

Abstract
Glial scar formation around implanted silicon neural probes compromises their ability to facilitate long-term recordings. One approach to modulate the tissue reaction around implanted probes in the brain is to develop probe coatings that locally release antiinflammatory drugs. In this study, we developed a nitrocellulose-based coating for the local delivery of the anti-inflammatory drug dexamethasone (DEX). Silicon neural probes with and without nitrocellulose-DEX coatings were implanted into rat brains, and inflammatory response was evaluated 1 week and 4 weeks post implantation. DEX coatings significantly reduced the reactivity of microglia and macrophages one week post implantation as evidenced by ED1 immunostaining. CS56 staining demonstrated that DEX treatment significantly reduced chondroitin sulfate proteoglycan (CSPG) expression one week post implantation. Both at one week and at four week time points, GFAP staining for reactive astrocytes and neurofilament (NF) staining revealed that local DEX treatment significantly attenuated astroglial response and reduced neuronal loss in the vicinity of the probes. Weak ED1, neurocan, and NG2 positive signal was detected four weeks post implantation for both coated and uncoated probes, suggesting a stabilization of the inflammatory response over time in this implant model. In conclusion, this study demonstrates that the nitrocellulose-DEX coating can effectively attenuate the inflammatory response to the implanted neural probes, and reduce neuronal loss in the vicinity of the coated probes. Thus anti-inflammatory probe coatings may represent a promising approach to attenuate astroglial scar and reduce neural loss around implanted neural probes.
Keywords: electrodes, inflammation, chondroitin sulfate proteoglycan, glial scar, coatings.
1. Introduction
Functional recording from the nervous system using silicon micromachined neural probes potentially aids patients with movement disorders by enabling the processing and decoding of recorded neural signals into movement commands (Donoghue, 2002;Otto et al., 2003;Sanchez et al., 2004). However, the long-term performance of the implanted neural probes is compromised by the formation of glial scar around the Simicroelectrodes, which is a typical consequence of the inflammatory tissue reaction to implantation-induced injury in the CNS. The glial scar is inhibitory to neurons and forms a barrier between the electrode and neurons in the surrounding brain tissue (Edell et al., 1992;Shain et al., 2003;Schwartz, 2004).
When a neural probe is inserted into the brain, neurons and glial cells are killed or injured during insertion, blood vessels are disrupted, and the blood-brain barrier (BBB) is damaged. The tissue injury and breakdown of BBB cause release of cytokines and neurotoxic free radicals, invasion of blood-borne macrophages, and edema (Fitch and Silver, 1997;Schwartz, 2004). The main cell types involved in tissue reaction to the brain injury are astrocytes, microglia/blood-borne macrophages, and oligodendrocyte precursors (OPCs) (Fawcett and Asher, 1999;Norton, 1999;Hampton et al., 2004). These cells express chondroitin sulfate proteoglycans (CSPGs), important inhibitory molecules in glial scar (Fawcett and Asher, 1999;Properzi and Fawcett, 2004). Astrocytes produce neurocan, phosphacan, and brevican; microglia/macrophages produce NG2; and OPCs produce neurocan, NG2, and versican (Fawcett and Asher, 1999;Tang et al., 2003;Hampton et al., 2004;Properzi and Fawcett, 2004;Tatsumi et al., 2005). It has recently been suggested that some of the NG2 positive cells proliferating in the injury site differentiate into the glial scar astrocytes (Alonso et al., 2005;Tatsumi et al. 2005). CSPGs and other glial scar associated inhibitory molecules create an inhibitory environment that blocks the regrowth of neural processes and may potentially cause the exclusion of neural cells by their presence. Although CSPG expression has been extensively studied in CNS injuries, its role in the tissue reactions to implanted neural electrodes has not been addressed.
