

Abstract
A systemic inflammatory response (SIR) occurs prior to and during the treatment of severe diabetic ketoacidosis (DKA). IL-1β, TNF-α and C5b-9 are components of SIR and have been speculated to be involved in the clinical brain edema (BE) of DKA. We studied IL- 1β, TNF-α, C5b-9, inducible nitric oxide (iNOS), ICAM-1, IL-10 and Hsp70 expression in the brains of two patients who died as the result of clinical BE during the treatment of DKA. IL- 1β was strongly expressed in the choroid plexus epithelium (CPE) and ependyma, and to a lesser extent in the hippocampus, caudate, white matter radiation of the pons, molecular layer of the cerebellum and neurons of the cortical gray matter. TNF-α was expressed to a lesser extent than IL-1 β, and only in the CP. C5b-9, previously shown to be deposited on neurons and oligodendrocytes, was found on CPE and ependymal cells. iNOS and ICAM-1 had increased expression in the CPE and ependyma. Hsp70 and IL-10 were also expressed in the CPE of the case with the shorter duration of treatment. Our data demonstrates the presence of a multifaceted neuroinflammatory cytotoxic insult of the CPE, which may play a role in the pathophysiology of the fatal brain edema of DKA.
Keywords: Brain Edema, C5b-9, Choroid Plexus, Diabetic Ketoacidosis, Ependyma, Hsp70, ICAM-1, IL-1β, IL-10, iNOS, Neuroinflammation, Systemic Inflammatory Response, TNF-α
INTRODUCTION
The catastrophic event of BE in diabetic ketoacidosis (DKA) was first reported more than 40 years ago (Fitzgerald et al., 1961). In spite of improved intensive care management and advances in our understanding of the pathophysiology of BE (Xi et al., 2002), the prevalence of fatal BE in children and adolescents in DKA remains a significant cause of morbidity and mortality. Progress in the understanding of the pathophysiology of the BE associated with DKA has been limited due to its relative infrequency (Edge et al., 2001) and limited autopsy data.
There is evidence of a systemic inflammatory response (SIR) occurring prior to and then increasing during the treatment of severe uncomplicated DKA in type 1 diabetes mellitus (T1DM) (Hoffman et al., 2003; Jerath et al., 2005). IL-1β and TNF-α, major components of SIR, are rapidly up regulated by numerous stimuli (Eriksson et al., 2000; DiLorento et al., 2004; Marques et al., 2006) and are involved in experimental BE (Holmin et al., 2000; Lazovic et al., 2005; Hua et al., 2006).
C5b-9 is the terminal component of the complement cascade that is assembled during the SIR of severe DKA (Jerath et al., 2005). C5b-9 can have a cytolytic effect and, at lower concentrations, can also activate pathways, which restrict injury and facilitate recovery of cells (Rus et al., 2006). C5b-9 has recently been reported to be deposited on neurons, oligodendrocytes (OLGs) and blood vessels in the fatal DKA (Hoffman et al., 2006). This finding supports the interaction between the immune and the central nervous systems and the hypothesis that in addition to the SIR, there is also a local neuroinflammatory component in the fatal cerebral complication of BE in DKA.
Hsp70-reactive T cells are present in patients with T1DM and express IL-10 upon exposure to extracellular hsp 70 (ehsp70) (Wendling et al., 2000; Abulafia-Lapid et al., 2003). Unlike the proinflammatory components, which increase during the treatment of DKA, hsp70 and IL-10 are maximally increased prior to treatment and then decrease during the treatment of DKA (Hoffman et al., 2003; Oglesbee et al., 2005). Intracellular hsp70 (ihsp70) have not been evaluated in the brains of DKA patients, although elevations might be anticipated (Ouyang et al., 2006). Increased ihsp70 levels serve cytoprotective functions and have been shown to preserve both neuronal and blood-brain barrier function at the time of various insults (Giffard et al., 2004; Lu et al., 2004; Wagner et al., 2006).
Our goal was to determine if the choroid plexus (CP), a critical site of physiologic and immunologic functions, is actively involved in a neuroinflammatory response of fatal BE in severe DKA. We selected known markers of an inflammatory response: IL-1β, TNF-α and C5b-9, iNOS, ICAM-1, IL-10 and hsp70. Immunohistochemistry was used the distribution of these markers of inflammatory response in the brains of two previously reported adolescent females with T1DM who died of BE during the treatment of severe DKA (Hoffman et al., 2006). Our data show the presence of IL-1β, TNF-α, iNOS, ICAM-1 and C5b-9 in CP indicating the involvement neuroinflammatory response in the fatal brain edema of DKA.
MATERIALS AND METHODS
Case 1
An adolescent female had a four-year history of poorly controlled T1DM, which resulted in recurrent hospitalization for DKA. The final admission was preceded by a 12-hour history of abdominal pain and several episodes of emesis. There was no history of fever or enteritis. On physical examination, she was oriented but drowsy. Her height was 163 cm; weight was 68.5 kg; blood pressure was 140/70 mm Hg; pulse was 140/min; respirations were 30/min; and temperature was 98.5°F. There was no evidence of infection. The mucous membranes were dry, the abdomen was diffusely tender and bowel pattern sounds were hypoactive. The Tanner stage was appropriate for age. Admission laboratory tests consisted of: pH 7.10; pCO2 15 mmHg; pO2 106 mmHg; glucose 810 mg/dl; Na 132 meq/L; K 5.7 meq/L; Cl 93 meq/L; HCO3 5 meq/L; and BUN 30 mg/dL. Treatment was in a Pediatric Intensive Care Unit and correction of the hyperglycemia and metabolic acidosis was unremarkable. Twelve hours following the initiation of treatment she developed sudden onset of labored respirations and had a cardio-respiratory arrest within 20 minutes. An emergency CT scan of the head showed sulcal effacement, cerebral and pontine edema with evidence of herniation. Efforts at resuscitation were unsuccessful and she was pronounced dead 1 ½ hours after the cardio -respiratory arrest.
