

Abstract
Studies of the anti-tumor activity of TNF-α in vivo have been hampered by the need to administer systemically toxic doses of the cytokine to obtain a curative response. To facilitate studies of the effect of high local concentrations of TNF-α on tumor growth and host immunity, a newly induced murine sarcoma was transduced with the gene for human TNF-α and the biologic characteristics of these cells were examined. We identified high and low TNF-producing tumor clones which exhibited stable TNF secretion over time. Significant amounts of membrane associated TNF were found in a high-TNF producing clone as well. No difference in the in vitro growth rates between TNF-producing and nonproducing cell lines was observed. In contrast, in vivo studies demonstrate that although unmodified parental tumor cells grew progressively when implanted s.c. in animals, tumor cells transduced with the TNF gene were found to regress in a significant number of animals after an initial phase of growth. This effect correlated with the amount of TNF produced and could be blocked with a specific anti-TNF antibody. Regressions of TNF-producing cells occurred in the absence of any demonstrable toxicity in the animals bearing these tumors. TNF-producing tumor cells could function in a paracrine fashion by inhibiting the growth of unmodified, parental tumor cells implanted at the same site. The ability of tumor cells to regress was abrogated by in vivo depletion of CD4+ or CD8+ T cell subsets and animals that had experienced regression of TNF-producing tumors rejected subsequent challenges of parental tumor. Our studies thus show that tumor cells elaborating high local concentrations of TNF regress in the absence of toxicity in the host and that this process requires the existence of intact host immunity. Studies of the lymphocytes infiltrating the gene modified tumors and attempts to use TNF gene modified tumor infiltrating lymphocytes to deliver high local concentrations of TNF to the tumor site without inducing systemic toxicity are underway.
TNF is a cytokine that possesses a wide variety of biologic activities including potent anti-tumor activity (1-8) and immunomodulatory properties such as enhancement of monocyte/macrophage (9, 10) and polymorphonuclear neutrophil (11) cytotoxic activities, increased T cell proliferation and IL-2R expression (12, 13) and augmentation of cytotoxic T lymphocyte (14, 15) and i.e. lymphokine activated killer LAK2 (16) development.
Previous work from our laboratory has focused on the potential use of TNF as an immunotherapeutic agent against cancer. We have reported that in vivo anti-tumor responses to systemically administered TNF were correlated with relative tumor immunogenicity (2) and that CD8+ effector lymphocytes played an important role in the in vivo TNF mediated regression of these tumors (17). These observations are consistent with reports from others that a thymus dependent host factor augments the in vivo activity of TNF (5) and that TNF is capable of causing necrosis, but not complete regression of an immunogenic tumor growing in T cell-deficient mice (18). In certain tumor systems, therefore, the therapeutic activity of TNF has been found to rely on immunologically dependent mechanisms.
To date, studies of TNF-mediated tumor regression in vivo in mice have been hampered by the need to systemically administer toxic doses to obtain a curative response (2, 18, 19). In addition, the incidence of complete tumor regression after the administration of toxic doses of TNF to tumor-bearing animals has been low (2, 18, 19). The problem of dose-limiting toxicity of TNF has been particularly apparent in human trials of the cytokine in which the maximal tolerated dose of TNF (10 μg/kg) was 40-fold less on a per kg basis than the doses that were required to generate a significant anti-tumor response in mice (400 μg/kg) (2, 20).
Because of the difficulties associated with the systemic administration of TNF, we sought to examine the effect of high local concentrations of the cytokine on tumor growth and host immunity using a retroviral construct containing the gene for human TNF-α (21) to produce murine tumor cell lines that constitutively produced TNF-α.
In this paper we report that high local concentrations of TNF are capable of causing regression of a murine tumor in vivo via immune mechanisms in the absence of demonstrable toxicity. The implications of these findings for immunotherapy using gene-altered TIL and the potential mechanisms responsible for the elimination of TNF-transduced tumor cells in vivo are discussed.
MATERIALS AND METHODS
Animals
Female C57BL/6 mice (B6). 10 to 12 wk old, were obtained from the Animal Production Colonies, Frederick Cancer Research Facility, National Institutes of Health (NIH), Frederick, MD.
Tumors
The weakly immunogenic MCA-205 sarcoma, syngeneic to B6 mice, was generated in our laboratory by i.m. injection of 0.1 ml of 0.1% MCA in sesame seed oil as described previously (22) and was maintained in vivo by serial passage. The WP-4 cell line that was used in these experiments is one of a series of clones derived from the MCA-205 tumor. Briefly, a single cell suspension of fresh MCA-205 tumor from the second transplant generation was prepared as described elsewhere (2). Tumor clones were then grown in 96-well flat-bottom tissue culture plates (Costar, Cambridge, MA) by limiting dilution technique at 0.3 cells/well. WP-4 was maintained as a monolayer culture in CM containing RPMI 1640 (Biofluids, Rockville, MD). 0.1 mM nonessential amino acids, 1.0 mM sodium pyruvate (both from Biofluids), 5 × 10−5 M 2-ME (Aldrich Chemical Co., Milwaukee, WI). 0.03% fresh glutamine, 100 U/ml penicillin (both from NIH media unit), 0.5 μg/ml amphotericin B (Flow Laboratories, McLean. VA), and 10% heat-inactivated FCS (Biofluids).
Gene transfer into cell cultures
WP-4 tumor cells were transduced with a TNF retroviral vector in the presence of polybrene (8 μg/ml) for 8 h at 37°C. The retroviral vector contained a human TNF cDNA under the transcriptional control of LTR from Moloney murine leukemia virus and the NeoR gene under transcriptional control of the SV-40 early region promoter and enhancer (21, 23). The transduced cells were selected for NeoR gene expression by exposure to the Neo analog G418 (GIBCO, Grand Island, NY) at a concentration of 500 to 600 μg/ml (active drug) for 12 to 15 days beginning 2 days after transduction. G418-resistant colonies were pooled to yield a bulk transduced culture designated TNF-B.
Transfections of WP-4 tumor cells were also carried out with CMV- LTR-neo, a construct containing the neomycin resistance gene without TNF. This plasmid, kindly supplied by I.S.Y. Chen (24) is based upon pSV2neo (25) with the SV40 early promoter replaced by a hybrid CMV/HTLV-I LTR promoter. Transfections were performed with calcium phosphate precipitation (26) without glycerol shock. Transfectants were selected in G418 (400 μg/ml) beginning two days after transfection. G418-resistant colonies were pooled to yield a bulk transfected culture designated NEO.
DNA and RNA blot analysis
For detection of vector DNA in TNF-transduced cells, total genomic DNA (15 μg) was isolated and digested to completion with EcoRI. The digested DNA was fractionated in 0.8% agarose gels, the framents were transferred to nylon membrane, and hybridized to a 32P-labeled TNF cDNA probe. In addition, for the expression studies on Northern blots, total cellular RNA (20 μg) was separated on a 1% agarose-formaldehyde gel, transferred to a nitrocellulose membrane, and hybridized to a 32P-labeled TNF cDNA.
