERM within the infected cell was directly correlated with the virus production detected by EGFP reporter expression and time dependent (data not shown) suggesting that reduction in ERM could eventually led to loss of membrane integrity as ERM is known to maintain the actin filament function (Kondo et al., 1997).
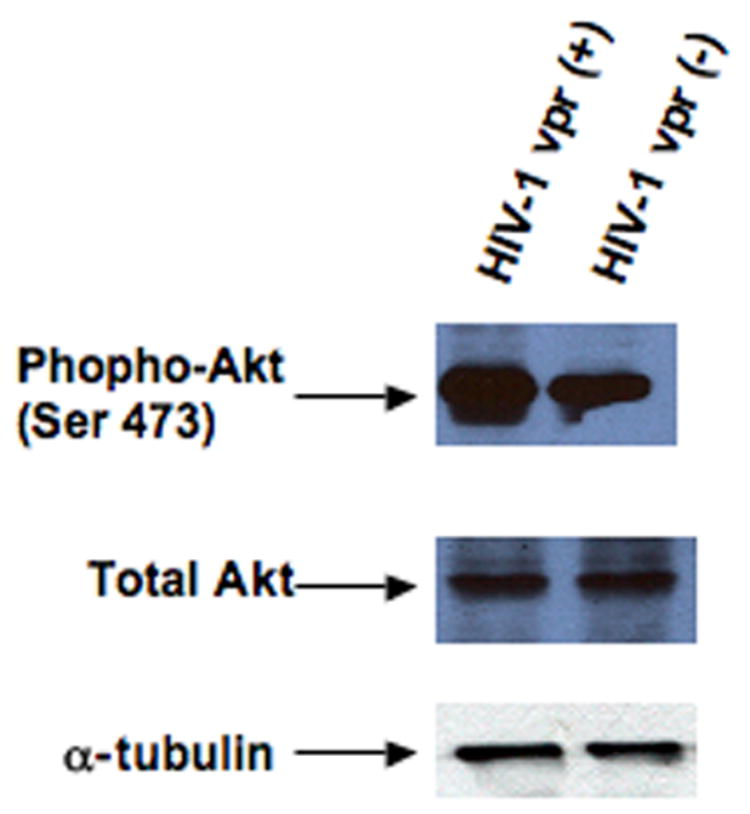
Figure 5. (A) Effect of Vpr on ERM expression in infected cells.
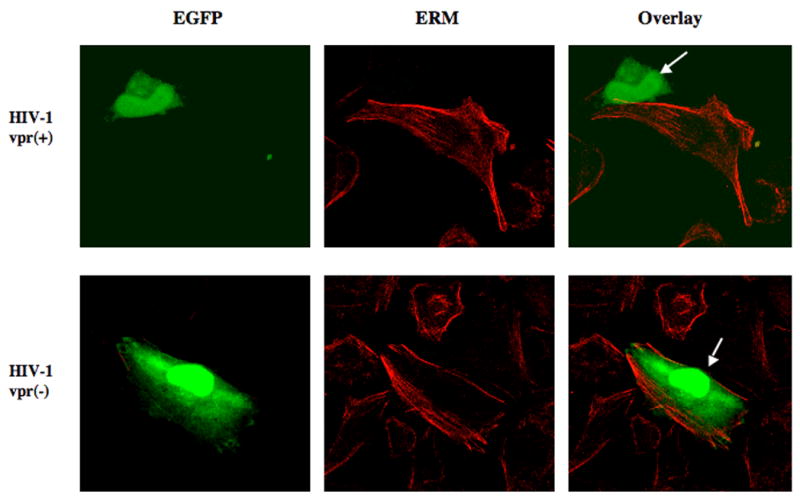
Confocal microscopy images of EGFP-reporter virus infected HeLa-T4 cells expressing ERM. Infected cells were directly visualized by EGFP (Green) and ERM by indirect immunofluorescence (Red). Arrows indicate virus-infected cells. The images are representative of three independent experiments.
