

Abstract
Chronic opioid treatment leads to agonist-specific effects at the mu opioid receptor. The molecular mechanisms resulting from chronic opioid exposure include desensitization, internalization and down-regulation of membrane-bound mu opioid receptors (MOP). The purpose of this study was to compare the cellular regulation of guinea pig, human and rat MOP expressed in Chinese hamster ovary (CHO) cells, following exposure to two clinically important opioids, morphine and methadone. MOP expressing CHO cells were treated in culture with methadone or morphine for up to 48 hours. Radioligand diprenorphine and [D-AIa2,N-Me-Phe4,Gly5-ol]-enkephalin (DAMGO)-stimulated GTPγS binding assays were carried out using paired control and opioid-exposed CHO cells. Methadone induced downregulation of the mu opioid receptor, while morphine induced desensitization of the receptor for all three species. Furthermore, morphine predominantly decreased the potency of DAMGO to stimulate GTPγS binding, whereas methadone primarily reduced its efficacy. Changes in DAMGO potency and efficacy differed among species and depended on the opioid used to treat the cells. Our results showed similarities between guinea pig and human MOP for morphine-induced desensitization, but identified differences between the two for methadone-induced desensitization. In contrast, human and rat MOP differed in response to morphine treatment, but were not distinct in their response to methadone treatment. The guinea pig is an excellent and established animal model to study opioid effects, but its molecular opioid pharmacology has not been investigated thus far. These results can assist in understanding species differences in the effects of opioid ligands activating the mu opioid receptor.
Keywords: G protein coupled receptor; down-regulation; desensitization; GTPγS binding; morphine; methadone
1. Introduction
Since the cloning of the rat mu opioid receptor (MOP) [1], many mammalian mu opioid receptor have been sequenced and cloned, including human [2], mouse [3], cow [4] and guinea pig [5]. Most of these receptors have been functionally characterized in cellular systems using different assays, e.g. binding of opioid agonists and antagonists, activation of GTPγS binding to G-coupled proteins, signaling to downstream effectors, inhibition of adenylyl cyclase or voltage-gated Ca2+ channels, or stimulation of G protein-coupled inwardly rectifying potassium channels. In vitro characterization of the guinea pig MOP though has not been done. The in vitro characterization studies of the cloned mu opioid receptors are difficult to compare among species, because comparative analyses of different species for any functional assay within the same experimental setup has not been done for the MOP.
Opioid receptors are regulated following agonist exposure. Receptor stimulation by agonists causes signaling through activation of the coupled Gi/o proteins followed by rapid phosphorylation of the receptor. This receptor stimulation leads to effective uncoupling of the Gi/o proteins from the receptor [6]. Chronic opioid exposure interrupts these acute regulatory processes and may contribute to tolerance and dependence. Thus, prolonged activation leads to adaptive changes in receptor number, receptor coupling to G proteins, and receptor signaling to effector proteins [7]. The molecular processes leading to cellular tolerance and long-term changes in mu opioid receptor activity remain controversial [6]. A reason for this controversy is a variety of different cell systems that are utilized for in vitro analyses of these processes, including heterologously expressed mu opioid receptor in Chinese hamster ovary cells [8,9], mouse pituitary tumor cells At-T20 [10,11], or endogenously found mu opioid receptor in human neuroblastoma cells [12] and neurons [13,14]. These different systems taken together with the different assays lead to the fact that reported observations and proposed mechanisms are strongly influenced by the chosen opioid agonist, the cellular or animal system, the time-scale of observation and the assay performed [15].
The guinea pig is a potent, important and well established animal model in gastro-intestinal, respiratory and opioid tolerance research [16-19]. It is the model of choice in these areas because of the closer anatomical, physiological, neurological and developmental similarities to human. Yet, there are no data available on the molecular properties of the guinea pig mu opioid receptor and how these compare to other animal models or human.
The purpose of this study is to perform an analysis of the newly cloned guinea pig [5], and compare it to the human and rat mu opioid receptors using the same Chinese hamster ovary in vitro cell system for all species. Our focus is on the changes in diprenorphine ligand binding and [D-AIa2,N-Me-Phe4,Gly5-ol]-enkephalin (DAMGO)-stimulated GTPγS binding following chronic exposure to morphine or methadone in cell culture to characterize the species dependent effects of these drugs.
2. Materials and methods
2.1. Materials
F12 Nutrient Mixture (Ham) with L-glutamine, Lipofectamin 2000, Opti-MEM reduced serum media and Trypsin/EDTA was purchased from Invitrogen Corporation (Carlsbad, CA, U.S.A.). Dulbecco’s Phosphate Buffered Saline (PBS) and geneticin G418 were purchased from Cellgro (Herndon, VA, U.S.A.). Fetalplex was purchased from Gemini Bioproducts (Woodland, CA, U.S.A.). [3H]-diprenorphine (50 Ci/mmol) and [35S]-GTPγS (1250 Ci/mmol) were purchased from Perkin Elmer Life Sciences (Boston, MA, U.S.A.). ScintiSafe 30 % scintillation cocktail was purchased from Fisher Scientific (Fair Lawn, NJ, U.S.A.). All opioid agonist drugs were supplied by the National Institute on Drug Abuse (NIDA) of the National Institutes of Health (Bethesda, MD, U.S.A.), except for (-)- and (+)-methadone (Eli Lilly, Indianapolis, IN, U.S.A.) and U50,488 (trans-(±)-3,4-dichloro-N-methyl-N-[2-(I-pyrrolidinyl)cyclohexyl]-benzeneacetamide) (Sigma Chemical Co., St. Louis, MO, U.S.A.). Complete Mini protease inhibitor cocktail tablets were purchased from Roche Diagnostics (Indianapolis, IN, U.S.A.), Dc Protein Assay was purchased from Bio-Rad Laboratories (Hercules, CA, U.S.A.). GF/C glass fiber filters were purchased from Whatman Inc. (Sanford, ME, U.S.A.). The opioid antagonist naloxone and all reagent grade chemicals were purchased from Sigma Chemical Co. (St. Louis, MO, U.S.A.), all salts and buffers were purchased from Fisher Scientific (Fair Lawn, NJ). CHO-K1 (CCL-61) cells were purchased from ATCC (Manassas, MA, U.S.A.). Plasmid cDNA for guinea pig mu opioid receptor (GenBank: AY166606) was cloned in our laboratory [5], human mu opioid receptor (GenBank: AY521028) was purchased from the University of Missouri Rolla (UMR) cDNA resource center (Rolla, MO, U.S.A.), and rat mu opioid receptor (GenBank: NM_013071) was kindly provided by Dr. S. Nagalla of Oregon Health & Science University.