Cloning of tumor cell cultures and assay of TNF activity
The bulk TNF-transduced culture of WP-4 tumor cells, TNF-B, was cloned by limiting dilution technique to yield several cell lines that were designated TNF-1 through TNF-29. The unmodified, parental WP-4 tumor culture was subcloned in a similar manner. All cell cultures were then tested for production of secreted TNF. One million cells of each culture were placed into individual wells of 24-well culture plates (Costar) along with 2 ml of fresh CM. After 24 h, the medium was removed from each well and centrifuged to remove any contaminating cells. TNF activity in the samples was then determined by a modified version of the L929 bioassay as described previously (2) or by a quantitative ELISA (R&D systems, Minneapolis, MN). Tumor clones producing high (TNF-12,26) and low (TNF-2,28) levels of TNF, along with TNF-B and WP-4.9, a non-TNF producing subclone of the unmodified parental culture, were chosen for further experiments.
G418 sensitivity of tumor cell lines
Single cell suspensions of the tumor cell lines were prepared from confluent cultures by brief trypsinization with a solution of 0.05% trypsin and 0.02% EDTA (trypsin/versine, Biofluids). Cells were then washed thrice in HBSS (Biofluids) and were resuspended at 2 × 105 cells/ml in CM. Aliquots (100 μl) of each cell suspension were dispensed into each well of 96-well flat bottom tissue culture plates. Solutions of G418 (G418 Sulfate, GIBCO) ranging in concentration from 0.2 to 2 mg/ml (active) in 0.2 mg increments were then prepared in CM and 100-μl aliquots of each solution were added to individual rows of the plates containing the cells (yielding a final concentration range of 0.1 to 1.0 mg/ml G418). In addition, two rows of cells received 100 μl of CM only and served as a control for cell viability in the absence of G418. The plates were then incubated for 4 days at 37°C in a humidified 7% CO2-air mixture. At the end of this culture period 100 μl of media in one of the rows that had CM without (G418 was removed and was replaced with 100 μl of 0.2% SDS. After the addition of SDS the plates were incubated for another 8 h at 37°C. After this incubation, the culture medium was removed by inverting and flicking the plate. The plates were then washed twice with phosphate buffered saline and the remaining adherent cells were fixed with a 3:1 methanol:acetic acid solution and then stained for 15 min with a solution of 10% ethanol with 0.5% crystal violet (NIH media unit). The plates were then washed with water to remove the excess dye and were allowed to dry. The remaining dye was then solubilized by placing 150 μl of a solution containing 10% acetic acid and 20% ethanol into each well and lightly vortexing the plates. The OD of each well was measured with a microplate spectrophotometer (Titertek Multiscan, Flow Laboratories, Helsinki, Finland] equipped with a 590-nm filter. The percent survival of cells exposed to G418 was calculated as follows: [(OD experimental wells − OD maximal cell death (SDS))]/(OD maximal viability (CM only) − OD maximal cell death)) × 100.
FMF anaIysis
Cell surface expression of TNF-α was determined by flow cytometry on a FACS 440 (Becton Dickinson, Mountain View, CA). Cell lines were harvested with 0.02% EDTA, washed, and then stained as described elsewhere (27) with either an antihuman TNF-α mAb (Olympus, Lake Success, NY) or an isotype-matched (IgGl) nonspecific control antibody (Becton Dickinson) in conjunction with a goat anti-mouse FITC conjugated antibody (Boehringer Mannheim Biochemicals, Indianapolis, IN).
In vivo tumor model
Confluent cultures of transduced and non-transduced cell lines were harvested with trypsin/EDTA, washed, and adjusted to the desired cell concentration. B6 mice were inoculated in the skin of the right flank with 8 × 106 to 1 × 107 viable tumor cells in 0.1 ml of HBSS (for tumor mixture experiments, 1 × 107 TNF transduced cells were combined with 1 × 106 nontransduced tumor cells). The tumor cells grew (s.c.) as confirmed by histologic analysis. After injection of tumor cells, the mice were ear tagged, randomized, and were monitored for tumor progression or regression. Tumor size was determined by measuring perpendicular tumor diameters with a vernier caliper. All measurements were done in a coded, blind fashion. Size was recorded as a product of perpendicular measurements.
In vivo depletion of T cell subsets
Two hybridomas producing rat IgG2b mAbs against the CD4 (GK1.5) and CD8 (2.43) T cell Ag were obtained from American Type Tissue Collection, Rockville, MD. The 2.43 mAb was harvested as ascites from sublethally irradiated (500 rad) DBA/2 mice. The GK1.5 mAb was purified from concentrated culture supernatants using ammonium sulfate. For in vivo depletion, each B6 mouse received one i.v. injection of 1.0 ml diluted mAb, in which 0.2 ml of 2.43 monoclonal ascitic fluid or 300 μg of GK1.5 mAb was mixed with HBSS. This procedure has been previously shown to be effective in producing long term (7 to 10 days) depletion of T cell subpopulations in vivo (28-30). Injection of diluted ascites containing an IgG2a mAb (1A14, a gift from Dr. Richard Alexander, National Cancer Institute, NIH). directed against the Thy-1.1 T cell Ag, served as an irrelevant control.
In vivo administration of anti-TNF antibodies
A murine mAb, TNF-E, which blocks the activity of human rTNF-α was kindly provided by Dr. Brian Fendly (Genentech Corporation, San Francisco, CA). TNF-E was protein A purified from ascites fluid and 100 μg of this mAb were injected i.p. q 3 to 4 days into mice bearing TNF-transduced tumors. The ability of this mAb to effectively block the activity of human TNF-α in vitro and in vivo has been demonstrated by others (31, 32).An isotype-matched murine mAb, 354(Genentech), which was raised to hamster tissue plasminogen activator was used as a negative control.
Evaluation of TNF production in vivo
TNF transduced tumors, approximately 1 cm in diameter, were excised from animals that had been immunodepleted with anti-CD4 antibody and were finely minced in 5 ml of HBSS. The resultant tumor suspensions were then centrifuged at 5000 rpm for 15 min and the supernatants were assayed for TNF contentusing a specific immunoassay. Tumors from immunodepleted animals were used in these experiments as they grew to a much larger size than TNF-producing tumors in immunocompetent animals and therefore provided more material for analysis.
RESULTS
lntegration and expression of TNF-α gene in tumor cells
The presence of the proviral sequences in transduced cells was confirmed by Southern blot analysis of EcoRI-digested DNA hybridized with the TNF cDNA probe. The expected 1-kb EcoRI fragment (21) containing the full length exogenous TNF gene was detectable in the transduced cultures (data not shown). The transduced cells also expressed the proviral (approximately 4 kb) transcribed mRNA that was undetectable in the non-transduced cells (not shown).