(B) Phospho-Akt in HIV-1 vpr(+) and HIV-1 vpr(−) virus infected PBMCs. Cell lysates generated from HIV-1 vpr(+) or HIV-1 vpr(−) virus infected cells were used in immunoblot for phospho-Akt and total Akt. Equivalent amount of protein (20μg) was separated by SDS-PAGE, transferred, and subjected to immunoblot with anti-phospho-Akt (ser 473) or anti-Akt. Alpha tubulin was used as a control for equivalent protein loading. Results are representative of four independent experiments.
Additionally, ERM proteins are also shown to promote cell survival by forming a signal complex with NHE1 and Akt (Wu et al., 2004). Lysates from PBMCs infected with HIV-1 vpr(+) or HIV-1 vpr(−) virus were immunoblotted using antibodies specific for the phosphorylated form of Akt, as well as, total Akt (Fig. 5B). Results indicate that the presence of Vpr correlates with a decreased level of phospho-Akt, whereas, no change was found in total Akt. Additionally, the decrease in phospho-Akt directly correlated with the increased infectivity and/or increased amount of Vpr as measured by increasing MOI of HIV-1 vpr(+) virus, while no change was observed with the highest dose of HIV-1 vpr(−) virus infected cells (data not shown). Similar results were observed in four different donors. A decrease level of NHE1 in correlation with loss of Akt-phosphorylation, further confirms the apoptotic role of Vpr.
HIV-1 Vpr induced NHE1 downregulation could be restored by glucocorticoids antagonist, mifepristone
Previous studies have shown that NHE1 transcription is regulated by the glucocorticoids. Specifically, the binding of glucocorticoids to their receptor, glucocorticoid receptor (GR) causes a decrease in NHE1 at the mRNA level (Muto et al., 2000). Previous studies by us and others have described a physical interaction between Vpr and GR and this Vpr-GR protein complex mediate the transcriptional regulation of genes through GRE (Kino et al., 2002; Thotala et al., 2004). In order to test whether this previously described function of Vpr plays a role in downregulation of NHE1, cells infected with HIV-1 vpr(+) virus or HIV-1 vpr(−) virus were treated with glucocorticoid antagonist, mifepristone, and analyzed for NHE1 level (Fig. 6). Considering no treatemnet group as 1, HIV-1 vpr(+) virus infected cells exhibited a 50% reduction in NHE1 level (0.5 fold), whereas HIV-1 vpr(−) virus infected did not show any difference. However, treatment with 1μM mifepristone increased the NHE1 by 70% and 5μM mifepristone fully restored NHE1 level. Results indicate that mifepristone inhibited Vpr-mediated NHE1 downregulation in a dose dependent manner, suggesting that the downregulation of NHE1 may, in part, be affected through the binding of Vpr to the GR, previously described (Sherman et al., 2000; Thotala et al., 2004). Together these results indicate that Vpr-mediated downregulation of NHE1 could be in part through Vpr-GR interaction.
Figure 6. Vpr mediated NHE1 downregulation is reversed by glucocorticoid antagonist, mifepristone.
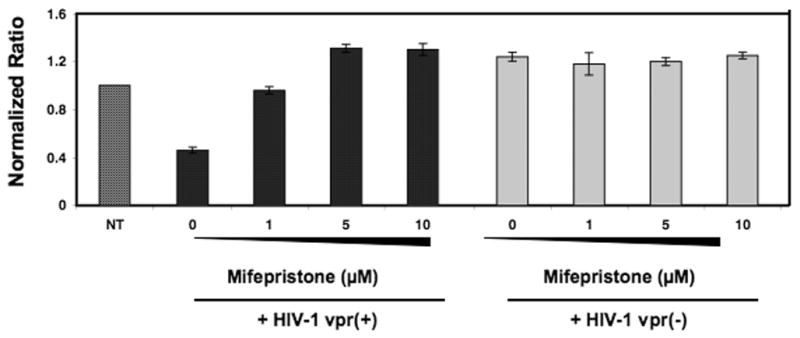
PBMCs were infected with HIV-1 vpr(+) or HIV-1 vpr(−) virus as described, and maintained in the presence or absence of different concentrations of mifepristone. Seventy-two hours post treatment cells were assessed for NHE1 mRNA expression via real-time RT-PCR. Figure represents the mean of three independent experiments and error bars represent standard deviation between experiments.