Case 2
An adolescent female had an eight-year history of poorly controlled T1DM, which had resulted in recurrent hospitalizations for DKA. The final admission was preceded by an 18-hour history of capillary blood glucoses of over 300 mg/dl, ketonuria, a 4-hour history of headache and several episodes of emesis. There was no history of fever or enteritis. On physical examination she was slightly confused and lethargic. Her height was 154 cm; weight was 45 kg; blood pressure was 135/68 mm/Hg; pulse was 132/min; respiration was 26/min; and temperature was 97°F. There were no signs of infection. Diffuse abdominal tenderness and decreased bowel sounds were present. The Tanner stage was appropriate for age. Admission laboratory tests consisted of: pH 7.16; pCO2 17 mm Hg; pO2 100 mmHg; blood glucose 581 mg/dl; Na 130 meq/L; K 4.8 meq/L; Cl 89 meq/L; HCO3 6 meq/L; and BUN 28 mg/dl. Treatment was in a Pediatric Intensive Care Unit and correction of the hyperglycemic and metabolic acidosis was unremarkable. Ten hours following initiation of treatment, she became unresponsive and was treated with mannitol, hyperventilation and placed on mechanical ventilation. A CT scan of the head showed diffuse cerebral edema and decreased intercaudate diameter. She was pronounced dead approximately 10 hours after the cardiorespiratory arrest.
Tissue analysis
Immunohistochemistry
Autopsy was performed within 6 h after death in both patients. Brains were fixed in10% neutral buffered formalin, paraffin–embedded and 5μm sections stained with hematoxylin and eosin using routine methods. Additional sections were mounted on positively charged slides for immunohistochemical staining. The slides were heated at 59° C and deparaffinized using xylenes; they were rehydrated in graded alcohol and distilled water. A methanol/3% H2O2 procedure was used to block endogenous peroxidase. The non-enzymatic antigen recovery step was performed in a microwave oven and treated with Citra Plus, pH of 6.0 (BioGenex, San Ramon, CA.). The slides were then thoroughly washed with distilled water and placed in a TBS buffer. Immunostaining was performed on the DAKO autostainer. An Avidin Biotin block was used for the IL-1β and TNF α to block out endogenous biotin. IL-1β and hsp70 were detected by labeled Streptavidin Biotin Peroxidase amplification system (DAKO). Dako LSAB+ detected TNF α. Diaminobenzidine was used for color visualization (DAKO) of IL-1β and DAB hydrogen peroxide for TNF alpha. The positive controls for IL-1β, TNF-α and hsp70 were lymph node, Cohn’s colon and breast tissue respectively. ICAM-1 was detected by a polymer- based detection system (DAKO, Carpanteria, CA). The positive control for ICAM-1 was tonsil. IL-10 was detected by a polymer-based detection system (Signet Aquity Polymer, Dedham, MA). The positive control for IL-10 was tonsil. INOS immunostaining was performed using pre-treatment and detection reagents with the automated analyzer from Ventana Medical Systems, Inc. (Tucson, AZ.). The positive control for iNOS was colon tissue. After staining was completed all slides were counterstained with Mayer’s hematoxylin (Sigma Chemical Co., St. Louis). Sources, dilutions and incubation times of the antibodies are: IL-1β (R&D Systems Inc., Minneapolis, MN) goat/IgG at 1:40 dilution, 60 minutes at room temperature (RT); TNFα polyclonal antibody (R&D Systems Inc., Minneapolis, MN) at 1:50 dilution, 2h at RT; Hsp70 (Santa Cruz Biotechnology, Santa Cruz, CA.) goat affinity purified polyclonal antibody, clone K-20 at 1:100 dilution, 30 minutes at RT; ICAM-1 (Novocastra, Newcastle Upon Tyne, United Kingdom) mouse antibody, clone 23G12 at 1:25 dilution, 60 minutes at RT; IL-10 mouse monoclonal (R&D Systems Inc., Minneapolis, MN), clone 23738 at 1:100 dilution, 30 minutes at RT; iNOS (Santa Cruz Biotechnology, Santa Cruz, CA.) mouse monoclonal antibody at 1:40 dilution. Specificity controls for the immunohistochemistry included omission of the primary antibody during the staining and the use of non-immune serum in place of the primary antibody. In addition, the specific immune deposits were abolished by preincubation of the primary antibody with the protein being stained for.
The methodology for C5b-9 has previously been described in detail (Hoffman et al., 2006). After xylene deparaffinisation and epitope retrieval using a Target Retrieval Solution (DAKO Carpanteria, CA), sections were treated with 3% H2O2 to remove endogenous peroxidase. Sections were incubated for 30 minutes with normal goat serum, then with the monoclonal antibody against C5b-9 (Quidel, SanDiego, CA) for 60 minutes at RT in a humid chamber. In separate experiments consecutive sections were incubated with mouse monoclonal anti-CD59 (clone MEM-43, Serotec, Oxford, UK). The sections were washed 3 times for 3 min at RT with PBS, and then incubated for 1 hr at RT with HPR-conjugated goat ant-mouse IgG (Jackson ImmunoResearch, West Grove, PA). After washing, the specific deposits were developed using NovaRed (Vector Laboratories, Burlingame, CA). The nuclei were counterstained with Mayer’s hematoxylin (Sigma Chemical Co, St. Louis). The positive control for C5b-9 was kidneys from patients with systemic lupus erythematous and for CD59 was normal brain. Controls for the specificity of each immunohistochemical reaction were performed by replacing the primary antibody with PBS, mouse, or rabbit IgG. Immunnostainings were independently evaluated by two investigators.