TNF production by the bulk TNF-transduced culture, TNF-B, and by clones of this culture was followed over time using a TNF-specific immunoassay (Fig. 1). TNF production by the different cell lines remained relatively stable over time. Low TNF-producing lines, TNF-2 and TNF-28, made an average of 418 ± 71 (n = 6)and 455 ± 109(n = 6) pg/106 cells/24 h, respectively, over a 63-day period with a range of production between 182 and 760 pg/l06 cells/24 h. High TNF producing clones, TNF-12 and TNF-26, made an average of 12,785 ± 843 (n = 7) and 9,214 ± 1,467 (n = 7) pg/l06 cells/24 h over the same time period and had a range of production between 3,800 and 15,200 pg/106/24 h. Bulk transduced tumor, TNF-B, was assayed over a 20-day period and showed an average production of 11,667 ± 667 (n = 3) pg/106 cells/24 h. Biologic activity of the TNF secreted by the bulk transduced tumor was confirmed in repeated L929 bioassays in which TNF-B cultures made between 150 and 250 U TNF/ml/106 cells in a 24-h period (data not shown). No TNF activity was detected in unmodified or neo-transfected WP-4 tumor cell lines in multiple assays.
Figure 1.
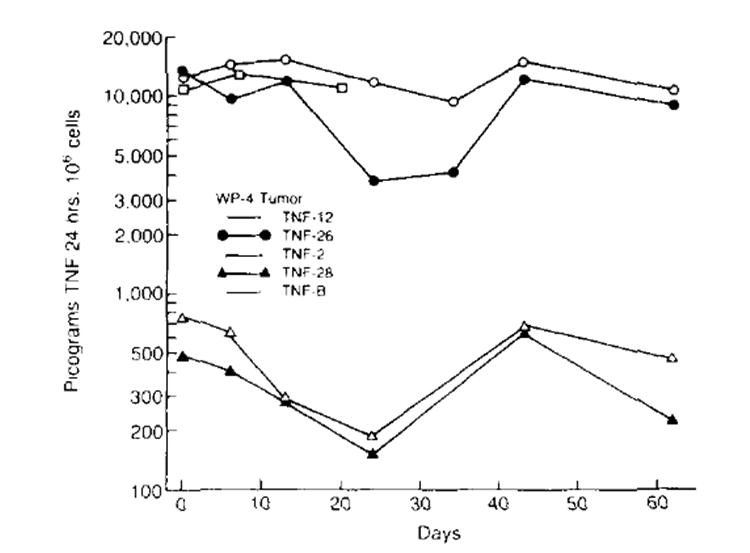
Secretion of TNF by transduced cell lines over time. TNF production of each cell line was measured by a specific immunoassay. TNF production of various cell lines remained relatively stable over time High TNF-producing cell lines. TNF-12, TNF-26 and TNF-B (bulk transduced WP-4 tumor) made an average of 12,785 ± 843 (n = 7), 9,214 ± 1,467 (n = 7) and 11,667 ± 667 (n= 3) pg/106 cells/24 h, respectively. Low producing TNF cell lines TNF-2 and TNF-28 made a n average of 418 ± 71 (n = 6) and 455 ± 109 (n = 6) pg/106 cells/24 h, respectively. No TNF activity was detected in unmodified or neo-transfected tumor cell lines.
Resistance of gene-modified tumor cell lines to G418
Gene-modified tumor cell lines were assayed for resistance to G418. Unmodified, parental WP-4 tumor cells were quite sensitive to low concentrations of G418, with more than 95% cell death at 0.2 mg/ml G418. In contrast, five WP-4 tumor cell lines, transduced with the construct coding for both neo resistance and human TNF (four cloned cell lines and the parental TNF-B), were highly resistant to G418 concentrations up to 1 mg/ml (data not shown). A neo-resistant control cell culture (transfected with the neo gene alone) showed lower, but still significant, resistance to high concentrations of G418. No obvious relationship between relative G418 sensitivity and tumor growth in vivo was observed.
Presence of membrane-associated human TNF on transduced murine tumor cells
A subclone of the unmodified WP-4 parental culture, designated WP 4.9, as well as two TNF producing clones TNF-12 and TNF-28, were examined by FMF analysis for presence of membrane associated human TNF. One of two consecutive experiments is shown in Figure 2. No evidence of membrane associated human TNF was observed on the non-transduced cell line, WP-4.9. In contrast, the high TNF producing clone, TNF-12, was found to have significant amounts of membrane associated TNF and >95% of the cells analyzed stained positively for the cytokine. In comparison, TNF-28, a low TNF producer, was found to express a very low amount of surface human TNF. A repeat of this experiment showed similar results. No TNF has been observed on the surface of neo-transfected control cells after a 24-h incubation in high concentrations (100 ng/ml) of added human rTNF (data not shown).
Figure 2.
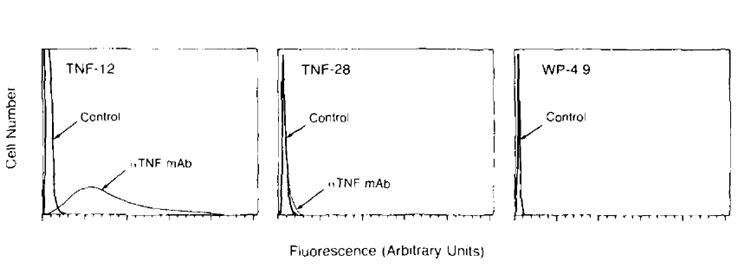
Flow microfluorimetric analysis of tumor cells stained with anti-human TNF antibody. A clone of the unmodified WP-4 parental culture. WP 4.9, as well as two TNF-producing clones, TNF-12 (high producer) and TNF-28 (low producer) were examined for the presence of membrane-associated human TNF. Fluorescence intensity was measured in arbitrary linear units. The 105 cells were analyzed in each frame. Negative controls stained with irrelevant antibody are represented by the sharp peaks to the far left. Cells staining positively for TNF are represented by the curves seen to the right of the controls. The high TNF producing clone was found to have significant amounts of membrane associated TNF (95% of the cells stained positively for TNF). In comparison, TNF-28, a low TNF secreter was found to express a very low amount of surface TNF. No evidence of membrane associated TNF was found on the nontransduced cell line WP 4.9.
In vivo growth of tumor
Figure 3 shows the results of a representative experiment in which the growth of unmodified and gene-modified tumor cells was measured. Unmodified and neotransfected WP-4 tumor cells grew progressively in animals over time and no regressions of these tumors were observed. In contrast, bulk TNF-transduced WP-4 cells typically grew progressively for an 8- to 12-day period at which point several went on to regress completely. In four consecutive experiments in which in vivo growth of these cell lines was observed, tumor incidence in the time period between 3 and 4 wk after injection was 24/24 (100%), 42/42 (100%), and 13/41 (31.7%) for unmodified, neo-transfected, and TNF-transduced WP-4 tumor cells, respectively. In the particular experiment shown, 91% of the TNF-transduced tumors regressed. In multiple experiments, no animals bearing TNF-transduced tumors have been observed to exhibit signs of TNF toxicity (lethargy, rigors). In addition, we have found no significant difference in body weight changes of animals bearing TNF-producing tumors compared with animals bearing unmodified WP-4 tumors in the first 28 days after tumor injection (data not shown).
Figure 3.