Discussion
Previous work in our laboratory has described the ability of HIV-1 Vpr to modulate host gene expression at the transcriptional level. Based upon these findings, we explored the effects of altered transcription on the cellular protein profile using the BD antibody array. This array provides a high throughput method for determining changes in known cellular protein levels. Of the 378 proteins that the array is able to detect, we report changes in 20 cellular proteins. Sixteen of these proteins were upregulated at a ratio >2.0, while six were downregulated at a ratio <0.6. Given the small number of proteins found to be regulated, we hypothesized that the diverse functions associated with Vpr may be controlled by convergence of pathways at some upstream mediators and that was further supported by the pathway analysis. We chose NHE1 for further study based on its previously described dual role in control of cell cycle and apoptosis, two functions that have been well described for Vpr.
NHE1 controls the exchange of intracellular hydrogen ions (H+) generated by metabolic processes with extracellular sodium ions (Na+), thereby maintaining intracellular pH and cell volume within physiological limits. Such maintenance has been linked to cell survival and cell cycle control (Putney and Barber, 2003; Rich et al., 2000). Regulation of NHE1 by Vpr would provide an upstream mediator of these two processes, potentially playing a role in cell cycle-dependent apoptosis induced by Vpr. Additionally, NHE1 has been shown to have a role in anti-apoptotic signaling completely separate from its ion channel functions through scaffolding cellular proteins at the plasma membrane (Wu et al., 2004). Thus, downregulation of NHE1 may play a role in apoptosis induction at several levels. This hypothesis was further supported by our results indicating that HIV-1 vpr(+) virus infected cells exhibited loss of ERM and decreased Akt phosphorylation. HIV-1 infection as well as viral protein (gp120) is known to induce loss and/or polarization of cytoskeleton protein and cell shrinkage (Matarrese and Malorni, 2005). Here we have shown that HIV-1 Vpr might also playa role in alteration in cell shape and maintenance which results in cell death either directly or through indirect mechanisms. These events could potentially initiate apoptosis that results in decreased level of prosurvival kinase, Akt phosphorylation through NHE1.
Results attained in this study indicate that Vpr-mediated downregulation of NHE1 is specific, as no regulation of closely related NHE3 expression was seen in the protein array. In addition, NHE1 downregulation was found to occur at the level of transcription. These results are not surprising, given our previous studies linking Vpr to changes in host cellular gene transcription (Janket et al., 2004). Utilizing AT-2 inactivated HIV-1 vpr(+) and HIV-1 vpr(−) we found that virion-associated Vpr also downregulated NHE1. These studies might explain the loss of bystander cells that are exposed to virus particle in the absence of productive infection. It has been documented that only <1% of the virus particles produced by the target cells are infectious, indicating that noninfectious particles might as well contribute to pathogenesis due to the presence of virion associated viral proteins. In support of this, studies by Benos et al. (1994) have shown that binding of gp120 to the cell surface stimulated the ion channel activities of NHE1 in astrocytes. Similarly, HIV-1 vpr and vpu gene products in the absence of other viral proteins are known to induce ion channel activities (Ewart et al., 1996; Piller et al., 1999; Schubert et al., 1996). Based on these observations it is possible to predict that HIV-1 Env, Vpu and Vpr might work in an additive manner in dysregulating the ion channel activities of bystander cells. These results in part might explain the bystander apoptosis observed in vivo. Furthermore ability of Vpr to regulate ion channels to induce apoptosis might have great impact on Vpr mediated neuronal dysregulation in HIV-1 dementia.