Analysis of Apoptosis
The “ApopTag” Peroxidase in Situ Apoptosis Detection Kit (Intergen Co., Purchase, NY) was used as previously described (Niculescu et al., 2004). This kit detects DNA strand breaks by measuring terminal deoxynucleotidyl transferase (TdT)-dependent incorporation of dUTP. The sections were incubated for 1 hr at 37°C with TdT enzyme, washed, and incubated with the anti-digoxigenin peroxidase-conjugated antibody. The reaction was developed using ImmuunoPure metal-enhanced diaminobenzidine tetrachloride (DAB) substrate kit (Pierce, Rockford, IL). Apoptosis was defined as TUNEL positive cells with nuclear fragmentation demonstrated by either karyorhexis or pyknosis.
Immunohistochemical staining profiles of the patient material was also compared to that observed on normal human brain tissue microarrays from Chemicon (Temecula, CA) and three age-matched controls with no cerebral pathology.
RESULTS
Gross and Histopathology
Brain edema was evident upon both gross and microscopic evaluation. Grossly, brains of both cases were swollen with flattening of gyri, narrowing of sulci, and uncal and tonsillar herniation. Coronal sectioning showed expansion of the white matter with compression of the lateral ventricles. Histologically, edematous change was supported by enlargement of perineuronal and perivascular spaces. Case 2 showed some congestive changes and regressive neuronal changes in the cortex and white matter features of a non-perfused brain. Neither case had evidence of necrosis in the CP. No leukocytic infiltration was evident in the cortex or white matter in either case, and there was no thrombosis of cerebral vessels.
Immunohistochemistry
IL-1β, TNF-α and IL-10 expression in CP
IL-1β immunohistochemistry revealed intense staining of the epithelial cells of the choroid plexus and ependymal cells (Fig. 1A). Staining in Case1 was somewhat more intense (Fig. 1 A) than in Case 2 (Table 1). IL-1β also had limited and less intense staining of the hilum and CA3-1 fields of the hippocampus, tail of the caudate, white matter radiations of the pons, and molecular layer of the cerebellum (data not shown). TNF-α was localized to the CP and only moderately expressed (Fig 1 C). IL-1β (Fig. 1B) and TNF-α (data not shown) immunostaining in control brains was negative. IL-10 was expressed in CP epithelium and blood vessels in Case 1 (Table 1). IL-10 immunostaining in Case 2 and the control brains was negative (Table 2).
Figure 1. Immunohistochemical localization of IL-1β and TNFα in DKA brains.
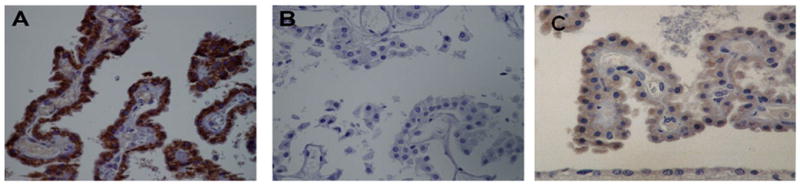
IL-1β and TNFα were localized using a Streptavidin Biotin Peroxidase amplification system. Epithelial cells of the CP expressed intense IL-1β staining (A) while control brains were negative (B). TNFα was abundantly expressed by epithelial cells of CP (C). Magnification × 400.
Table 1.
Quantification of Immunostaining in the CP of DKA and Controls.
| Case No. | IL-1β | TNFα | hsp70 | iNOS | ICAM-1 | IL-10 | C5b-9 | CD59 | TUNEL |
|---|---|---|---|---|---|---|---|---|---|
| 1 | +++ | ++ | +++ | +++ | ++ | ++ | +++ | − | − |
| 2 | ++ | + | + | + | ++ | − | +++ | − | ++ |
| 3–5 Controls | − | − | + | − | − | − | − | + | − |
− negative; +, slightly positive; ++, positive; +++, highly positive.
iNOS, ICAM-1 and hsp70 expression in CP
iNOS (Fig. 2A) and ICAM-1 (Fig. 2B) were expressed almost exclusively to the epithelial cells of the CP and ependyma with the control brains being negative (Fig. 2C).
Figure 2. Immunohistochemical localization of iNOS and ICAM-1 in DKA brains.
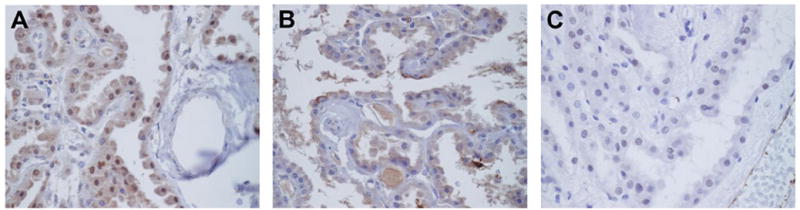
Epithelial cells of the CP and ependyma expressed iNOS (A) and ICAM-1 (B) while controls were negative (C). Magnification × 400.
Hsp70 showed the most diffuse and intense immunoreactivity in the epithelial cells of the choroid plexus of Case1 (Fig. 3A). Expression of hsp70 in Case 2 (Fig. 3B) was faint and comparable to that observed in control brains. Ependymal staining was greater in Case1 (Fig. 3A) than in Case 2 (data not shown) or the control tissue (Fig. 3B), although the magnitude of the difference was less than that in the CPE. Normal brain tissue exhibited moderate constitutive expression of hsp70 in neuronal cytoplasm (Fig. 3C). Constitutive expression of inducible 70 kDa heat shock protein is a characteristic of the human brain (Pardue et al., 2006). Case 1 exhibited marked intranuclear hsp70 immunoreactivity within the hippocampus/dentate gyrus, accompanied by increased cytoplasmic staining of axonal processes (Fig. 3D). Increased expression of hsp70 was also observed in sporadic glial cells exhibiting both astrocytic and oligodendroglial morphologies (Fig. 3E). Small blood vessels, most prominently in the hippocampus, exhibited increased nuclear and cytoplasmic hsp70 staining of endothelial and adventitial cells in Case 1 (Fig. 3E) relative to Case 2 (data not shown) and the control brains (Fig. 3 C).