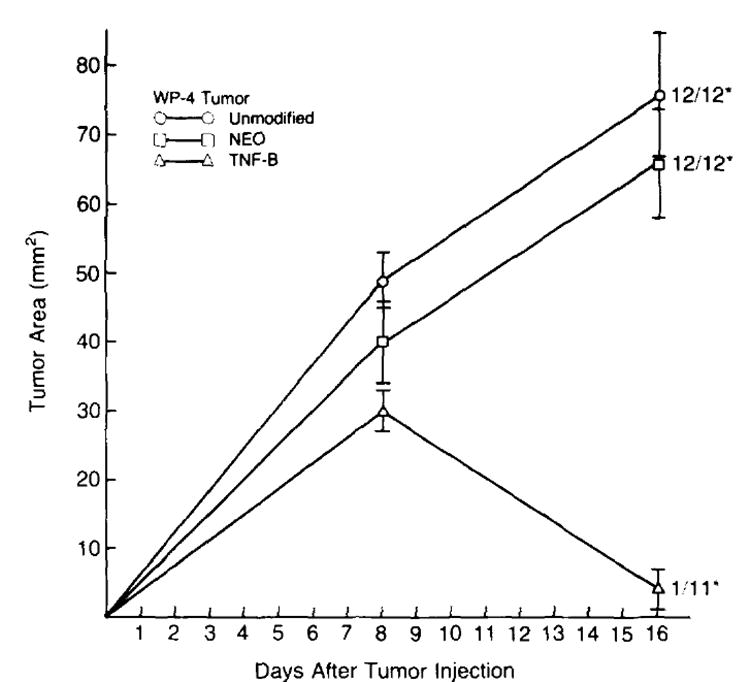
In vivo growth of unmodified and gene modified tumor cells. B6 animals were injected s.c. with 1 × 107 neo-transfected (NEO) , TNF-transduced (TNF-B).or unmodified WP-4 tumor cells on day 0 and tumor growth was followed over time. Unmodified and neo-transfected WP-4 tumors grew progressively over time and no regressions were observed. In contrast, bulk TNF-transduced tumor cells typically grew progressively over an 8- to 12-day period at which point approximately 70% regressed completely. In the representative experiment shown here, 91% of the TNF-transduced tumors regressed. (* number with tumor/total at day 28. Tumor area measurements are plotted for each time point and are expressed as mean ± SEM.)
The 10 animals that experienced regression of TNF-transduced tumor in the experiment shown in Figure 3 were rechallenged 2 wk after tumor disappearance with 1 × 106 viable unmodified WP-4 tumor cells at the s.c. site distant from the site of the original tumor. Ten normal control animals were challenged in a similar fashion After 40 days of observation, none of the animals that had their TNF tumors grow and regress developed tumor. In contrast, 10/10 of the control animals developed palpable tumor nodules.
Tumor progression/regressionin vivo is related to TNF production in vitro and in vivo
Figure 4 shows two consecutive experiments in which the in vivo growth of high and low TNF-producing WP-4 tumor clones was assessed. In both experiments, low TNF-producing tumor clones (TNF-2 and TNF-28, making an average of 418 and 455 pg TNF/106 cells/24 h, respectively) grew progressively over time in all but one animal. In contrast, animals bearing high TNF-producing tumor clones (TNF-12 and TNF-26, making an average of 12,785 and 9,214 pg TNF/106 cells/24 h , respectively) experienced significant reductions in average tumor size after an initial period of tumor growth, and several animals experienced complete tumor regression. In four consecutive experiments in which the in vivo growth of these clones was observed, tumor incidence in the time period between 3 and 4 wk after tumor injection was 16/56 (28.6%) and 54/56 (96.4%) for high and low TNF producing clones, respectively (p = <0.01). In contrast to these in vivo experiments, no significant difference in in vitro growth rates of TNF transduced, neo-transfected or unmodified WP-4 tumor cultures have been observed (data not shown).
Figure 4.
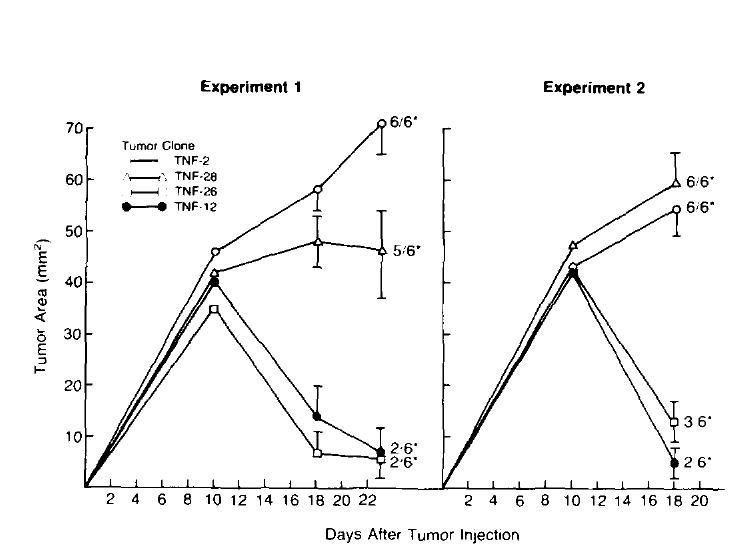
Growth of TNF producing tumor clones in mice. Two consecutive experiments in which the in vivo growth of different TNF-producing clones was assessed. On day 0, B6 mice received 1 × 107 tumor cells s.c. and tumor growth was followed over time. In both experiments, low TNF-producing clones (TNF-2 and TNF-28) grew progressively in all but one animal. In contrast, animals bearing high TNF producing tumors (TNF-12. TNF-26) experienced significant reductions in average tumor size after an initial period of tumor growth, and several animals experienced complete tumor regression. (* number with tumor/total at day 28, Tumor area measurements are plotted for each time point and are shown mean ± SEM.)
To further determine the role of TNF in the regression of transduced tumors, experiments were performed in which the growth of human TNF transduced cells was observed in animals given control or specific anti-human TNF antibody. Figure 5 shows one of two consecutive experiments. Administration of anti-TNF antibody abrogated the regression of TNF-transduced tumors. In contrast, control antibody had no effect on the tumor’s ability to regress after an initial period of growth. Thus, regression of TNF transduced tumors was related to the local elaboration of TNF.
Figure 5.