2.2. Cell culture and transfection
cDNAs encoding the guinea pig, human, and rat mu opioid receptor were ligated separately into the mammalian expression vector pcDNA3.1 (Invitrogen Corporation, Carlsbad, CA, U.S.A.). Chinese hamster ovary (CHO) cells were cultured in F12-HAM media supplemented with 10 % Fetalplex serum at 37 °C in a humidified atmosphere with 5 % CO2. Cells were seeded at 2x105 cells per plate on 6-well plates and 48 h later transfected with 2 μg of the expression vectors using Lipofectamin 2000 transfection kit according to the manufacturer’s manual. The next day cells were split and transferred into 25 cm2 culture flasks. Selective media containing 700 μg/ml geneticin was added 48 h post transfection and cells were cultured until the appearance of stable colonies. Single colonies were isolated by transferring 200 μl cell suspension at 2.5 cells/ml into 96-well plates. Wells containing single colonies were grown and expression of the mu opioid receptors was analyzed by radioligand diprenorphine binding. CHO cell lines stably expressing mu opioid receptors were cultivated in the presence of 300 μg/ml geneticin. For cellular tolerance studies cells were seeded 24 h prior to the earliest treatment time-point. Drugs (20 μM morphine, 10 μM rac-methadone, or 5 μM (-)- or (+)methadone) were added to the culture media without media change at the indicated times prior to harvesting the cells. Dosage was determined in a pilot experiment as the minimal concentration needed to induce the maximum observed effect on GTPγS binding. Control cells received an equal volume of phosphate buffered saline (PBS) pH 7.4 only.
2.3. Protein assay
The Bio-Rad DC Protein assay was used to measure the protein concentration of the membrane preparation according to the manufacturer’s microplate assay protocol. The standard curve was established using bovine serum albumin. The sample concentration was determined in triplicates and measured using the PowerWavex 340 microplate reader (Bio-Tek Instruments, Inc., Winooski, VT, U.S.A.).
2.4. Radioligand binding assay
For saturation and competition binding, 4×106 cells were cultivated on 150 mm culture plates for 48 h. For cellular tolerance studies 8×105 cells were seeded on 100 mm plates and grown for 72 h. Drugs for the cellular tolerance studies were added to the culture media up to 48 h before harvesting. For all experiments, cells were washed with 10 ml cold PBS buffer (pH 7.4), collected using a cell scraper, and centrifuged at 500g for 10 min at 4 °C. The cell pellets were rinsed twice with cold PBS buffer and precipitated as above. Cells were re-suspended in binding buffer (50 mM Tris-HCl pH 7.7) containing a protease inhibitor cocktail mix (Roche Diagnostics, Indianapolis, IN, U.S.A.). Cells were disrupted with a Polytron homogenizer for 10 s at setting 4.5 (Brinkmann Instruments, Inc., Westbury, NY, U.S.A.), incubated 10 min on ice, and membranes isolated by high-speed centrifugation (20,000g, 10 min, 4 °C). Membranes were thoroughly re-suspended in 3 ml (2 ml for 100 mm cultures) binding buffer per plate and protein concentration was determined. [3H]-diprenorphine saturation binding was conducted to determine the dissociation constant (Kd) and maximum binding (Bmax) for all mu opioid receptor expressing cell lines. 30-60 μg of membranes were incubated with increasing concentrations of radioligand at 25 °C for 75 min in a total volume of 500 μl binding buffer. Non-specific binding was determined in the presence of 50 μM naloxone. Bound radioligand was trapped on GF/C glass fiber filters, presoaked with 0.5 % polyethyleneimine in binding buffer for 2 h, by rapid filtration through a 48-well cell harvester (Brandel, Gaithersburg, MD, U.S.A.). Filters were washed three times with 4 ml of ice-cold binding buffer for 1 min, placed in glass scintillation vials with 4 ml scintillation cocktail and counted with a Beckman LS6500 coulter counter for 4 min per sample. Inhibition constants (Ki) for guinea pig mu opioid receptor were determined for tested compounds by competition binding displacing 0.3 nM radioligand with 10 different concentrations (10-13-10-2.5 M) of each competitor. For cellular tolerance studies 0.6 nM radioligand was employed (approximately equivalent to 2×Kd). Assays were performed as above. Each reaction was performed in triplicates and repeated at least three times.
2.5. GTPγS binding assay
Mu opioid receptor expressing Chinese hamster ovary cells (4×106) were cultivated on 150 mm culture plates and grown for 48 h. For cellular tolerance studies drugs were added to the culture media at the indicated times prior to harvesting. Membranes were harvested, washed and homogenized as above, except cells were re-suspended in assay buffer (50 mM Tris-HCl pH 7.4, 100 mM NaCl, 3 mM MgCl2, 0.2 mM EGTA) instead of binding buffer. Assay conditions were initially tested for optimal protein concentration (10-60 μg) and incubation time (30 min – 2 h) for all species to get maximum GTPγS binding within the linear response range. GTPγS binding was performed with 12-18 µg of total membrane protein. Membranes were incubated for 2 h with 10 µM GDP, 100 pM [35S]-GTPγS, and increasing concentrations of opioid agonist (10-10 to 10-4 M) at 30 °C in a final volume of 500 μl assay buffer. Each drug concentration was performed in duplicate and non-specific binding was measured in the presence of 50 μM naloxone. Basal stimulation was determined in the absence of agonist. Bound GTPγS was filtered and counted as above using Whatman GF/C glass fiber filters, pre-wetted in ice-cold wash buffer (50 mM Tris-HCl pH 7.4), except filters were washed three times, very rapidly using 3 ml ice-cold wash buffer. Experiments were repeated at least three times for each assay.
2.6. Data analysis
Saturation and competition radioligand binding curves as well as opioid-stimulated GTPγS dose response curves were generated using GraphPad Prism 3.0 (GraphPad Software Inc., San Diego, CA, U.S.A.). Kd, Bmax and Ki were calculated using the built in equations for one site binding hyperbola and one site competition binding, respectively. Using the GraphPad Prism F test for comparison of two equations, one site competition binding was found to be the best fit model for all compounds tested. The maximum effect (Emax) and potency (EC50) for opioid-stimulated GTPγS binding curves were calculated using sigmoidal dose response curve fit. EC50 was considered to represent the potency of each agonist, and the Emax reflected the efficacy of the agonist. An increase in EC50 indicated a decrease in potency, whereas a decrease in Emax specified a decrease in efficacy. Analysis of data was always performed in a paired fashion of control and treated cells assayed at the same time to resolve the small differences on the logarithmic dose response curve. Data for maximum effect was normalized in two ways: In relation to the maximum receptor binding for comparison between cell lines and in relation to DAMGO-stimulation as a standard for comparisons between drugs. Efficiency of GTPγS binding was calculated as Emax/Bmax for each cell line. The number of experiments for each condition was at least three.