Figure 3. Immunohistochemical localization of Hsp70 in DKA brains.
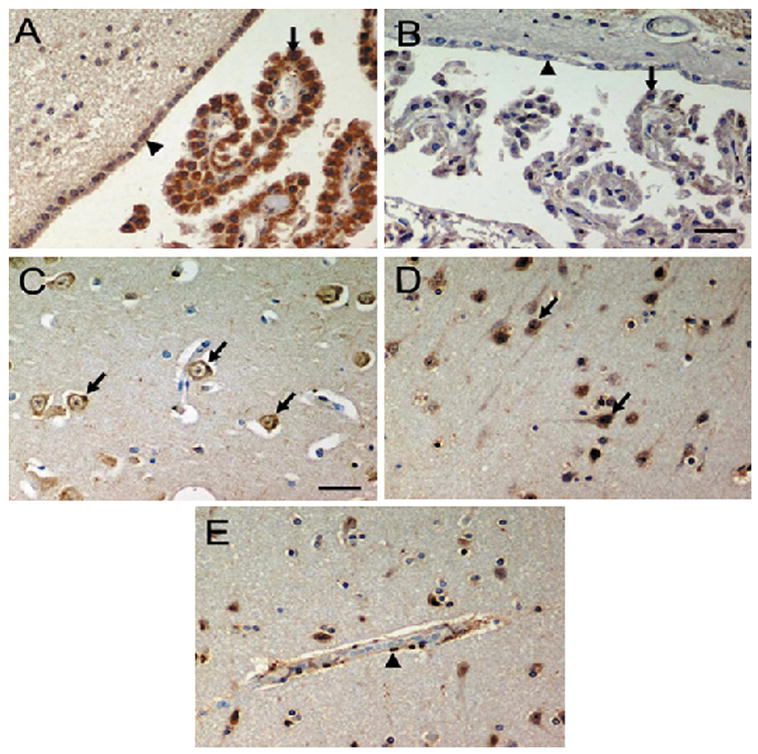
Epithelial cells of the CP (A, arrows) in Case 1 expressed intense hsp70 staining. In the control brain (B, arrows) hsp70 is expressed in the epithelial cells of the CP only faintly but in the same pattern as in Case 1. Staining of the ependymal cell cytoplasm and nuclei (A, arrow heads) was also more intense in Case 1 relative to the control brains (B, arrow heads). Hsp70 immunoreactivity in the cortex was restricted to the cytoplasm of neurons in controls (C, arrows). Staining intensity of neurons in Case 1 was more intense, evident as increased staining of neuronal processes, and in the cytoplasm together with intranuclear localization (D, arrows), consistent with a cellular stress response. The blood vessels of Case 1 also exhibited intense immunoreactivity, with expression in the cytoplasm and nuclei of endothelial (E, arrow heads) and adventitial cells. Magnification × 400.
C5b-9 and CD59 immunoreactive deposits in CP
C5b-9 showed intense staining of the epithelial cells of the CP (Fig. 4A) and ependymal cells of the lateral, third, and fourth ventricles in both cases of DKA (Fig. 4 A, B). In the same brains as previously reported, up to 60–80% of neurons and OLGs displayed C5b-9 deposits (Hoffman et al., 2006). C5b-9 deposits were absent in the normal brains examined (data not shown). CD59 expression was absent on epithelial cells of the choroid plexus and ependymal cells (Fig. 4C). As previously reported, CD59 expression was absent on the neuron, blood vessels and OLGs in the DKA brains (Hoffman et al., 2006). In normal brains, CD59 expression was found on neurons and blood vessels (as previously described (Singhrao et al., 1999)) and on the CP epithelial cells and ependymal cells (data not shown). Controls of immunoperoxidase reaction were negative (Fig 4 D).
Figure 4. Immunohistochemical localization of C5b-9, CD59 and apoptotic cells in choroid plexus.
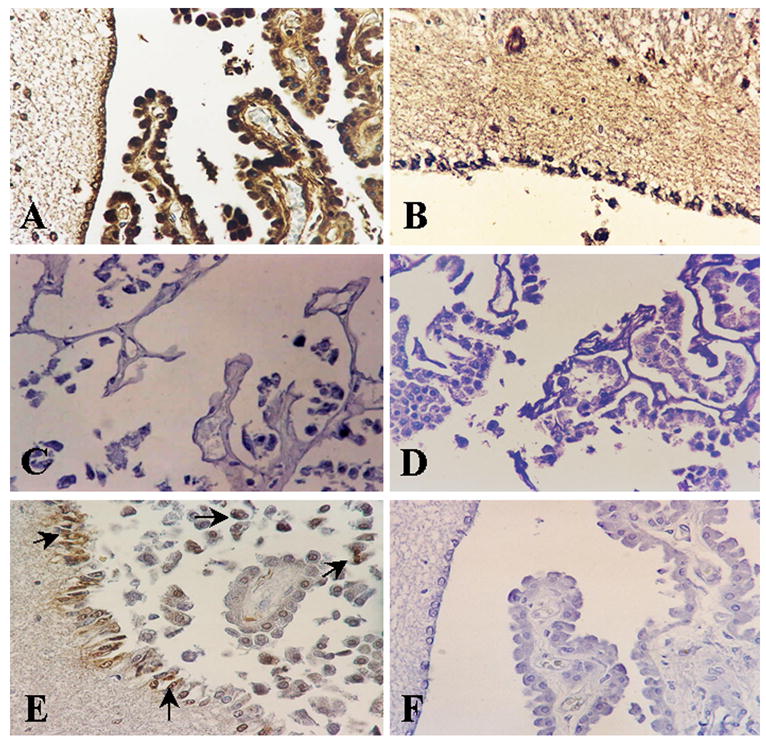
C5b-9 and CD59 were localized by indirect immunoperoxidase (A, B). Choroid plexus epithelial cells show strong immunoreactivity for C5b-9 deposits in case 1 (A) and on ependyma in case 2 (B); (C) CD59 was absent in the choroids plexus. (D) Corresponding controls for the immunoperoxidase reaction were negative. (E, F) Apoptotic cells were localized by TUNEL method. TUNEL positive cells were found both in choroids plexus epithelium and ependyma in case 1 (arrows). Magnification ×400.