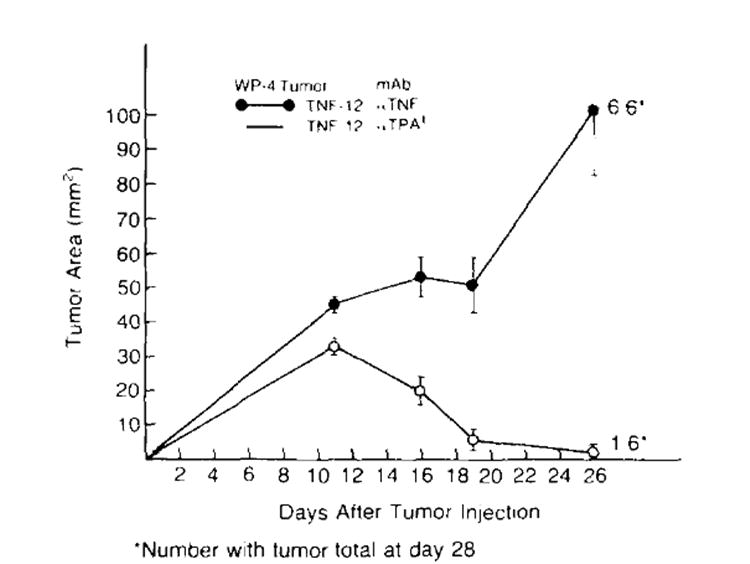
Anti-TNF antibody blocks the regression of a TNF-producing tumor. TNF-12 cells (1 × 107) (a high TNF-producing clone) were inoculated s.c. into B6 mice on day 0. The animals were then divided into two experimental groups. Group one was given 100 μg of a specific anti-human TNF antibody i.p. q 3 to 4 days. Group two was treated in the same fashion with an identical quantity of control irrelevant antibody (+mouse anti-hamster tissue plasminogen activator). Tumor growth was followed. Administration of anti-TNF antibody resulted in progressive growth of TNF-producing tumors. In contrast, control antibody had no effect on the ability of these tumors to regress after an initial period of growth. (*number with tumor/total at day 28. Tumor area measurements are plotted for each time point and are shown mean ± SEM.)
In vivo depletion of T cell subsets abrogates regression of TNF-transduced tumors
Treatment of B6 mice with one intravenous dose of anti-CD4 or anti-CD8 mAb produced a pronounced reduction of spleen cells bearing the CD4 or CD8 phenotype, respectively (28-30) (data not shown). In contrast, administration of anti-Thy-1.1 mAb had no effect on the number of Thy-1.2 cells in the spleen. Figure 6 shows two consecutive experiments in which animals treated with anti-Thy-1.1, anti-CD4, or anti-CD8 mAb were subsequently given s.c. injections of TNF-producing cells and tumor growth was monitored over time. Animals treated with control mAb. Thy-1.1, experienced significant reductions in tumor sizes after an initial phase of positive tumor growth and 9 of 12 animals went on to experience complete tumor regression. In contrast, TNF-transduced tumors in animals given anti-CD4 or anti-CD8 mAb grew progressively overtime and none of these tumors regressed. In related experiments, TNF-transduced tumors grew progressively in mice acutely immunosuppressed by a sublethal (500 rad) dose of whole body irradiation before tumor injection (data not shown).
Figure 6.
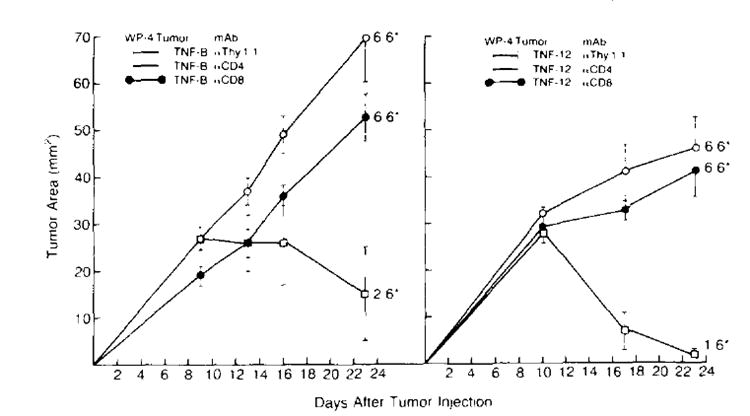
In vivo depletion of CD4+ or CD8+ T cell subsets eliminates regression of TNF-producing tumors. B6 mice received a single i.v. dose of either anti-CD4. anti-CD8 or anti-Thy-1.1 mAb on day 0. Two hours after treatment with mAb, mice were inoculated s.c. with 1 × 107 TNF-producing tumor cells (in experiment 1, TNF-B: in experiment 2. TNF-12) and tumor growth was followed. In both experiments, elimination of CD4+ or CD8+ cells abrogated the ability of TNF-producing cells to regress. In contrast, animals treated with anti-Thy-l.1 control mAb experienced significant reductions in tumor sizes after an initial phase of tumor growth and 9/12 animals went on to experience complete tumor regression. (*number with tumor/total at day 28. Tumor area measurements are plotted for each time point and are shown mean ± SEM.)
TNF-transduced tumors that grew in immunosuppressed mice were producing TNF
Table I shows the results of three experiments in which tumors from immunodepleted animals were analyzed for TNF content (tumors from immunodepleted animals were used for these experiments because they grew to a much larger size than did similar tumors growing in immunocompetent animals and therefore provided more material for analysis). Significant levels of TNF could be detected in the TNF-transduced tumors and no TNF was found in the neo-transfected tumor preparations. Low-TNF producing cell lines were not examined. No TNF has been detected in the serum of animals bearing TNF-transduced tumors.
TABLE I.
TNF content of fresh tumor preparationsa
| Tumor | TNF Content of Supernatants (pg/ml)b | ||
|---|---|---|---|
| Expt. 1 | Expt. 2 | Expt. 3 | |
| TNF-B | 399 | 30 | 126 |
| Neo | 0 | 0 | 0 |
Progressively growing tumors were taken from animals that had been treated with anti-CD4 antibody as these tumors grew to larger sizes than TNF-producing tumors in immunocompetent animals and therefore provided more material for analysis.
Fresh tumors were minced in a small volume of HBSS. The resultant preparation was then centrifuged and the supernatants were assayed for TNF content using a specific immunoassay.
TNF-producing tumor cells inhibit growth of unmodified parental tumor cells injected at same, but not different site
Figure 7 shows two experiments in which 1 × 107 TNF-producing tumor cells inhibited the growth of 1 × 106 unmodified WP-4 tumor cells when the two were mixed and coinjected s.c. In contrast, 1 × 106 non-transduced cells, inoculated alone, were found to grow progressively in all mice. Neo-transfected control tumor cells had no effect on the growth of unmodified WP-4 cells (data not shown). The effect of TNF-producing tumor cells on non-cross-reacting tumor cells has not been examined. TNF-transduced WP-4 tumor cells growing s.c. had no effect on the growth of unmodified WP-4 tumor cells growing at a distant s.c. site, nor did they have an effect on the growth of 3-day pulmonary metastases from that tumor (data not shown).
Figure 7.