2.7. Statistical analysis
Statistical analysis was performed using SigmaStat 3.1 software (Systat Software Inc., Richmond, CA, U.S.A.). Data are reported as mean ± S.E.M. Opioid-stimulated GTPγS binding was analyzed by two-way ANOVA with factors of species and drug. Potencies were compared between species for each drug by one-way ANOVA. Multiple comparisons were done on significant effects using the Holm-Sidak method. A value of P < 0.05 was considered significant.
To compare results of cellular tolerance studies, the data was normalized as percent of untreated control within the same experiment. Both radioligand binding and opioid-stimulated GTPγS binding were initially analyzed using three-way analysis of variance (ANOVA) with factors of species, drug and time of drug treatment. To analyze the effects of drug treatment on GTPγS binding, two-way ANOVA with factors of species and time were performed for each drug separately. Two-way ANOVA with factors of drug and time of treatment was also used to compare the enantiomers of methadone. Post hoc analysis was done as described above.
3. Results
3.1. Stable mu opioid receptor expression in CHO cells
Mu opioid receptor cDNAs encoding guinea pig, human and rat species variants were stably transfected into Chinese hamster ovary cells. Greater than 20 clonal populations were screened for each species through equilibrium receptor binding studies using the nonselective opioid antagonist [3H]-diprenorphine and three clones were selected, one for each species, with similar dissociation constants (Kd) and maximum specific binding (Bmax) of 1-3 pmol/mg protein [20] (Table 1).
Table 1. Kdand Bmax for stable mu opioid receptor CHO cell lines.
| Species | Kd[nM] | Bmax[fmol/mg] |
|---|---|---|
| Guinea Pig | 0.30±0.03 | 1210± 30 |
| Human | 0.38±0.05 | 1030± 80 |
| Rat | 0.26±0.01 | 2480± 20 |
Saturation binding of [3H]-diprenorphine was performed using membrane preparations from CHO cells expressing guinea pig, human or rat mu opioid receptor. Dissociation constant (Kd) and maximum binding (Bmax) were calculated as described in materials and methods. Data are expressed as mean ± S.E.M (n=3).
3.2. Competition binding analysis of the guinea pig mu opioid receptor
In a prior study we reported the sequence of the guinea pig mu opioid receptor [5]. In order to verify the mu-specific binding profile of the cloned receptor we used competition binding of [3H]-diprenorphine with a series of opioid peptides and alkaloids (Figure 1). The rank order of all tested compounds for the ability to displace [3H]-diprenorphine from the mu opioid receptor was levorphanol > DAMGO = (-)-methadone > morphine > rac-methadone > morphine-6-β-D-glucuronide >> (+)-methadone >morphine-3-β-D-glucuronide >>> U50,488, dextrorphan, [D-Pen2,5]-enkephalin (DPDPE) (Table 2). The mu-specific agonists DAMGO (3.4 ± 0.6 nM), levorphanol (1.2 ± 0.2 nM), methadone (7.2 ± 1.1 nM) and morphine (5.5 ± 0.5 nM) were most potent. Also consistent with the binding profile of a mu opioid receptor, the kappa and delta specific opioid agonists U50,488 and DPDPE exhibited a low displacement capacity.
Figure 1.
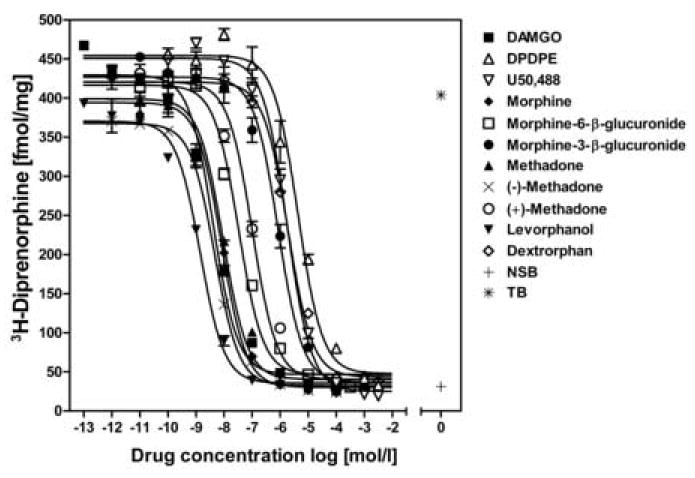
Competition of specific [3H]-diprenorphine binding (0.3 nM) by unlabeled compounds was measured using homogenized membranes prepared from guinea pig mu opioid receptor CHO cells. Each curve was plotted using data from one representative experiment with each measurement determined in triplicates. Each competitor was evaluated in three independent experiments. A Ki value was calculated for each ligand and is represented in Table 2. (NSB, non-specific binding, TB, total binding)
Table 2. Inhibition constants for different agonists at the cloned guinea pig mu opioid receptor.
| Agonist | Ki[nM] |
|---|---|
| DAMGO | 3.4±0.6 |
| U50,488 | 930±90 |
| DPDPE | 3300±500 |
|
| |
| Methadone | 7.2±1.1 |
| (−)-Methadone | 3.4±0.9 |
| (+)-Methadone | 105±30 |
|
| |
| Morphine | 5.5±0.5 |
| Morphine-6-β-D-glucuronide | 30±6 |
| Morphine-3-β-D-glucuronide | 560±50 |
|
| |
| Levorphanol | 1.2±0.2 |
| Dextrorphan | 1740±360 |
Competition binding was performed with CHO membrane preparations. The Ki values reflect the potency of each unlabeled competitor to displace 0.3 nM [3H]-diprenorphine. One site analysis was the best fit model for all compounds tested. Non-specific (NSB) and total binding (TB) was determined in the absence of competitor. Data are mean ± S.E.M of three separate experiments conducted in triplicates.
3.3. Functional studies of the stable mu opioid receptor CHO cell lines
DAMGO, methadone and morphine induced stimulation of GTPγS binding was used to describe the activation the cloned mu opioid receptors (Figure 2A-C). The rank of potencies for the stimulation of GTPγS binding to the guinea pig receptor was DAMGO > morphine > methadone, while for human and rat receptor the rank order of morphine and methadone was reversed (Table 3). Methadone was equipotent for all three species. DAMGO and morphine were significantly more potent at the guinea pig mu opioid receptor than at the human or rat receptor (P < 0.05). DAMGO was also more potent at the human compared to the rat mu opioid receptor (P < 0.01).
Figure 2.