Though Vpu and Env have been implicated in altering ion channel, the involvement of host cellular proteins are not established, whereas Vpr is known to dysregulate NHE1. However, the mechanism(s) by which Vpr alters the NHE1 level is not fully understood. Based on the results on Vpr induced downregulation of NHE1 transcripts, it is possible to predict that Vpr might act as a transcriptional regulator of NHE1 through its interaction with transcription factors. Other stimulants, such as glucocorticoids, also stimulate the ion channel activity of NHE1 at short time intervals (3 hours), but eventually induce downregulation of NHE1 at the transcriptional level. Because Vpr does mimic the effects of glucocorticoids, regulation of NHE1 may be in part through GR mediated pathway, thus contributing to its transcriptional regulation. Results attained in this study suggest that Vpr regulation of NHE1 may present one of the potential mechanism(s) underlying Vpr-mediated apoptosis. In particular, it appears that NHE1 downregulation may be mediated through the action of Vpr on the glucocorticoid receptor. Restoration of NHE1 by glucocorticoid antagonist, mifepristone further confirms the involvement of Vpr-GR in NHE1 regulation. Further studies are necessary to fully delineate the mechanism(s) by which Vpr mediates NHE1 downregulation and its role in apoptosis. Understanding the viral-host cellular protein interactions and the cellular pathways regulated by this interaction is necessary for development of additional antiviral not only to block virus replication, but to restore host cell functions and antiviral activity.
Acknowledgments
We thank Dr. Ronald Collman, University of Pennsylvania for his kind gift of 89.6 wt and 89.Δvpr constructs. TZM-bl cells were obtained from Dr. John Kappes through AIDS RRRP, NIH. We thank Dr. John Kappes (University of Alabama at Birmingham) for anti-Vpr antibody. This work was supported by grants AI50463 from NIAID, NIH to V.A. We thank Dr. Charles R. Rinaldo for the use of his flow facility, Dr. Anusman Chattopadhyay, for pathway analysis, and Dr. Simon Watkins for the use of confocal microscopy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alimonti JB, Ball TB, Fowke KR. Mechanisms of CD4+ T lymphocyte cell death in human immunodeficiency virus infection and AIDS. J Gen Virol. 2003;84(Pt 7):1649–61. doi: 10.1099/vir.0.19110-0. [DOI] [PubMed] [Google Scholar]
- Amendola A, Gougeon ML, Poccia F, Bondurand A, Fesus L, Piacentini M. Induction of “tissue” transglutaminase in HIV pathogenesis: evidence for high rate of apoptosis of CD4+ T lymphocytes and accessory cells in lymphoid tissues. Proc Natl Acad Sci U S A. 1996;93(20):11057–62. doi: 10.1073/pnas.93.20.11057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen JL, Zimmerman ES, DeHart JL, Murala S, Ardon O, Blackett J, Chen J, Planelles V. ATR and GADD45alpha mediate HIV-1 Vpr-induced apoptosis. Cell Death Differ. 2005;12(4):326–34. doi: 10.1038/sj.cdd.4401565. [DOI] [PubMed] [Google Scholar]
- Arunagiri C, Macreadie I, Hewish D, Azad A. A C-terminal domain of HIV-1 accessory protein Vpr is involved in penetration, mitochondrial dysfunction and apoptosis of human CD4+ lymphocytes. Apoptosis. 1997;2(1):69–76. doi: 10.1023/a:1026487609215. [DOI] [PubMed] [Google Scholar]
- Azad AA. Could Nef and Vpr proteins contribute to disease progression by promoting depletion of bystander cells and prolonged survival of HIV-infected cells? Biochem Biophys Res Commun. 2000;267(3):677–85. doi: 10.1006/bbrc.1999.1708. [DOI] [PubMed] [Google Scholar]