TUNEL staining in DKA brains
Case 2 showed the presence of TUNEL positive epithelial cells of the CP and ependymal cells (Fig 4 E). Some of the apoptotic cells on consecutive sections were also C5b-9 positive (Fig. 4A and 4E). In contrast, only a few focal areas of the brain in case 2 had a small number of apoptotic neurons and OLGs (Hoffman et al., 2006). TUNEL positive cells were not found in Case 1 (Fig. 4 F).
DISCUSSION
These results extend the previous report of a neuroinflammatory response during the fatal BE of DKA (Hoffman et al., 2006) by demonstrating for the first time the presence of multiple inflammatory mediators in CP. The numerous physiologic (Emerich et al., 2005) and immunologic (Chodobski et al., 2001) functions of the CP make it a candidate to play a role in the fatal BE of DKA. While we have no information on the onset or extent of the initial insult to the CP or the sequence of progression, we hypothesize that these neuroinflammatory factors are not involved in the initiation or early progression of subclinical BE (Hoffman et al., 1988; Durr et al., 1992), but rather during the clinical BE of DKA and may be the critical factor in a progressively edematous brain. This later insult would be in keeping with: 1) the direct perturbation of cerebral capillary endothelial cells by the ketone bodies serving as the initiating factor of subclinical BE (Isales et al., 1999; Hoffman et al 2002); 2) a pattern of inflammation is similar to the accentuated SIR which follows the initiation of treatment of DKA (Hoffman et al., 2003; Jerath et al., 2005); and 3) the relatively infrequent occurrence of fatal clinical BE in DKA in comparison to the frequency of subclinical BE (Hoffman et al., 1988; Durr et al., 1992; Edge et al., 2001).
IL-1β and TNF-α in the CPE may reflect either local synthesis due to a peripheral stimulus (Marques et al., 2006) or sequestration after systemic production (Maness et al., 1998). The demonstration of iNOS, one of the mediators of IL-1β and TNF- α (Kim et al., 2004), favors the local synthesis of these inflammatory cytokines by the CPE. If the CPE is the site of cytokines production, the selectivity of expression in the CPE relative to other regions of the brain may reflect a variable response to systemic proinflammatory stimuli (Quan et al., 1999). Such a response would be similar to the variability in the degree and duration of metabolic and oxidative stress and the degree and duration of dysregulation of the neuroinflammatory cytokines, as was reported for the plasma inflammatory cytokines during the correction of DKA (Hoffman et al., 2003). Regardless of the site of synthesis of IL-1β and TNF-α, treatment of the CP with IL-1β results in a significant reduction in the efficiency of the CPE to clear the CSF of organic anions (Strazielle et al., 2003). With regard to localization of IL-1β in areas of the brain other than the CPE, it was reported that following intracerebroventricular injection, IL-1β rapidly enters the periventricular tissue and then spreads along white matter fibers via volume transmission, thereby acting as a neuromodulator of neurons and glial cells (Konsman et al., 2000). It is important to note that IL-1 has recently been suggested to have an important role in several of the acute complications of DKA (Eisenhut, 2006). IL-10 an anti-inflammatory and an immunosuppressive cytokine, was found in small amounts only in Case 1. These data might suggest a predominance of proinflammatory cytokines in CP of patients with fatal DKA. C5b-9 may play a role in the expression of IL-1β in the CP (Casarsa et al., 2003), since it induces transcriptional activation of IL-1α (Brunn et al., 2006) and the intracerebroventricular injection of the terminal complement complex result in an increase of IL-1β in the periventricular areas and CSF. Since we did not find any signs of necrosis in the DKA brains is possible that C5b-9 could result in a sublethal attack and a protective effect on CPE. The potential protective effect of sublethal attacks by C5b-9, may explain the infrequent occurrence of clinical BE in DKA (Edge et al., 2001) despite the significant prevalence of subclinical BE (Hoffman et al., 1988; Durr et al., 1992; Glaser et al., 2004).
In addition to being a mediator of IL-1β and TNF –α effect, iNOS also mediates free radical damage and has been reported in several forms of CNS insult (Nag et al., 2001; Togashi et al., 2001). Its potential importance during DKA is extended by the recent report that DKA activates iNOS in lymphocytes and monocytes (Iori et al., 2002), and by our demonstration here of iNOS expression in CPE. The nitric oxide generated by iNOS could also be directly involved in the initiation and progression of the BE (Sharma et al., 2006) and/or cerebral hyperemia (Brian et al., 1996). Increased iNOS in the CPE also has the potential to disrupt ion transport, given the fact that the Na-K-Cl cotransporter is regulated by inflammatory cytokines (Topper et al., 1997). In contrast, the up regulation of ICAM-1 is unlikely to be mediated by iNOS (Hickey et al., 2006); but likely to be the result of a direct activation by DKA. This is suggested by the ketone body acetoacetate increasing the expression of ICAM-1 in an in-vitro human cerebral microvascular system (Hoffman et al., 2002).