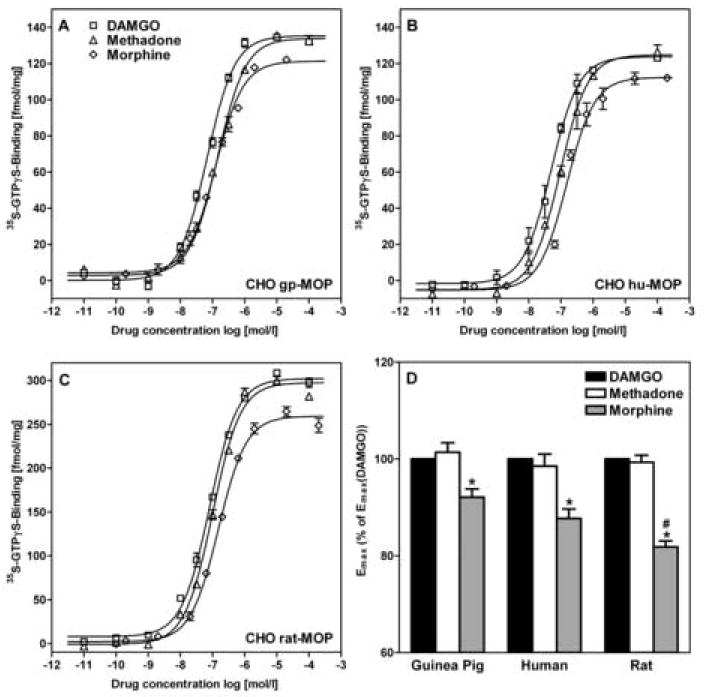
A-C:Opioid-stimulated GTPγS binding was performed using DAMGO, methadone and morphine (Table 3) for guinea pig (A), human (B), and rat (C) mu opioid receptor in membranes isolated from CHO cells. Curves are plotted using data from one representative experiment. D: The efficacies of the different opioids were normalized to the efficacy of the DAMGO-stimulated assay of the same experiment. Emax of morphine was significantly lower than Emax of DAMGO and methadone for all species. Emax of morphine for rat mu opioid receptor was significantly lower than for guinea pig and human. Data are mean ± S.E.M of three independent experiments (D).
* P < 0.05 vs. DAMGO and methadone
# P < 0.05 vs. guinea pig and human
Table 3. Potency and efficacy for activation of GTPγS binding to the mu opioid receptors in membranes isolated from CHO cells.
| Guinea Pig | Human | Rat | ||
|---|---|---|---|---|
| EC50[nM] | DAMGO | 46±3 a | 58±3 b | 80±7 |
| Methadone | 134±4 | 128±31 | 124±19 | |
| Morphine | 106±5 a | 184±13 | 166±16 | |
|
| ||||
| Emax[fmol/mg] | DAMGO | 133±16 | 128±10 | 302±19 |
| (Emax/Bmax)[%] | (11±1) | (12±1) | (12±1) | |
| Methadone | 135±16 | 126±10 | 300±24 | |
| (11±1) | (12±1) | (12±1) | ||
| Morphine | 122±15 | 112±6 | 247±15 | |
| (10±1) | (10±1) | (10±1) | ||
Potency (EC50) and efficacy (Emax) values were calculated for all guinea pig, human and rat mu opioid expressing CHO cell lines using sigmoidal dose response curve fit. Data are mean ± S.E.M of a minimum of three experiments done in duplicates for each data point. The maximum effects are reported as Emax [fmol/mg] and as Emax/Bmax [%] relative to the Bmax for [3H]-diprenorphine binding. Statistical analysis of this ratio revealed that there were no significant differences between species for the efficiency (Emax/Bmax) of GTPγS binding. a P < 0.05 vs. human and rat, b P < 0.01 vs. rat.
To compare the opioid-stimulated GTPγS binding among the mu opioid receptor expressing cell lines, the Emax values were normalized to the number of receptors (Bmax) expressed in each cell line (Table 3). The resulting ratio (Emax/Bmax) was defined as efficiency of opioid-stimulated GTPγS binding. There were no significant differences in efficiencies among cell lines by this analysis. To compare the efficacies for the different stimulating agonists DAMGO, methadone and morphine, experiments were always performed in a paired fashion of all three drugs within the same set of experiment and the maximum effects were evaluated in relation to the Emax of DAMGO (relative Emax) (Figure 2D). A significant difference was found between the Emax of morphine and the Emax of both DAMGO and methadone for all species (P < 0.01). The relative Emax value of morphine for the rat mu opioid receptor was significantly lower than that for guinea pig and human (P < 0.05).
For CHO cells expressing the guinea pig mu opioid receptor we extended our study and analyzed both methadone enantiomers separately. The potency for (-)-methadone-stimulated GTPγS binding (52 ± 6 nM) was 2.5-fold stronger than for rac-methadone (134 ± 4 nM) and 23-fold greater than for (+)-methadone (1.2 ± 0.3 µM). The maximum effect was not statistically different for rac-, (-)- and (+)-methadone.
3.4. Receptor binding after chronic drug treatment
We next examined the chronic effects of different opioid drugs on receptor binding by exposing our stably expressing mu opioid receptor CHO cells to 20 μM morphine or 10 μM methadone for up to 48 h (Figure 3). No significant differences in receptor binding were found among species. Methadone induced a time-dependent reduction of receptor binding while morphine treatment did not change binding compared to control (P < 0.001 for methadone vs. morphine). Receptor binding of cells treated with methadone for 12 or more hours was maximally reduced by 65-73 % for all species (Figure 3).
Figure 3.
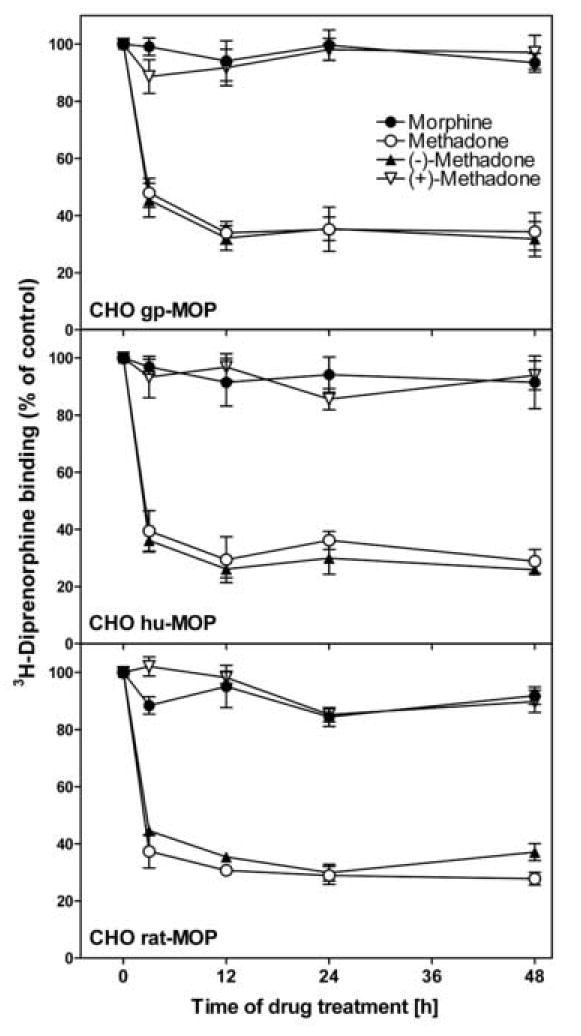
Receptor binding in opioid treated mu opioid receptor expressing CHO cells. Cells were exposed in culture to 20 μM morphine, 10 μM methadone, or 5 μM (-)- or (+)-methadone for 3 h to 48 h. Homogenized membrane preparations were assayed for their ability to bind [3H]-diprenorphine (0.6 nM). Receptor binding is plotted as percent of untreated control. Data is mean ± S.E.M of three independent experiments performed in triplicates. No significant changes in receptor binding were observed for chronic morphine or (+)-methadone treatment. Both rac- and (-)-methadone caused reduced receptor binding after 3 h of exposure that was sustained up to 48 h (P < 0.001 vs. control). No statistical differences were found after 12 h of treatment for each drug.