- Azuma A, Matsuo A, Suzuki T, Kurosawa T, Zhang X, Aida Y. Human immunodeficiency virus type 1 Vpr induces cell cycle arrest at the G(1) phase and apoptosis via disruption of mitochondrial function in rodent cells. Microbes Infect. 2006;8(3):670–9. doi: 10.1016/j.micinf.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Balliet JW, Kolson DL, Eiger G, Kim FM, McGann KA, Srinivasan A, Collman R. Distinct effects in primary macrophages and lymphocytes of the human immunodeficiency virus type 1 accessory genes vpr, vpu, and nef: mutational analysis of a primary HIV-1 isolate. Virology. 1994;200(2):623–31. doi: 10.1006/viro.1994.1225. [DOI] [PubMed] [Google Scholar]
- Baumgartner M, Patel H, Barber DL. Na(+)/H(+) exchanger NHE1 as plasma membrane scaffold in the assembly of signaling complexes. Am J Physiol Cell Physiol. 2004;287(4):C844–50. doi: 10.1152/ajpcell.00094.2004. [DOI] [PubMed] [Google Scholar]
- Benos DJ, McPherson S, Hahn BH, Chaikin MA, Benveniste EN. Cytokines and HIV envelope glycoprotein gp120 stimulate Na+/H+ exchange in astrocytes. J Biol Chem. 1994;269(19):13811–6. [PubMed] [Google Scholar]
- Bond J, Varley J. Use of flow cytometry and SNARF to calibrate and measure intracellular pH in NS0 cells. Cytometry A. 2005;64(1):43–50. doi: 10.1002/cyto.a.20066. [DOI] [PubMed] [Google Scholar]
- Bukrinsky M, Adzhubei A. Viral protein R of HIV-1. Rev Med Virol. 1999;9(1):39–49. doi: 10.1002/(sici)1099-1654(199901/03)9:1<39::aid-rmv235>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Cohen EA, Terwilliger EF, Jalinoos Y, Proulx J, Sodroski JG, Haseltine WA. Identification of HIV-1 vpr product and function. J Acquir Immune Defic Syndr. 1990;3(1):11–8. [PubMed] [Google Scholar]
- Collman R, Hassan NF, Walker R, Godfrey B, Cutilli J, Hastings JC, Friedman H, Douglas SD, Nathanson N. Infection of monocyte-derived macrophages with human immunodeficiency virus type 1 (HIV-1). Monocyte-tropic and lymphocyte-tropic strains of HIV-1 show distinctive patterns of replication in a panel of cell types. J Exp Med. 1989;170(4):1149–63. doi: 10.1084/jem.170.4.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewart GD, Sutherland T, Gage PW, Cox GB. The Vpu protein of human immunodeficiency virus type 1 forms cation-selective ion channels. J Virol. 1996;70(10):7108–15. doi: 10.1128/jvi.70.10.7108-7115.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliegel L. The Na+/H+ exchanger isoform 1. Int J Biochem Cell Biol. 2005;37(1):33–7. doi: 10.1016/j.biocel.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Gibellini D, Vitone F, Schiavone P, Re MC. HIV-1 tat protein and cell proliferation and survival: a brief review. New Microbiol. 2005;28(2):95–109. [PubMed] [Google Scholar]
- Jacotot E, Ferri KF, El Hamel C, Brenner C, Druillennec S, Hoebeke J, Rustin P, Metivier D, Lenoir C, Geuskens M, Vieira HL, Loeffler M, Belzacq AS, Briand JP, Zamzami N, Edelman L, Xie ZH, Reed JC, Roques BP, Kroemer G. Control of mitochondrial membrane permeabilization by adenine nucleotide translocator interacting with HIV-1 viral protein rR and Bcl-2. J Exp Med. 2001;193(4):509–19. doi: 10.1084/jem.193.4.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janket ML, Manickam P, Majumder B, Thotala D, Wagner M, Schafer EA, Collman RG, Srinivasan A, Ayyavoo V. Differential regulation of host cellular genes by HIV-1 viral protein R (Vpr): cDNA microarray analysis using isogenic virus. Biochem Biophys Res Commun. 2004;314(4):1126–32. doi: 10.1016/j.bbrc.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Kino T, Gragerov A, Slobodskaya O, Tsopanomichalou M, Chrousos GP, Pavlakis GN. Human immunodeficiency virus type 1 (HIV-1) accessory protein Vpr induces transcription of the HIV-1 and glucocorticoid-responsive promoters by binding directly to p300/CBP coactivators. J Virol. 2002;76(19):9724–34. doi: 10.1128/JVI.76.19.9724-9734.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kino T, Pavlakis GN. Partner molecules of accessory protein Vpr of the human immunodeficiency virus type 1. DNA Cell Biol. 2004;23(4):193–205. doi: 10.1089/104454904773819789. [DOI] [PubMed] [Google Scholar]