Expression of hsp70 in the CPE, like IL-1β, TNF-α and IL-10 may reflect either local production or sequestration from systemic sources. Our finding of only cytoplasmic hsp70 staining in the CPE suggests the possibility that the hsp70 was sequestered from the serum. In support of this, the differences in CPE expression of hsp70 in Case 1 versus Case 2 are in agreement with the kinetics of serum hsp70 levels during treatment of DKA. Serum hsp70 levels are increased prior to treatment of DKA and do not change significantly at 6 hours after initiation of treatment, but decrease to within baseline values by 24 h (Oglesbee et al., 2005). Elevated CPE expression of hsp70 in Case 1 may thus reflect the shorter treatment interval of 12 h, an interval at which serum levels of hsp70 are still elevated. By contrast, decreased expression in Case 2 may reflect the occurrence of death following 20 h of treatment, a time when serum levels of hsp70 have returned to baseline (Oglesbee et al., 2005).
The increased nuclear and cytoplasmic expression of hsp70 in the neurons and blood vessels in Case 1 indicates an active production as part of a cellular stress response, which could provide protection against the prothrombotic state of severe DKA (Hoffman et al., 1999; Carl et al., 2003). In support of a protective effect, both hsp70 and its constitutively expressed isoform have been implicated in protection against complement-mediated cell lysis (Fishelson et al., 2001).
This study indicate that dysregulation of the inflammatory response in the fatal BE of DKA involves not only deposition of C5b-9 (Hoffman et al., 2006), but also expression of inflammatory cytokines and hsp70. By expressing both pro- and anti-inflammatory molecules, the CP seems to be involved in the pathophysiology of the fatal BE of DKA by compromising the numerous physiologic and immunologic functions of the CP. Studying the detailed balance between both types of signals is certainly essential to understand the final response of the brain to an inflammatory insult, and the CP must be considered a relevant player in such processes. Further studies are needed to clarify the role of neuroinflammation in the multifaceted cerebral insult, a potentially fatal complication of DKA.
Acknowledgments
The authors are grateful to Dr. J. Cannon for his advice, Dr. R. Gala for his critical review of the manuscript and Sarah Albertin for technical assistance. This work was supported in part by U.S. Public Health Grants, RO1 NS42011 (to HR) and by the T M fund (WH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abulafia-Lapid R, Gillis D, Yosef O, Atlan H, Cohen IR. T cells and autoantibodies to human HSP70 in type 1 diabetes in children. J Autoimmunity. 2003;20:313–321. doi: 10.1016/s0896-8411(03)00038-6. [DOI] [PubMed] [Google Scholar]
- Brian JE, Jr, Faraci FM, Heistad DD. Recent insights into the regulation of cerebral circulation. Clin Exp Pharmacol Physiol. 1996;23:449–457. doi: 10.1111/j.1440-1681.1996.tb02760.x. [DOI] [PubMed] [Google Scholar]
- Brunn GJ, Saadi S, Platt JL. Differential regulation of endothelial cell activation by complement and interleukin 1alpha. Circ Res. 2006;98:793–800. doi: 10.1161/01.RES.0000216071.87981.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carl GF, Hoffman WH, Passmore GG, Truemper EJ, Lightsey AL, Cornwell PE, Jonah MH. Diabetic ketoacidosis promotes a prothrombotic state. Endocr Res. 2003;29:73–82. doi: 10.1081/erc-120018678. [DOI] [PubMed] [Google Scholar]
- Casarsa C, De Luigi A, Pausa M, DeSimoni MG, Tedesco F. Intracerebroventricular injection of the terminal complement complex causes inflammatory reaction in the rat brain. Eur J Immunol. 2003;33:1260–1270. doi: 10.1002/eji.200323574. [DOI] [PubMed] [Google Scholar]
- Chodobski A, Szmydynger-Chodobska J. Choroid plexus: target for polypeptides and site of their synthesis. Microsc Res Tech. 2001;52:65–82. doi: 10.1002/1097-0029(20010101)52:1<65::AID-JEMT9>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Di Lorento S, Caracciolo V, Colafarina S, Sebastiani P, Gasbarri A, Amicarelli F. Methylglyoxal induces oxidative stress-dependent cell injury and up-regulation of interleukin-1 beta and nerve growth factor in cultured hippocampal neuronal cells. Brain Res. 2004;1006:157–167. doi: 10.1016/j.brainres.2004.01.066. [DOI] [PubMed] [Google Scholar]
- Durr JA, Hoffman WH, Sklar AH, el Gammal T, Steinhart CM. Correlates of brain edema in uncontrolled IDDM. Diabetes. 1992;41:627–632. doi: 10.2337/diab.41.5.627. [DOI] [PubMed] [Google Scholar]
- Edge JA, Hawkins NM, Winter DL, Dunger DB. The risk and outcome of cerebral oedema developing during diabetic ketoacidosis. Arch Dis Child. 2001;85:16–22. doi: 10.1136/adc.85.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerich DF, Skinner SJ, Borlongan CV, Vasconcellos AV, Thanos CG. The choroid plexus in the rise, fall and repair of the brain. Bioessays. 2005;27:262–274. doi: 10.1002/bies.20193. [DOI] [PubMed] [Google Scholar]