In addition we analyzed the two enantiomers of methadone separately for all species. Cells were treated with either 5 μM (-)- or (+)-methadone. The effects of methadone treatment were solely due to its active (-)-enantiomer, while (+)-methadone treatment at this concentration had no effect on receptor binding (Figure 3).
3.5. Opioid-stimulated GTPγS binding after chronic drug treatment
To determine the effects of chronic opioid exposure on the activation of G proteins by the mu opioid receptor, DAMGO-stimulated GTPγS binding curves were generated using DAMGO concentrations ranging from 10-10-10-4 M. As there were no changes in receptor binding after 12 h, we treated cells with morphine or methadone for only 3 or 12 h (Figure 4). Experiments were always performed in controlled sets and normalized to the untreated control of the same experiment. Effects on potency and efficacy were not different between 3 h and 12 h treatment except for the EC50 of the rat mu opioid receptor following morphine treatment (P < 0.05) (Figure 4A).
Figure 4.
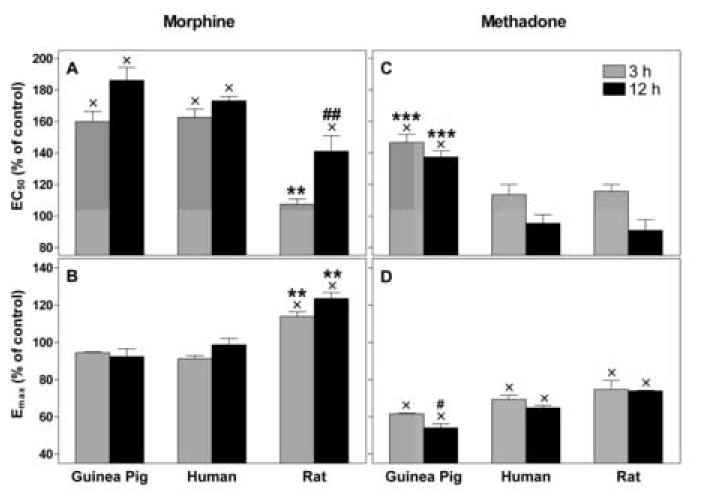
DAMGO-stimulated GTPγS binding in opioid treated CHO cells. Cells were treated in culture as in Figure 3 with either morphine or methadone for 3 or 12 h. Dose response curves for DAMGO-stimulated GTPγS binding with homogenized membranes were generated. EC50 (panel A,C) and Emax (panel B,D) were calculated and are graphed as percent of paired control. Data is mean ± S.E.M of three independent experiments performed in duplicates for each concentration. Methadone-induced tolerance was different for the guinea pig mu opioid receptor compared to the other two species, as was the case for the rat receptor following morphine treatment. Significant time-dependent changes for both treatments are indicated.
x P < 0.05 vs. untreated control
** P < 0.01 vs. guinea pig and human
## P < 0.01 vs. guinea pig
# P = 0.05 vs. rat
*** P < 0.001 vs. human and rat
Changes in potency (EC50) and efficacy (Emax) were significantly different for both species and drug (P < 0.001 for both comparisons). To isolate these effects each drug was analyzed separately. Morphine treatment significantly increased the EC50 of the DAMGO-stimulated G protein binding for all species by 60 ± 11 % (guinea pig), and 63 ± 9 % (human) after 3 h, and by 86 ± 14 % (guinea pig), 73 ± 5 % (human), and 41 ± 20 % (rat) after 12 h (P < 0.01 for all comparisons). The potency decrease of DAMGO at the rat mu opioid receptor was only significant for the 12 h time-point. Changes in potency of DAMGO at the guinea pig and human receptor were considerably greater than for the rat (P < 0.01 vs. guinea pig and human at 3 h, and vs. guinea pig at 12 h, (Figure 4A). Morphine treatment increased the maximum effect (Emax) of DAMGO for the rat mu opioid receptor by 18 ± 5 % (3 h) and 24 ± 6 % (12 h) (P < 0.05 vs. control, Figure 4B), but did not significantly change Emax for guinea pig- or human mu opioid receptor.
In contrast, methadone treatment reduced the potency of DAMGO-stimulated G protein binding to the guinea pig mu opioid receptor by 47 ±10 % (3h), and 37 ± 8 % (12h) (P < 0.01), but did not affect the potency (EC50) of DAMGO at the human or rat mu opioid receptor (Figure 4C). Methadone significantly reduced the efficacy of DAMGO-stimulated G protein binding for all species by 25-38 % after 3 h and 26-44 % after 12 h compared to control. A significant difference in efficacy reduction between species was only found for guinea pig and rat at 12 h of methadone treatment (P < 0.01, Figure 4D).
For the guinea pig receptor we evaluated both methadone isomers and found that only the active (-)-enantiomer was responsible for the observed changes in potency and efficacy of DAMGO-stimulated G protein binding to the receptor (Figure 5). There were no statistical differences between rac- and (-)-methadone, while both were different from (+)-methadone (P ≤ 0.001). (+)-Methadone increased the EC50 slightly, but this tendency is not statistically significant (P = 0.09).
Figure 5.
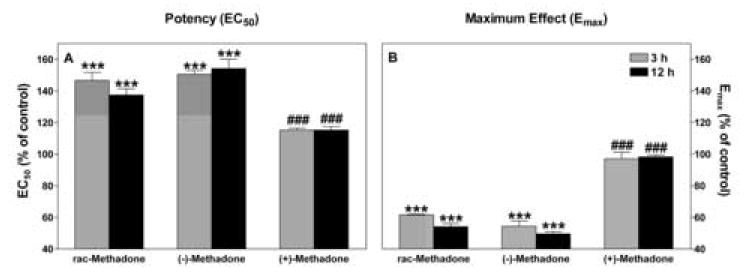
Effects of methadone isomers on GTPγS binding at the guinea pig mu opioid receptor. DAMGO-stimulated GTPγS binding curves were generated using membrane preparations from guinea pig mu opioid receptor expressing-CHO cells pretreated with 10 μM rac-methadone, or 5 μM (-)- or (+)-methadone for the times indicated. EC50 (Figure 5A) and Emax (Figure 5B) are plotted as percent of control. Data is mean ± S.E.M of three or four independent experiments performed in duplicates for each concentration of DAMGO. Two-way ANOVA for factors of drug and time for both values revealed no significant differences between rac- and (-)-methadone. Both drugs significantly decreased potency and efficacy compared to control after 3 h (P < 0.001 for all comparisons). No significant change in either value with time was observed for (+)methadone treated cells. Differences between methadone isomers are denoted.