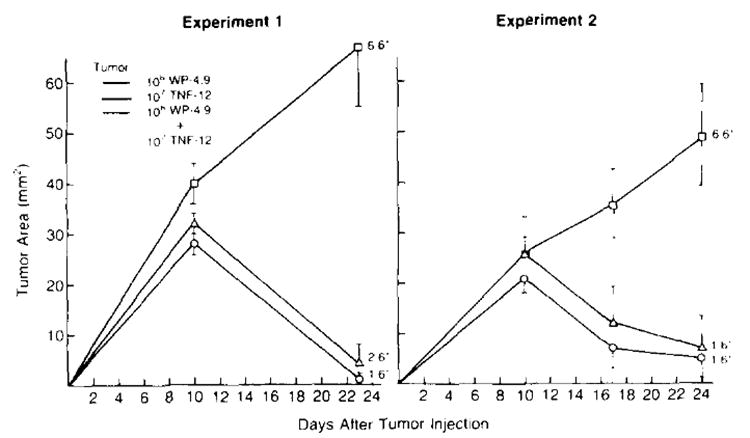
TNF-producing tumor cells inhibit the growth of unmodified tumor cells inoculated at the same site. The 1 × 106 WP-4.9 tumor cells (a clone of the unmodified parental culture WP-4) and 1 × 107 TNF-12 tumor cells (a high TNF-producing clone) were injected s.c. on day 0 into B6 animals either alone or mixed together and tumor growth was followed. Although WP-4.9 tumor cells injected alone grew progressively in all animals, animals bearing TNF-12 cells or the mixture of TNF producing and nonproducing cells experienced regression of tumor after an initial phase of tumor growth. (*number with tumor/total at day 28. Tumor measurements are plotted for each time point and are shown mean ± SEM)
DISCUSSION
Recently, investigators have described the biology of tumor cells into which cDNA coding for a variety of cytokine genes has been inserted (33-36). A potential benefit of such systems is that they can allow for the provision of high local concentrations of cytokines while avoiding the systemic toxicity that can be associated with parenteral administration of the cytokine. Because toxic doses of TNF are often required to observe significant anti-tumor effects in vivo (2, 18, 19). we introduced the gene for human TNF-α into murine tumor cells in order to more thoroughly characterize the antitumor and immunomodulatory effects of this cytokine.
In our studies tumor cells transduced with the TNF gene grew and regressed in a significant number of animals; unmodified tumor cells grew progressively. This effect appeared to correlate with the amount of TNF produced by the tumor cells and could be blocked with the systemic administration of a mAb directed against human TNF-α. Tumor regression occurred in the absence of obvious host toxicity. TNF transduced cells functioned in a paracrine fashion by inhibiting the growth of unmodified, parental tumor cells implanted at the same site. Additionally, the ability of TNF transduced cells to grow and regress was dependent on an intact host immune system.
We have detected TNF in tumors transduced with the TNF gene but not in the serum of animals bearing these tumors. These observations may explain why TNF-producing tumor cells used in these experiments influenced the growth of unmodified tumor cells growing locally, but not at distant sites and suggest that high intratumoral concentrations of TNF are important for tumor elimination. By increasing TNF production by these cells (i.e., by introducing multiple copies of the TNF gene) one could potentially observe an effect on the growth of distant tumors. Such an approach, however, could lead to the development of TNF-induced toxicity because previous studies with TNF-transfected tumors (making approximately 250 times the amount of TNF our tumor cells can) growing i.p. in mice and generating high systemic levels of TNF led to severe cachexia(37).
Although high local concentrations of TNF appear to be important for tumor cell elimination in vivo, the data presented here suggest that indirect mechanism(s) are responsible for this process because immunosuppression of animals eliminated the ability of TNF-transduced tumors to regress. Moreover, no differences were noted in the in vitro growth rates of TNF-producing and nonproducing cell lines, and even the addition of high concentrations of recombinant TNF (up to 100 ng/ml) in vitro was not toxic to tumor cells (data not shown).
The observations that depletion of T cell subsets prevents the regression of TNF-transduced tumors, and animals experiencing complete regression of TNF-transduced tumors were immune to subsequent challenge normal tumor, are consistent with previous reports from our laboratory (17) and others (18) that described the requirement of T cell-mediated immunity in the elimination of tumors by exogenously administered TNF. The precise role of T cells in TNF-mediated elimination of tumors, however, remains to be determined. Other investigators have described modulation of T cell and LAK cell responses by TNF in other systems (12-16). It is possible, therefore, that TNF mediates regression of tumors by enhancing host T cell responses.
The ability of TNF-transduced cells to inhibit the growth of nontransduced cells implanted in the same vicinity demonstrates that the effect exerted by these cells can be transmitted locally and is not entirely restricted to the transduced cells themselves. It is possible, however, that TNF-transduced cells possess qualities that make them more susceptible to attack in vivo such as the surface expression of immunogenic foreign gene products coded by the expression vector. We have, in fact, observed occasional regressions of tumor cell lines transduced with a different expression vector composed of the NeoR gene with a CMV promoter (data not shown). It is also possible that the selection process itself may have given rise to highly immunogenic clones that would regress irrespective of TNF secretion. Such explanations are unlikely, however, in the case of high TNF-producing tumors because their regression was completely inhibited by the presence of an anti-TNF antibody: in contrast, anti-TNF antibodies had no effect on the tendency of CMV-neo transduced clones to regress (not shown). Further, low TNF-producing cells, transduced with the identical expression vector used to generate high producers and exhibiting equivalent G418 resistance, grew progressively in virtually all animals tested. We are currently investigating the mechanism(s) responsible for the regression of certain CMV-neo-transduced tumor cell lines.
Although high TNF producer tumor clones regress and low producer clones do not, it is not clear that secreted TNF is solely responsible for the anti-tumor response in vivo. The retroviral vector used in these experiments has been demonstrated to code for the production of both the secretory 17- and the 26-kDa trans-membrane forms of TNF (21).In addition, FMF analyses reported here confirm the presence of membrane associated TNF on the surface of TNF-transduced tumor cells. Other investigators have recently demonstrated the existence of membrane-bound TNF on activated CTL (38, 39) and on monocytes (21) raising the possibility that this form of TNF plays a role in modulating host immunity.
TNF does not appear to exert its effect on transduced cells by changing their inherent lysability by nonspecific effector cells as we have observed no difference in LAK lysis of these cells compared to nontransduced controls in 4-h Cr51 release assays (data not shown). Moreover, although cytokine pretreatment of tumor cells in vivo or in vitro (40) has been shown to up-regulate MHC class I expression, we have not observed any demonstrable changes in class I expression on TNF-transduced cells compared with unmodified or neo-transfected controls (data not shown). We are presently attempting to determine whether TNF-transduced tumors are more susceptible to cytotoxic T cell killing in vitro.
Although regression of TNF-transduced tumors appears to be dependent on TNF, the final effector mechanism(s) leading to tumor elimination remain unknown. Secreted or membrane-bound TNF may not be the only factors responsible for regression of TNF-transduced tumors in vivo. Earlier studies in our laboratory showing that IL-6 is produced in animals after systemic administration of TNF (41, 42) and that exogenous IL-6-mediates tumor regression in vivo (43), suggest that other cytokines may also be involved in this process.
The finding that TNF-transduced cells grew to a diameter of approximately 5 to 6 mm before they regressed is reminiscent of observations made previously (1, 44) demonstrating a size-dependent response of tumor to exogenously administered TNF. Although it is not clear how the size of TNF-transduced tumors relates to their ability to regress, it is possible that a critical tumor mass is required for the induction of a host immune response. Another possibility is that size is important for the maturation stage of blood vessels growing within the tumor as exogenously administered TNF is well known to cause extensive central hemorrhagic necrosis in tumors growing s.c in experimental animals (1, 2, 4, 5, 7). However, in histologic analyses in which we have examined TNF- transduced tumors taken from animals at days 8 and 15 after tumor injection we have observed no evidence of central hemorrhagic necrosis despite the fact that many tumors were obviously regressing (data not shown). In these same studies, TNF-transduced tumors were to contain increased numbers of accompanying lymphocytes as compared with unmodified and neo-transduced tumors. No significant differences in other inflammatory cell types were noted. Further histologic examinations at intermediate time points are being performed in an effort to determine whether TNF-transduced tumors show evidence of earlier hemorrhagic necrosis or possessa characteristic inflammatory infiltrate.