- Eriksson C, Nobel S, Winblad B, Schultzberg M. Expression of interleukin 1 alpha and β, and interleukin 1 receptor antagonist mRNA in the rat central nervous system after peripheral administration of lipopolysaccharides. Cytokine. 2000;12:423–431. doi: 10.1006/cyto.1999.0582. [DOI] [PubMed] [Google Scholar]
- Eisenhut M. Interleukin-1 and the constellation of pulmonary oedema, and cerebral infarctions and brain oedema in diabetic ketoacidosis. Diabet Med. 2006;23:1386. doi: 10.1111/j.1464-5491.2006.02002.x. [DOI] [PubMed] [Google Scholar]
- Fishelson Z, Hochman I, Greene LE, Eisenberg E. Contribution of heat shock proteins to cell protection from complement-mediated lysis. Int Immunol. 2001;13:983–991. doi: 10.1093/intimm/13.8.983. [DOI] [PubMed] [Google Scholar]
- Fitzgerald MG, O’Sullivan DJ, Malins JM. Fatal diabetic ketoacidosis. Br Med J. 1961;1:247–250. doi: 10.1136/bmj.1.5221.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giffard RG, Xu L, Zhao H, Carrico W, Ouyang Y, Qiao Y, Sapolsky R, Steinberg G, Hu B, Yenari MA. Chaperones, protein aggregation, and brain protection from hypoxic/ischemic injury. J Exp Biol. 2004;207:3213–3220. doi: 10.1242/jeb.01034. [DOI] [PubMed] [Google Scholar]
- Glaser NS, Wootton-Gorges SL, Marcin JP, Buonocore MH, Dicarlo J, Neely EK, Barnes P, Bottomly J, Kuppermann N. Mechanism of cerebral edema in children with diabetic ketoacidosis. J Pediatr. 2004;145:164–171. doi: 10.1016/j.jpeds.2004.03.045. [DOI] [PubMed] [Google Scholar]
- Hickey MJ, Granger DN, Kubes P. Inducible nitric oxide synthase (iNOS) and regulation of leucocyte/endothelial cell interactions: studies in iNOS-deficient mice. Acta Physiol Scand. 2001;173:119–126. doi: 10.1046/j.1365-201X.2001.00892.x. [DOI] [PubMed] [Google Scholar]
- Hoffman WH, Steinhart CM, el Gammal T, Steele S, Cuadrado AR, Morse PK. Cranial CT in children and adolescents with diabetic ketoacidosis. Am J Neuroradiol. 1988;9:733–739. [PMC free article] [PubMed] [Google Scholar]
- Hoffman WH, Casanova MF, Bauza JA, Passmore GG, Sekul EA. Computer analysis of magnetic resonance imaging of the brain in children and adolescents after treatment of diabetic ketoacidosis. J Diabetes Complications. 1999;13:176–181. doi: 10.1016/s1056-8727(99)00042-2. [DOI] [PubMed] [Google Scholar]
- Hoffman WH, Cheng C, Passmore GG, Carroll JE, Hess D. Acetoacetate increases the expression of intercellular adhesion molecule-1 (ICAM-1) in human microvascular endothelial cells. Neurosci Lett. 2002;334:71–74. doi: 10.1016/s0304-3940(02)00816-9. [DOI] [PubMed] [Google Scholar]
- Hoffman WH, Burek CL, Waller JL, Fisher LE, Khichi M, Mellick LB. Cytokine response to diabetic ketoacidosis and its treatment. Clin Immunol. 2003;108:175–181. doi: 10.1016/s1521-6616(03)00144-x. [DOI] [PubMed] [Google Scholar]
- Hoffman WH, Cudrici CD, Zafranskaia E, Rus H. Complement activation in diabetic ketoacidosis brains. Exp Mol Pathol. 2006;80:283–288. doi: 10.1016/j.yexmp.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Holmin S, Mathiesen T. Intracerebral administration of interleukin-1β and induction of inflammation, apoptosis and vasogenic edema. J Neurosurg. 2000;92:108–120. doi: 10.3171/jns.2000.92.1.0108. [DOI] [PubMed] [Google Scholar]
- Hua Y, Wu J, Keep RF, Nakamura T, Hoff JT, Xi G. Tumor necrosis factor-alpha increases in the brain after intracerebral hemorrhage and thrombin stimulation. Neurosurgery. 2006;58:542–550. doi: 10.1227/01.NEU.0000197333.55473.AD. [DOI] [PubMed] [Google Scholar]
- Iori E, Calo L, Valbusa D, Ceolotto G, Milani M, Pengo V, de Kreutzenberg SV, Tiengo A, Avogaro A. Diabetic ketosis activates lymphocyte-inducible nitric oxide synthase. Diabet Med. 2002;19:777–783. doi: 10.1046/j.1464-5491.2002.00787.x. [DOI] [PubMed] [Google Scholar]
- Isales CM, Min L, Hoffman WH. Acetoacetate and β-hydroxybutyrate differentially regulate endothelin-1 and vascular endothelial growth factor in mouse brain microvascular endothelial cells. J Diabetes Complications. 1999;13:91–97. doi: 10.1016/s1056-8727(99)00030-6. [DOI] [PubMed] [Google Scholar]
- Jerath RS, Burek CL, Hoffman WH, Passmore GG. Complement activation in diabetic ketoacidosis and its treatment. Clin Immunol. 2005;116:11–17. doi: 10.1016/j.clim.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Kim SH, Kim J, Sharma RP. Inhibition of p38 and ERK MAP kinases blocks endotoxin-induced nitric oxide production and differentially modulate cytokine expression. Pharmacol Res. 2004;49:433–439. doi: 10.1016/j.phrs.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Konsman JP, Tridon V, Dantzer R. Diffusion and action of the intracerebroventricularly injected interleukin-1 in the CNS. Neuroscience. 2000;101:957–967. doi: 10.1016/s0306-4522(00)00403-6. [DOI] [PubMed] [Google Scholar]
- Lazovic J, Basu A, Lin HW, Rothstein RP, Krady JK, Smith MB, Levison SW. Neuroinflammation and both cytotoxic and vasogenic edema are reduced in interleukin type 1 receptor-deficient mice conferring neuroprotection. Stroke. 2005;36:2226–2231. doi: 10.1161/01.STR.0000182255.08162.6a. [DOI] [PubMed] [Google Scholar]