*** P < 0.001 vs. untreated control
### P ≤ 0.001 vs. rac- and (-)-methadone
4. Discussion
The purpose of this study was to characterize the cellular pharmacology of the guinea pig mu opioid receptor and compare it to the rat, and human receptor following chronic opioid exposure. Both guinea pig and rat are important animal models for opioid research. We chose to analyze the guinea pig mu opioid receptor because the neonatal guinea pig is a better model for studying respiratory effects caused by chronic in utero or acute administration of opioid drugs than the rat [17,21]. The rat is the most common animal model for analysis of opioid induced cellular as well as whole animal tolerance. Our results show that based on the pharmacology of mu opioid receptor expressing CHO cells the guinea pig is equally suitable as an animal model as the rat for opioid-induced development of tolerance, dependence and withdrawal.
The mu opioid receptor is mainly implicated in mediating the cellular responses of clinically used analgesic opioids, such as morphine or methadone. Morphine is different from methadone in that it induces significantly less mu opioid receptor internalization in vitro [10,11,22-24]. Differential cellular effects of chronic treatment with both drugs are reported for several species [10,23,25,26], but have never been performed in a comparison of multiple species within a single assay system in order to delineate species-dependent differences.
In order to do these comparisons, we first needed to show that the newly cloned guinea pig mu opioid receptor [5] has a classical mu opioid ligand binding and activation profile comparable to other cloned mu opioid receptors [1-4]. Both one site [3,4] and two site [27,28] binding models have been used to analyze displacement curves for opioid ligands. In our assay system the one site model produced the best fit for all tested drugs. We established that the guinea pig receptor has ligand binding properties similar to previous species studied.
Using a single biological system (CHO cells) to express the mu opioid receptor from each of the three species, we excluded effects due to different host cell lines and avoided complications due to the presence of other opioid receptors. Similar studies described species differences between the human and rat kappa opioid receptor for U50,488 induced kappa opioid receptor regulation in CHO cells [20,29]. The expression level variation of approximately 2.5-fold among the three cell lines used in this study is nearly identical to the expression range in the kappa opioid receptor species study [20] and comparable to a mu opioid receptor mutant study [30]. This variation is small compared to the expression level difference of mu opioid receptor expressing cell systems found in the literature which vary more than 40-fold between studies [14,23]. In addition, normalization of the data shows that the expression range does not affect the comparison among species tested. Furthermore, we used supramaximal levels [23,31] of drugs for the study of long-term adaptations of mu opioid receptor to minimize the influence of spare receptors [19]. To our knowledge, this report is the first species comparison for the mu opioid receptor within a single assay.
Opioid-stimulated GTPγS binding was used to compare the potency and maximum effect of different drugs at the three mu opioid receptors. In all cases DAMGO was the most potent drug. Methadone was equally potent for all species, but was more potent than morphine for human and rat and less potent compared to morphine for guinea pig. Both orders of potency have been observed for different species and different functional assays looking at the mu opiod receptor, but are difficult to compare as assay and system variations complicate comparisons [10,23,31,32]. One study reported one rank for potency to stimulate GTPγS binding for mouse mu opioid receptor in CHO cells and the opposite rank of potency for methadone and morphine in rat thalamus neurons [33].
We compared the maximum effect of opioid-stimulated GTPγS binding of DAMGO, methadone and morphine among species by relating it to the maximum receptor binding (Bmax) and observed no statistical differences. This verifies that GTPγS binding at least in untreated cells was not dependent on the receptor number present in our three mu opioid receptor expressing CHO cell lines. Although the S.E.M. of efficacy between experiments ranged from 6 to 12 % of the mean, we were able to exclude variations that result from different membrane preparations [34] by normalization of the maximum effect to an internal control performed always within the same experiment (DAMGO-stimulation). As a result the comparison of all drugs showed a clear rank order of efficacy for all species within any one experiment where DAMGO and methadone are equally efficacious and morphine’s efficacy is significantly lower. Therefore we conclude that our three CHO cell lines have similar ligand and GTPγS binding profiles.
Many studies have shown that morphine and methadone have different agonist profiles at the mu opioid receptor with respect to the development of tolerance [6,15]. Our results suggest that the cellular processes in response to chronic exposure are not only drug, but also species dependent with a clear distinction between receptor binding and GTPγS binding.
Changes in receptor ligand binding were not distinct among species. A rapid reduction in receptor numbers was observed in all methadone exposed cells, while no changes in receptor binding were detected in all morphine treated cells over time. Even though we did not perform full dose response curves for this set of experiment, we concluded from previous reports [4,8,35] that our observed changes were predominantly due to changes in receptor number rather than changes in receptor binding affinity. This observation was in agreement with multiple previous studies that showed that morphine has a low potential to internalize and remove mu opioid receptors from the cell membrane [10,24,25] in most cells and tissues whereas methadone exhibited a high internalization rate [11,31]. For both drug treatments, the new guinea pig MOP receptor is not distinct from the rat or human MOP receptor with respect to diprenorphine binding.
In contrast to the receptor binding, the activation of GTPγS binding was differentially regulated for the three species following chronic exposure to methadone or morphine. Morphine treatment of the guinea pig and human mu opioid receptor expressing CHO cells resulted in a significant decrease of potency of DAMGO-stimulated GTPγS binding, but did not alter its maximum effect. The rat mu opioid receptor cell line displayed a smaller and slower decrease in potency and showed an increase in maximum effect, significantly differing from the other two.
In the case of methadone treated cells, the maximum effect of stimulation was reduced for all species, with effects on the guinea pig mu opioid receptor being greater than for the rat. The potency of DAMGO to stimulate GTPγS binding at both the human and rat receptor was not altered significantly. Only the potency of DAMGO at the guinea pig mu opioid receptor was significantly reduced and therefore distinct from the other two species.
For all experiments with the newly characterized guinea pig mu opioid receptor we included the two stereoisomers of methadone in the study to show that all effects resulting from chronic treatment with rac-methadone stemmed from the active (-)-isomer. We showed that changes in ligand receptor binding are due to the (-)-isomer only (also shown for human and rat mu opioid receptor), and that changes in potency and efficacy of DAMGO-stimulated GTPγS binding occured only in rac- or (-)-methadone treated cells. The small decrease in potency of DAMGO to stimulate GTPγS binding induced by (+)-methadone at this concentration is not statistically significant and should therefore play no role in the development of cellular tolerance.