- Kondo T, Takeuchi K, Doi Y, Yonemura S, Nagata S, Tsukita S. ERM (ezrin/radixin/moesin)-based molecular mechanism of microvillar breakdown at an early stage of apoptosis. J Cell Biol. 1997;139(3):749–58. doi: 10.1083/jcb.139.3.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JA. HIV pathogenesis: knowledge gained after two decades of research. Adv Dent Res. 2006;19(1):10–6. doi: 10.1177/154407370601900104. [DOI] [PubMed] [Google Scholar]
- Li M, Gao F, Mascola JR, Stamatatos L, Polonis VR, Koutsoukos M, Voss G, Goepfert P, Gilbert P, Greene KM, Bilska M, Kothe DL, Salazar-Gonzalez JF, Wei X, Decker JM, Hahn BH, Montefiori DC. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J Virol. 2005;79(16):10108–25. doi: 10.1128/JVI.79.16.10108-10125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahalingam S, Ayyavoo V, Patel M, Kieber-Emmons T, Weiner DB. Nuclear import, virion incorporation, and cell cycle arrest/differentiation are mediated by distinct functional domains of human immunodeficiency virus type 1 Vpr. J Virol. 1997;71(9):6339–47. doi: 10.1128/jvi.71.9.6339-6347.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder B, Janket ML, Schafer EA, Schaubert K, Huang XL, Kan-Mitchell J, Rinaldo CR, Jr, Ayyavoo V. Human immunodeficiency virus type 1 Vpr impairs dendritic cell maturation and T-cell activation: implications for viral immune escape. J Virol. 2005;79(13):7990–8003. doi: 10.1128/JVI.79.13.7990-8003.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matarrese P, Malorni W. Human immunodeficiency virus (HIV)-1 proteins and cytoskeleton: partners in viral life and host cell death. Cell Death Differ. 2005;12(Suppl 1):932–41. doi: 10.1038/sj.cdd.4401582. [DOI] [PubMed] [Google Scholar]
- Moon HS, Yang JS. Role of HIV Vpr as a regulator of apoptosis and an effector on bystander cells. Mol Cells. 2006;21(1):7–20. [PubMed] [Google Scholar]
- Muto S, Ebata S, Okada K, Saito T, Asano Y. Glucocorticoid modulates Na+/H+ exchange activity in vascular smooth muscle cells by nongenomic and genomic mechanisms. Kidney Int. 2000;57(6):2319–33. doi: 10.1046/j.1523-1755.2000.00092.x. [DOI] [PubMed] [Google Scholar]
- Paxton W, Connor RI, Landau NR. Incorporation of Vpr into human immunodeficiency virus type 1 virions: requirement for the p6 region of gag and mutational analysis. J Virol. 1993;67(12):7229–37. doi: 10.1128/jvi.67.12.7229-7237.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfettini JL, Castedo M, Roumier T, Andreau K, Nardacci R, Piacentini M, Kroemer G. Mechanisms of apoptosis induction by the HIV-1 envelope. Cell Death Differ. 2005;12(Suppl 1):916–23. doi: 10.1038/sj.cdd.4401584. [DOI] [PubMed] [Google Scholar]
- Piller SC, Ewart GD, Jans DA, Gage PW, Cox GB. The amino-terminal region of Vpr from human immunodeficiency virus type 1 forms ion channels and kills neurons. J Virol. 1999;73(5):4230–8. doi: 10.1128/jvi.73.5.4230-4238.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piller SC, Ewart GD, Premkumar A, Cox GB, Gage PW. Vpr protein of human immunodeficiency virus type 1 forms cation-selective channels in planar lipid bilayers. Proc Natl Acad Sci U S A. 1996;93(1):111–5. doi: 10.1073/pnas.93.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putney LK, Barber DL. Na-H exchange-dependent increase in intracellular pH times G2/M entry and transition. J Biol Chem. 2003;278(45):44645–9. doi: 10.1074/jbc.M308099200. [DOI] [PubMed] [Google Scholar]