Because of the involvement of host T cell immunity in the elimination of TNF-transduced tumors, we are currently studying the properties of TIL derived from TNF- transduced tumors to determine whether these cells possess heightened anti-tumor activity compared to TIL from normal tumor. TNF-transduced tumors may be a potential source of TIL useful in the adoptive immunotherapy of unmodified tumor. Further, these studies have implications for the use of gene-modified TIL for the treatment of human cancer. Recently, we have succeeded in introducing the TNF cDNA into human tumor infiltrating lymphocytes and studies of the in vivo properties of these cells have begun. It is hoped that these genetically altered cells will provide a mechanism by which therapeutic local concentrations of TNF can be achieved in growing tumors without inducing systemic toxicity in patients with advanced cancers.
Acknowledgments
The authors thank Ellen Bolton and Liliana Guedez for performing flow microfluorometric analyses and Diane Butler for secretarial assistance
Abbreviations used in this paper
- LAK
lymphokine-activated killer cell
- TIL
tumor infiltrating lymphocytes
- Neo
neomycin
- MCA
methyl-cholanthrene
- CM
complete medium
- FMF
flow microfluorimetry
- NeoR
neomyrin resistance
References
- 1.Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci USA. 1975;72:3666. doi: 10.1073/pnas.72.9.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asher AL, Mulé JJ, Reichert CM, Shiloni E, Rosenberg SA. Studies on the anti-tumor efficacy of systemically administered recombinant tumor necrosis factor against several murine tumors in vivo. J Immunol. 1987;138:963. [PubMed] [Google Scholar]
- 3.Haranaka K, Satomi N. Cytotoxic activity of tumor necrosis factor (TNF) on human cancer cells in vitro. Jpn J Exp Med. 1981;51:191. [PubMed] [Google Scholar]
- 4.Creasy AA, Reynolds TR, Laird W. Cures and partial regression of murine and human tumors by recombinant tumor necrosis factor. Cancer Res. 1986;46:5687. [PubMed] [Google Scholar]
- 5.Haranaka K, Satomi N, Sakurai A. Antitumor activity of murine tumor necrosis factor (TNF) against transplanted murine tumors and hetero-transplanted human tumors in nude mice. Int J Cancer. 1984;34:263. doi: 10.1002/ijc.2910340219. [DOI] [PubMed] [Google Scholar]
- 6.Nelson L, Green S, Carswell E, Old LJ. Effect of tumor necrosis factor on cultured human melanoma cells. Nature. 1975;258:731. doi: 10.1038/258731a0. [DOI] [PubMed] [Google Scholar]
- 7.Helson L, Helson C, Green S. Effects of murine tumor necrosis factor on heterotransplanted human tumors. Exp Cell Biol. 1979;47:53. doi: 10.1159/000162922. [DOI] [PubMed] [Google Scholar]
- 8.Sugarman BJ, Aggarwal BB, Hass PE, Figari IS, Palladino MA, Shepard HM. Recombinant human tumor necrosis factor-alpha: effects on proliferation of normal and transformed cells in vitro. Science. 1985:230–943. doi: 10.1126/science.3933111. [DOI] [PubMed] [Google Scholar]
- 9.Philip R, Epstein L. Tumor necrosis factor as immunomodulator and mediator of monocyte toxicity induced by itself, gamma-interferon and interleukin-1. Nature. 1986;323:86. doi: 10.1038/323086a0. [DOI] [PubMed] [Google Scholar]
- 10.Talmadge JE, Phillips H, Schneider M, Rowe T, Pennington R, Bowersox O, Lenz B. Immunomodulatory properties of recombinant murine and human tumor necrosis factor. Cancer Res. 1988;48:544. [PubMed] [Google Scholar]
- 11.Shalaby MR, Aggarwal BB, Rinderknecht E, Svedersky LP, Finkle BS, Palladino MA. Activation of human polymorphonuclear neutrophil functions by interferon-gamma and tumor necrosis factors. J Immunol. 1985;135:2069. [PubMed] [Google Scholar]
- 12.Scheurich P, Thoma B, Ucer V, Pfizenmaier K. Immunoregulatory activity of recombinant human tumor necrosis factor (TNF)-alpha: Induction of TNF receptors on human T cells and TNF-alpha mediated enhancement of T cell responses. J Immunol. 1987;138:1786. [PubMed] [Google Scholar]
- 13.Hackett RJ, Davis LS, Lipsky PE. Comparative effects of tumor necrosis factor and IL-l beta on mitogen-induced T cell activation. J Immunol. 1988:140–2639. [PubMed] [Google Scholar]
- 14.Ranges GE, Figari IS, Espevik T, Palladino MA. Inhibition of cytotoxic T cell development by transforming growth factor-beta and reversal by recombinant tumor necrosis factor-alpha. J Exp Med. 1987;166:991. doi: 10.1084/jem.166.4.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robinet E, Branelec D, Termijtelen AM, Blay JY, Gay F, Chouaib S. Evidence for tumor necrosis factor-alpha involvement in the optimal induction of class I allospecific cytotoxic T cells. J Immunol. 1990;144:4555. [PubMed] [Google Scholar]
- 16.Owen-Schaub LB, Gutterman JU, Grimm EA. Synergy of tumor necrosis factor and interleukin-2 in the activation of human cytotoxic lymphocytes: Effect of tumor necrosis factor alpha and interleukin-2 inthegeneration of human lymphokine-activated killer cell cytotoxicity. Cancer Res. 1988;48:788. [PubMed] [Google Scholar]
- 17.Asher AL, Mulé JJ, Rosenberg SA. Recombinant human tumor necrosis factor mediates regression of a murine sarcoma in vivo via Lyt2+ cells. Cancer Immunol Immunother. 1989;28:153. doi: 10.1007/BF00199117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Havell EA, Fiers W, North RJ. The antitumor function of tumor necrosis factor (TNF) I. Therapeutic action of TNF against an established murine sarcoma is indirect, immunologically dependent and limited by severe toxicity. J Exp Med. 1988;167:1067. doi: 10.1084/jem.167.3.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palladino MA, Jr, Shalaby MR, Kramer SM, Ferraiolo BL, Baughman RA, Deleo AH, Crase D, Marifino B, Aggarwal BB, Figari IS, Liggitt D, Patton JS. Characterization of the anti-tumor activities of tumor necrosis factor-alpha and the comparison with other cytokines: induction of tumor specific immunity. J Immunol. 1987;138:4023. [PubMed] [Google Scholar]