In summary, the above data led to the conclusion that morphine exposure didn’t reduce the number of receptors, but did reduce the ability of the receptor to transmit the signal through activation of GTPγS binding. This effect was observed in parallel for both time and magnitude in the case of the guinea pig and human mu opioid receptor. The somewhat lesser effect on potency decrease on the rat mu opioid receptor could be attributed in part to the higher number of receptors in the rat mu opioid receptor expressing cell line. In contrast, methadone exposure reduced the number of receptors for all species. This reduction in receptor number is mainly responsible for the decrease in the maximum effect of GTPγS binding. The remaining membrane bound receptor show no decrease in potency to stimulate GTPγS binding, following the ligand signal with the exception of the guinea pig mu opioid receptor.
The species divergence was somewhat surprising considering the high amino acid sequence homology among the mu opioid receptors. The same dose of morphine seemed to have a greater ability to induce changes in receptor activity in CHO cells expressing the guinea pig or human receptor than it did with the rat receptor. On the other hand guinea pig mu opioid expressing CHO cells were more susceptible to methadone treatment than human and rat mu opioid receptor expressing CHO cells. This data suggests that some of the differences among species in development of tolerance and sensitivity to certain drugs may be due to the binding and signaling properties of the mu opioid receptor. Therefore it is crucial to understand the interactions of drugs and receptor on the cellular level of each species to fully appreciate the mode of action in humans or whole animals.
Acknowledgments
The authors like to thank Rosemary T. Nettleton for her help with the statistical analysis. Funding for this project was provided by the National Institute on Drug Abuse Grant DA007912. J. M. Mulvaney was awarded a Murdock Charitable Trust Fellowship, H. S. Hernandez was supported by the National Institute on Child Health & Human Development Grant HD46420. Preliminary results on the functional characterization of the guinea pig mu opioid receptor were presented at the 2005 International Narcotic Research Conference, Annapolis, MD and the 2006 Annual Meeting of the Society for Neuroscience, Atlanta, GA.
Abbreviations
- Bmax
maximum receptor binding
- CHO
Chinese hamster ovary
- DAMGO
[D-AIa2,N-Me-Phe4,Gly5-ol]-enkephalin
- DPDPE
[D-Pen2,5]-enkephalin
- Emax
maximum effect
- gp
guinea pig
- hu
human
- Ki
inhibition constant
- M3G
morphine-3-β-D-glucuronide
- M6G
morphine-6-β-D-glucuronide methadone refers to rac- methadone
- MOP
mu opioid receptor
- MOP-CHO
mu opioid receptor expressing Chinese hamster ovary cells
- U50,488
trans-(±)-3,4-dichloro-N-methyl-N-[2-(I-pyrrolidinyl)cyclohexyl]-benzeneacetamide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chen Y, Mestek A, Liu J, Hurley JA, Yu L. Molecular cloning and functional expression of a mu-opioid receptor from rat brain. Mol Pharmacol. 1993;44:8–12. [PubMed] [Google Scholar]
- 2.Wang JB, Johnson PS, Persico AM, Hawkins AL, Griffin CA, Uhl GR. Human mu opiate receptor. cDNA and genomic clones, pharmacologic characterization and chromosomal assignment. FEBS Lett. 1994;338:217–222. doi: 10.1016/0014-5793(94)80368-4. [DOI] [PubMed] [Google Scholar]
- 3.Kaufman DL, Keith DE, Jr, Anton B, Tian J, Magendzo K, Newman D, et al. Characterization of the murine mu opioid receptor gene. J Biol Chem. 1995;270:15877–15883. doi: 10.1074/jbc.270.26.15877. [DOI] [PubMed] [Google Scholar]
- 4.Onoprishvili I, Andria ML, Vilim FS, Hiller JM, Simon EJ. The bovine mu-opioid receptor: cloning of cDNA and pharmacological characterization of the receptor expressed in mammalian cells. Brain Res Mol Brain Res. 1999;73:129–137. doi: 10.1016/s0169-328x(99)00249-1. [DOI] [PubMed] [Google Scholar]
- 5.Smith SA, Stupfel JT, Ilias NA, Olsen GD. Guinea pig mu opioid receptor: brainstem expression in the morphine-exposed neonate. Neurotoxicol Teratol. 2004;26:121–129. doi: 10.1016/j.ntt.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 6.Waldhoer M, Bartlett SE, Whistler JL. Opioid receptors. Annu Rev Biochem. 2004;73:953–990. doi: 10.1146/annurev.biochem.73.011303.073940. [DOI] [PubMed] [Google Scholar]
- 7.Bailey CP, Connor M. Opioids: cellular mechanisms of tolerance and physical dependence. Curr Opin Pharmacol. 2005;5:60–68. doi: 10.1016/j.coph.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 8.Kato S, Fukuda K, Morikawa H, Shoda T, Mima H, Mori K. Adaptations to chronic agonist exposure of mu-opioid receptor-expressing Chinese hamster ovary cells. Eur J Pharmacol. 1998;345:221–228. doi: 10.1016/s0014-2999(98)00023-5. [DOI] [PubMed] [Google Scholar]
- 9.Harrison C, McNulty S, Smart D, Rowbotham DJ, Grandy DK, Devi LA, et al. The effects of endomorphin-1 and endomorphin-2 in CHO cells expressing recombinant mu-opioid receptors and SH-SY5Y cells. Br J Pharmacol. 1999;128:472–478. doi: 10.1038/sj.bjp.0702798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borgland SL, Connor M, Osborne PB, Furness JB, Christie MJ. Opioid agonists have different efficacy profiles for G protein activation, rapid desensitization, and endocytosis of mu-opioid receptors. J Biol Chem. 2003;278:18776–18784. doi: 10.1074/jbc.M300525200. [DOI] [PubMed] [Google Scholar]
- 11.Celver J, Xu M, Jin W, Lowe J, Chavkin C. Distinct domains of the mu-opioid receptor control uncoupling and internalization. Mol Pharmacol. 2004;65:528–537. doi: 10.1124/mol.65.3.528. [DOI] [PubMed] [Google Scholar]
- 12.Elliott J, Smart D, Lambert DG, Traynor JR. Characterisation of mu-opioid receptors on SH-SY5Y cells using naloxonazine and beta-funaltrexamine. Eur J Pharmacol. 1994;268:447–450. doi: 10.1016/0922-4106(94)90072-8. [DOI] [PubMed] [Google Scholar]