- Putney LK, Denker SP, Barber DL. The changing face of the Na+/H+ exchanger, NHE1: structure, regulation, and cellular actions. Annu Rev Pharmacol Toxicol. 2002;42:527–52. doi: 10.1146/annurev.pharmtox.42.092001.143801. [DOI] [PubMed] [Google Scholar]
- Rasola A, Gramaglia D, Boccaccio C, Comoglio PM. Apoptosis enhancement by the HIV-1 Nef protein. J Immunol. 2001;166(1):81–8. doi: 10.4049/jimmunol.166.1.81. [DOI] [PubMed] [Google Scholar]
- Rich IN, Worthington-White D, Garden OA, Musk P. Apoptosis of leukemic cells accompanies reduction in intracellular pH after targeted inhibition of the Na(+)/H(+) exchanger. Blood. 2000;95(4):1427–34. [PubMed] [Google Scholar]
- Rossio JL, Esser MT, Suryanarayana K, Schneider DK, Bess JW, Jr, Vasquez GM, Wiltrout TA, Chertova E, Grimes MK, Sattentau Q, Arthur LO, Henderson LE, Lifson JD. Inactivation of human immunodeficiency virus type 1 infectivity with preservation of conformational and functional integrity of virion surface proteins. J Virol. 1998;72(10):7992–8001. doi: 10.1128/jvi.72.10.7992-8001.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roumier T, Vieira HL, Castedo M, Ferri KF, Boya P, Andreau K, Druillennec S, Joza N, Penninger JM, Roques B, Kroemer G. The C-terminal moiety of HIV-1 Vpr induces cell death via a caspase-independent mitochondrial pathway. Cell Death Differ. 2002;9(11):1212–9. doi: 10.1038/sj.cdd.4401089. [DOI] [PubMed] [Google Scholar]
- Schneider D, Gerhardt E, Bock J, Muller MM, Wolburg H, Lang F, Schulz JB. Intracellular acidification by inhibition of the Na+/H+-exchanger leads to caspase-independent death of cerebellar granule neurons resembling paraptosis. Cell Death Differ. 2004;11(7):760–70. doi: 10.1038/sj.cdd.4401377. [DOI] [PubMed] [Google Scholar]
- Schubert U, Ferrer-Montiel AV, Oblatt-Montal M, Henklein P, Strebel K, Montal M. Identification of an ion channel activity of the Vpu transmembrane domain and its involvement in the regulation of virus release from HIV-1-infected cells. FEBS Lett. 1996;398(1):12–8. doi: 10.1016/s0014-5793(96)01146-5. [DOI] [PubMed] [Google Scholar]
- Sherman MP, de Noronha CM, Pearce D, Greene WC. Human immunodeficiency virus type 1 Vpr contains two leucine-rich helices that mediate glucocorticoid receptor coactivation independently of its effects on G(2) cell cycle arrest. J Virol. 2000;74(17):8159–65. doi: 10.1128/jvi.74.17.8159-8165.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart SA, Poon B, Jowett JB, Chen IS. Human immunodeficiency virus type 1 Vpr induces apoptosis following cell cycle arrest. J Virol. 1997;71(7):5579–92. doi: 10.1128/jvi.71.7.5579-5592.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thotala D, Schafer EA, Tungaturthi PK, Majumder B, Janket ML, Wagner M, Srinivasan A, Watkins S, Ayyavoo V. Structure-functional analysis of human immunodeficiency virus type 1 (HIV-1) Vpr: role of leucine residues on Vpr-mediated transactivation and virus replication. Virology. 2004;328(1):89–100. doi: 10.1016/j.virol.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Tungaturthi PK, Sawaya BE, Singh SP, Tomkowicz B, Ayyavoo V, Khalili K, Collman RG, Amini S, Srinivasan A. Role of HIV-1 Vpr in AIDS pathogenesis: relevance and implications of intravirion, intracellular and free Vpr. Biomed Pharmacother. 2003;57(1):20–4. doi: 10.1016/s0753-3322(02)00328-1. [DOI] [PubMed] [Google Scholar]
- Wieder ED, Hang H, Fox MH. Measurement of intracellular pH using flow cytometry with carboxy-SNARF-1. Cytometry. 1993;14(8):916–21. doi: 10.1002/cyto.990140810. [DOI] [PubMed] [Google Scholar]
- Wu KL, Khan S, Lakhe-Reddy S, Jarad G, Mukherjee A, Obejero-Paz CA, Konieczkowski M, Sedor JR, Schelling JR. The NHE1 Na+/H+ exchanger recruits ezrin/radixin/moesin proteins to regulate Akt-dependent cell survival. J Biol Chem. 2004;279(25):26280–6. doi: 10.1074/jbc.M400814200. [DOI] [PubMed] [Google Scholar]