- Lu TS, Chen HW, Huang MH, Wang SJ, Yang RC. Heat shock treatment protects osmotic stress-induced dysfunction of the blood-brain barrier through preservation of tight junction proteins. Cell Stress Chaperones. 2004;9:369–377. doi: 10.1379/CSC-45R1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maness LM, Kastin AJ, Banks WA. Relative contributions of a CVO and the microvascular bed to delivery of blood-borne IL-1 alpha to the brain. Am J Physiol. 1998;275:E207–212. doi: 10.1152/ajpendo.1998.275.2.E207. [DOI] [PubMed] [Google Scholar]
- Marques F, Sousa JC, Correia-Neves M, Oliveira P, Sousa N, Palha JA. The choroids plexus response to peripheral inflammatory stimulus. Neuroscience. 2007;144:424–430. doi: 10.1016/j.neuroscience.2006.09.029. [DOI] [PubMed] [Google Scholar]
- Nag S, Picard P, Stewart DJ. expression of nitric oxide synthases and nitrotyrosine during blood-brain barrier breakdown and repair after cold injury. Lab Invest. 2001;81:41–49. doi: 10.1038/labinvest.3780210. [DOI] [PubMed] [Google Scholar]
- Niculescu F, Niculescu T, Rus H. C5b-9 terminal complement complex assembly on apoptotic cells in human arterial cell wall with atherosclerosis. Exp Mol Pathol. 2004;76:17–23. doi: 10.1016/j.yexmp.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Oglesbee MJ, Herdman AV, Passmore GG, Hoffman WH. Diabetic ketoacidosis increases extracellular levels of the major inducible 70-kDa heat shock protein. Clin Biochem. 2005;38:900–904. doi: 10.1016/j.clinbiochem.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Ouyang YB, Xu LJ, Sun YJ, Giffard RG. Overexpression of inducible heat shock protein 70 and it’s mutants in astrocytes is associated with maintance of mitochondrial physiology during glucose deprivation stress. Cell Stress Chaperones. 2006;11:180–186. doi: 10.1379/CSC-182R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardue S, Wang S, Miller MM, Morrison-Bogorad M. Elevated levels of inducible heat shock 70 proteins in human brain. Neurobiol Aging. 2007;28:314–324. doi: 10.1016/j.neurobiolaging.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Quan N, Stern EL, Whiteside MB, Herkenham M. Induction of pro-inflammatory cytokine mRNAs in the brain after peripheral injection of subseptic doses of lipopolysaccharide in the rat. J Neuroimmunol. 1999;93:72–80. doi: 10.1016/s0165-5728(98)00193-3. [DOI] [PubMed] [Google Scholar]
- Rus H, Cudrici C, David S, Niculescu F. The complement system in the central nervous system diseases. Autoimmunity. 2006;39:395–402. doi: 10.1080/08916930600739605. [DOI] [PubMed] [Google Scholar]
- Sharma HS, Wiklund L, Badgaiyan RD, Mohanty S, Alm P. Intracerebral administration of neuronal nitric oxide synthase antiserum attenuates traumatic brain injury-induced blood-brain barrier permeability, brain edema formation, and sensory motor disturbances in the rat. Acta Neurochir Suppl. 2006;96:288–294. doi: 10.1007/3-211-30714-1_62. [DOI] [PubMed] [Google Scholar]
- Singhrao SK, Neal JW, Rushmere NK, Morgan BP, Gasque P. Differential expression of individual complement regulators in the brain and choroid plexus. Lab Invest. 1999;79:1247–1259. [PubMed] [Google Scholar]
- Strazielle N, Khuth ST, Murat A, Chalon A, Giraudon P, Belin MF, Ghersi-Egen JF. Pro-inflammatory cytokines modulate matrix metalloproteinase secretion and organic anion transport at the blood-cerebrospinal fluid barrier. J Neuropathol Exp Neurol. 2003;62:1254–1264. doi: 10.1093/jnen/62.12.1254. [DOI] [PubMed] [Google Scholar]
- Togashi H, Mori K, Itoh Y, Matsumoto M, Ueno K, Ohashi S, Otani H, Yoshioka M. Involvement of interleukin-1beta/nitric oxide pathway in the post ischemic impairment of long- term potentiation of the rat hippocampus. Neurosci Lett. 2001;313:133–136. doi: 10.1016/s0304-3940(01)02271-6. [DOI] [PubMed] [Google Scholar]
- Topper JN, Wasserman SM, Anderson KR, Cai J, Falb D, Gimbrone MA. Expression of the bumetanide-sensitive Na-K-Cl cotransporter BSC2 is differentially regulated by fluid mechanical and inflammatory cytokine stimuli in vascular endothelium. J Clin Invest. 1997;99:2941–2949. doi: 10.1172/JCI119489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner KR, Beiler S, Beiler C, Kirkman J, Casey K, Robinson T, Larnard D, de Courten-Myers GM, Linke MJ, Zuccarello M. Delayed profound local brain hypothermia markedly reduces interleukin-1 beta gene expression and vasogenic edema development in a porcine model of intracerebral hemorrhage. Acta Neurochir Suppl. 2006;96:177–182. doi: 10.1007/3-211-30714-1_39. [DOI] [PubMed] [Google Scholar]
- Wendling U, Paul L, van der Zee R, Prakken B, Singh M, van Eden W. A conserved mycobacterial heat shock protein (hsp)70 sequence prevents adjuvant arthritis upon nasal administration and induces IL-10-producing T cells that cross react with the mammalian self-hsp70 homologue. J Immunol. 2000;164:2711–2717. doi: 10.4049/jimmunol.164.5.2711. [DOI] [PubMed] [Google Scholar]
- Xi G, Keep RF, Hoff JT. Pathophysiology of brain edema formation. Neurosurg Clin N Am. 2002;13:371–383. doi: 10.1016/s1042-3680(02)00007-4. [DOI] [PubMed] [Google Scholar]