- 13.Alvarez VA, Arttamangkul S, Dang V, Salem A, Whistler JL, von Zastrow M, et al. mu-Opioid receptors: Ligand-dependent activation of potassium conductance, desensitization, and internalization. J Neurosci. 2002;22:5769–5776. doi: 10.1523/JNEUROSCI.22-13-05769.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bailey CP, Couch D, Johnson E, Griffiths K, Kelly E, Henderson G. Mu-opioid receptor desensitization in mature rat neurons: lack of interaction between DAMGO and morphine. J Neurosci. 2003;23:10515–10520. doi: 10.1523/JNEUROSCI.23-33-10515.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Connor M, Osborne PB, Christie MJ. Mu-opioid receptor desensitization: is morphine different? Br J Pharmacol. 2004;143:685–696. doi: 10.1038/sj.bjp.0705938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gray RE, Munks MW, Haynes RR, Olsen GD. Mu opioid receptor efficacy and potency of morphine-6-glucuronide in neonatal guinea pig brainstem membranes: comparison with transfected CHO cells. Brain Res Bull. 2001;54:499–505. doi: 10.1016/s0361-9230(01)00427-0. [DOI] [PubMed] [Google Scholar]
- 17.Hunter MA, Vangelisti GR, Olsen GD. Chronic intermittent in utero exposure to morphine: effects on respiratory control in the neonatal guinea pig. Biol Neonate. 1997;72:293–304. doi: 10.1159/000244496. [DOI] [PubMed] [Google Scholar]
- 18.Olsen GD, Murphey LJ. Effects of morphine and cocaine on breathing control in neonatal animals: a minireview. NIDA Res Monogr. 1995;158:22–39. [PubMed] [Google Scholar]
- 19.Chavkin C, Goldstein A. Opioid receptor reserve in normal and morphine-tolerant guinea pig ileum myenteric plexus. Proc Natl Acad Sci U S A. 1984;81:7253–7257. doi: 10.1073/pnas.81.22.7253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J, Li JG, Chen C, Zhang F, Liu-Chen LY. Molecular basis of differences in (-)(trans)-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidiny)-cyclohexyl]benzeneacetamide-induced desensitization and phosphorylation between human and rat kappa-opioid receptors expressed in Chinese hamster ovary cells. Mol Pharmacol. 2002;61:73–84. doi: 10.1124/mol.61.1.73. [DOI] [PubMed] [Google Scholar]
- 21.Murphey LJ, Olsen GD. Morphine-6-beta-D-glucuronide respiratory pharmacodynamics in the neonatal guinea pig. J Pharmacol Exp Ther. 1994;268:110–116. [PubMed] [Google Scholar]
- 22.Keith DE, Murray SR, Zaki PA, Chu PC, Lissin DV, Kang L, et al. Morphine activates opioid receptors without causing their rapid internalization. J Biol Chem. 1996;271:19021–19024. doi: 10.1074/jbc.271.32.19021. [DOI] [PubMed] [Google Scholar]
- 23.Blake AD, Bot G, Freeman JC, Reisine T. Differential opioid agonist regulation of the mouse mu opioid receptor. J Biol Chem. 1997;272:782–790. doi: 10.1074/jbc.272.2.782. [DOI] [PubMed] [Google Scholar]
- 24.Johnson EA, Oldfield S, Braksator E, Gonzalez-Cuello A, Couch D, Hall KJ, et al. Agonist-selective mechanisms of mu-opioid receptor desensitization in human embryonic kidney 293 cells. Mol Pharmacol. 2006;70:676–685. doi: 10.1124/mol.106.022376. [DOI] [PubMed] [Google Scholar]
- 25.Keith DE, Anton B, Murray SR, Zaki PA, Chu PC, Lissin DV, et al. mu-Opioid receptor internalization: opiate drugs have differential effects on a conserved endocytic mechanism in vitro and in the mammalian brain. Mol Pharmacol. 1998;53:377–384. [PubMed] [Google Scholar]
- 26.Yang JC, Shan J, Ng KF, Pang P. Morphine and methadone have different effects on calcium channel currents in neuroblastoma cells. Brain Res. 2000;870:199–203. doi: 10.1016/s0006-8993(00)02369-6. [DOI] [PubMed] [Google Scholar]
- 27.Bunzow JR, Zhang G, Bouvier C, Saez C, Ronnekleiv OK, Kelly MJ, et al. Characterization and distribution of a cloned rat mu-opioid receptor. J Neurochem. 1995;64:14–24. doi: 10.1046/j.1471-4159.1995.64010014.x. [DOI] [PubMed] [Google Scholar]
- 28.Brown GP, Yang K, Ouerfelli O, Standifer KM, Byrd D, Pasternak GW. 3H-morphine-6beta-glucuronide binding in brain membranes and an MOR-1-transfected cell line. J Pharmacol Exp Ther. 1997;282:1291–1297. [PubMed] [Google Scholar]
- 29.Zhang F, Li J, Li JG, Liu-Chen LY. (-)U50,488H [(trans)-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)-cyclohexyl]benzeneace tamide] induces internalization and down-regulation of the human, but not the rat, kappa-opioid receptor: structural basis for the differential regulation. J Pharmacol Exp Ther. 2002;302:1184–1192. doi: 10.1124/jpet.302.3.1184. [DOI] [PubMed] [Google Scholar]
- 30.Bot G, Blake AD, Li S, Reisine T. Fentanyl and its analogs desensitize the cloned mu opioid receptor. J Pharmacol Exp Ther. 1998;285:1207–1218. [PubMed] [Google Scholar]
- 31.Whistler JL, Chuang HH, Chu P, Jan LY, von Zastrow M. Functional dissociation of mu opioid receptor signaling and endocytosis: implications for the biology of opiate tolerance and addiction. Neuron. 1999;23:737–746. doi: 10.1016/s0896-6273(01)80032-5. [DOI] [PubMed] [Google Scholar]
- 32.Ivarsson M, Neil A. Differences in efficacies between morphine and methadone demonstrated in the guinea pig ileum: a possible explanation for previous observations on incomplete opioid cross-tolerance. Pharmacol Toxicol. 1989;65:368–371. doi: 10.1111/j.1600-0773.1989.tb01190.x. [DOI] [PubMed] [Google Scholar]
- 33.Selley DE, Liu Q, Childers SR. Signal transduction correlates of mu opioid agonist intrinsic efficacy: receptor-stimulated [35S]GTP gamma S binding in mMOR-CHO cells and rat thalamus. J Pharmacol Exp Ther. 1998;285:496–505. [PubMed] [Google Scholar]
- 34.Costa T, Herz A. Antagonists with negative intrinsic activity at delta opioid receptors coupled to GTP-binding proteins. Proc Natl Acad Sci U S A. 1989;86:7321–7325. doi: 10.1073/pnas.86.19.7321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Elliott J, Guo L, Traynor JR. Tolerance to mu-opioid agonists in human neuroblastoma SH-SY5Y cells as determined by changes in guanosine-5’-O-(3-[35S]-thio)triphosphate binding. Br J Pharmacol. 1997;121:1422–1428. doi: 10.1038/sj.bjp.0701253. [DOI] [PMC free article] [PubMed] [Google Scholar]