The failure of implanted neural probes over time can be attributed to neuronal loss around the probe including both neuron cell body loss and neural process loss (Liu et al., 1999;Schwartz, 2004;Spataro et al., 2005). Therefore, to maintain long-term recording stability, reactive gliosis and other inflammatory processes around the electrode need to be minimized. One approach to modulate the inflammatory response around neural probes is to develop coatings that modify the neural probe surfaces to achieve better integration of the neural probes with brain tissue. Dexamethasone (DEX) is a synthetic glucocorticoid hormone that is used to treat many inflammatory responses. In the CNS, systematic injection of DEX has been shown to reduce tissue reaction around neural implants (Shain et al., 2003;Spataro et al., 2005). Addition of DEX to activated microglia-neuron cocultures protects neurons by down-regulating nitric oxide (NO) production (Golde et al., 2003). However, systemic administration of DEX may cause serious side effects including myopathy and diabetes (Twycross, 1994;Koehler, 1995;Kaal and Vecht, 2004). Therefore local delivery of DEX is a promising strategy to minimize the side effects. In this study, a nitrocellulose-based coating that is capable of sustained local release of DEX from implanted neural probes was designed. Scar/inflammation related cellular and molecular responses to neural probes with and without DEX coatings were characterized.
2. Results
2.1. Assessing reactive microglia and macrophages around the probes
Immunostaining for ED1 around the probe site revealed reactive microglia/macrophages. One week post implantation, for both uncoated and DEX coated probes, ED1 staining was concentrated around the probe-brain interface (Fig. 1A and B). The ED1 positive cells were small, and amoeboid in appearance. Quantitative analysis of histological data reviewed that both ED1 peak intensity and reactive area for coated probes were reduced compared with uncoated probes at 1 week (Fig. 1E), and total ED1 intensity for coated probes was significantly lower than uncoated probes (Fig. 1F).
Fig. 1.
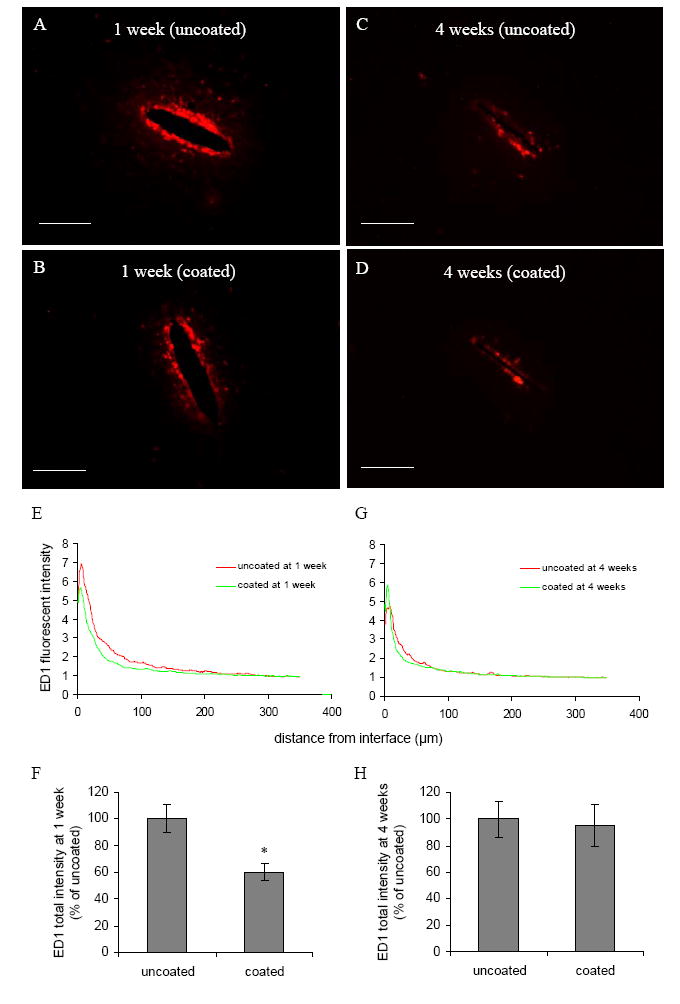
(A)–(D) Representative images of ED1 staining for reactive microglia and macrophages in the horizontal brain sections 1 week and 4 weeks post implantation for both uncoated and DEX coated probes. Scale bar = 100 μm. (E, G) ED1 fluorescent intensity profiles as a function of distance 1 week and 4 weeks post implantation. (F, H) Quantification of total ED1 fluorescent intensity 1 week and 4 weeks post implantation (n = 4). * P < 0.05 compared with uncoated probes.