- 20.Rosenberg SA, Lotze M, Yang J, Aebersold P, Linehan WM, Seipp C, White D. Experience with the use of high dose interleukin-2 in the treatment of 652 cancer patients. Ann Surg. 1989;210:474. doi: 10.1097/00000658-198910000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kriegler M, Perez C, DeFay K, Albert I, Lu SD. A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: Ramifications for the complex physiology of TNF. Cell. 1988;53:45. doi: 10.1016/0092-8674(88)90486-2. [DOI] [PubMed] [Google Scholar]
- 22.Wexler H, Rosenberg SA. Pulmonary metastases from autochthonous 3-methylcholathrene-induced murine tumors. J Natl Cancer Inst. 1979;63:1393. [PubMed] [Google Scholar]
- 23.Kasid A, Morecki S, Aebersold P, Cornetta K, Culver K, Freeman S, Director E, Lotze MT, Blaise RM, Anderson WF, Rosenberg SA. Human gene transfer: characterization of human tumor infiltrating lymphocytes as vehicles for retroviral-mediated gene transfer in man. Proc Natl Acad Sci. 1990;87:473. doi: 10.1073/pnas.87.1.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cann AJ, Koyanagi Y, Chen ISY. High efficiency transfection of primary human lymphocytes and studies of gene expression. Oncogenes. 1988;3:123. [Google Scholar]
- 25.Southern PJ, Berg P. Transformation of mammalian cells to antibiotic resistance with a bacterial gene under control of the SV40 early region promoter. J Molec Appl Genet. 1982;1:327. [PubMed] [Google Scholar]
- 26.Wigler MA, Pellicer A, Silverstein S, Axel R, Urlaub G, Chasin L. DNA-mediated transfer of the adenine phosphoribosyltransferase locus into mammalian cells. Proc Natl Acad Sci USA. 1979;76:1373. doi: 10.1073/pnas.76.3.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Topalian SL, Muul LM, Solomon D, Rosenberg SA. Expansion of human tumor infiltrating lymphocytes for use in immunotherapy trials. J Immunol Methods. 1987;102:127. doi: 10.1016/s0022-1759(87)80018-2. [DOI] [PubMed] [Google Scholar]
- 28.Cobbold SP, Jayasuriya A, Nash A, Prospero TD, Waldman H. Therapy with monoclonal antibodies by elimination of T cell subsets in vivo. Nature. 1986;323:164. doi: 10.1038/312548a0. [DOI] [PubMed] [Google Scholar]
- 29.Cobbold SP, Martin G, Qin S, Waldman H. Monoclonal antibodies to promote marrow engraftment and tissue graft tolerance. Nature. 1986;323:164. doi: 10.1038/323164a0. [DOI] [PubMed] [Google Scholar]
- 30.Mulé JJ, Yang JC, Lafreniere R, Shu S, Rosenberg SA. Identification of cellular mechanisms operational in vivo during the regression of established pulmonary metastases by the systemic administration of high-dose recombinant interleukin-2. J Immunol. 1987;139:285. [PubMed] [Google Scholar]
- 31.Bringman TS, Aggarwal BB. Monoclonal antibodies to human tumor necrosis factors alpha and beta: application for affinity purification, immunoassays, and as structural probes. Hybridoma. 1987;6:489. doi: 10.1089/hyb.1987.6.489. [DOI] [PubMed] [Google Scholar]
- 32.Teng MN, Park BH, Koeppen HKW, Schreiber H. Antibodies to TNF reverse and prevent cachexia but increase tumor growth. FASEB J. 1990;4:1867. [Google Scholar]
- 33.Fearon ER, Pardoll D, Itaya T, Golumbek P, Levitsky HI, Simons JW, Karasuyama H, Vogelstein B, Frost P. Interleukin-2 production by tumor cells bypasses T-helper function in the generation of an anti-tumor response. Cell. 1990;60:397. doi: 10.1016/0092-8674(90)90591-2. [DOI] [PubMed] [Google Scholar]
- 34.Tepper RI, Pattengale RK, Leder P. Murine interleukin-4 displays potent anti-tumor activity in vivo. Cell. 1989;57:503. doi: 10.1016/0092-8674(89)90925-2. [DOI] [PubMed] [Google Scholar]
- 35.Watanabe Y, Kuribayashi K, Miyatake J, Nishihara K, Nakayama E, Taniyama T, Sakata T. Exogenous expression of mouse interferon-gamma cDNA in mouse neuroblastoma C1300 cells results in reduced tumorigenicity by augmented anti-tumor immunity. Proc Natl Acad Sci USA. 1989;86:9456. doi: 10.1073/pnas.86.23.9456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Torre-Amione G, Beauchamp RD, Koeppen H, Park BH, Schreiber H, Moses HI, Rowley DA. A highly immunogenic tumor transfected with a murine transforming growth factor type Beta, cDNA escapes immune surveillance. Proc Natl Acad Sci USA. 1990;87:1486. doi: 10.1073/pnas.87.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oliff A, Defeo-Jones D, Boyer M, Martinez D, Kiefer D, Vuocolo A, Wolfe G, Socher JH. Tumors secreting TNF/cachectin induce cachexia in mice. Cell. 1987;50:555. doi: 10.1016/0092-8674(87)90028-6. [DOI] [PubMed] [Google Scholar]
- 38.Kinkhabwala M, Sehajpal P, Skolnik E, Smith D, Sharma VK, Vlassara H, Cerami A, Suthanthiran M. A novel addition to the T cell repertory: Cell surface expression of tumor necrosis factor/cachectin by activated normal human T cells. J Exp Med. 1990:941. doi: 10.1084/jem.171.3.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu C, Detmers PA, Jiang S, Young JD. Identification and characterization of a membrane-bound cytotoxin of murine cytotoxic lymphocytesthat is related to tumor necrosis factor/cachectin. Proc Natl Acad Sci USA. 1989;86:3286. doi: 10.1073/pnas.86.9.3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weber JS, Rosenberg SA. Modulation of murine tumor major histocompatibility antigens by cytokines in vivo and in vitro. Cancer Res. 1988;48:5818. [PubMed] [Google Scholar]
- 41.McIntosh JK, Mulé JJ, Jablons DM, Nordan RP, Rudikoff S, Lotze MT, Rosenberg SA. The kinetics of interleukin-6 induction by the systemic administration of rhTNF-alpha in mice. Ann NY Acad Sci. 1989;577:572. [Google Scholar]
- 42.McIntosh JK, Jablons DM, Mulé JJ, Nordan RP, Rupikoff S, Lotze MT, Rosenberg SA. In vivo induction of IL-6 by administration of exogenous cytokines and detection of de novo serum levels of IL-6 in tumor-bearing mice. J Immunol. 1989;143:162. [PubMed] [Google Scholar]
- 43.Mulé JJ, McIntosh JK, Jablons DM, Rosenberg SA. Antitumoractivity of recombinantinterleukin-6 in mice. J Exp Med. 1990:171–629. doi: 10.1084/jem.171.3.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mulé J, Asher A, McIntosh J, Lafreniere R, Shiloni E, Leffor A, Reichert CM, Rosenberg SA. Antitumor effect of recombinant tumor necrosis factor-alpha against murine sarcomas at visceral sites: Tumor size influences the response to therapy. Cancer Immunol Immunother. 1988;26:202. doi: 10.1007/BF00199930. [DOI] [PMC free article] [PubMed] [Google Scholar]