At the end of 4 weeks, ED1 staining was reduced compared with 1 week for both uncoated and coated probes (Fig. 1A and C). The difference of total intensity between the uncoated probes and DEX coated probes at 4 weeks was not statistically significant (Fig. 1H).
2.2. Characterizing reactive astrocytes in vicinity of implanted probes
GFAP staining was used to identify reactive astrocytes. One week post implantation, extensive GFAP elevation extending over a 400 μm radius around the uncoated probes was observed. GFAP intensity was relatively weak in the region within 40 μm from the probe-brain interface (Fig. 2A and E), as there were few GFAP-positive cells/processes in this region. It is noteworthy that this region was correlated with intensive ED1 staining (Fig. 1A and E). GFAP intensity started to increase from about 40 μm, peaked between 60 to 110 μm, and decreased rapidly after 200 μm (Fig. 2E). Most astrocytes in the GFAP intensive area (40–200 μm) appeared hypertrophic (Fig. 2A, inset), and the processes of astrocytes were interwoven to form a dense meshwork. In contrast, GFAP positive astrocytes located further away were more stellate in appearance, and the processes were less densely distributed.
Fig. 2.
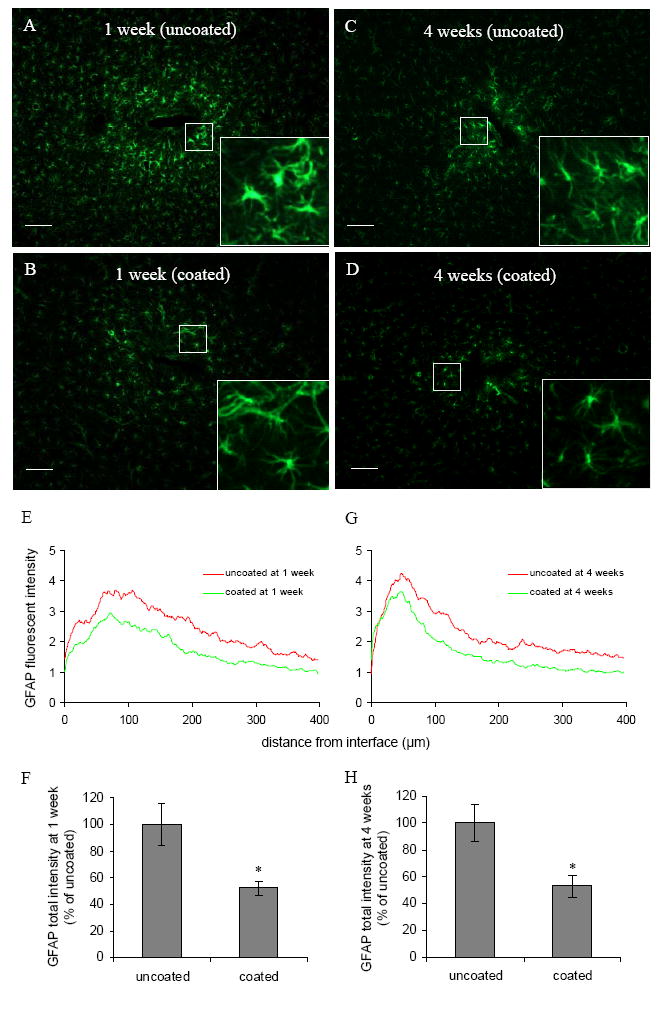
(A)–(D) Representative images of GFAP staining for astrocytes in the horizontal brain sections 1 week and 4 weeks post implantation for both uncoated and DEX coated probes. Scale bar = 100 μm. Inset denotes higher magnification (four-fold) of identified area showing the morphology of reactive astrocytes. (E, G) GFAP fluorescent intensity profiles as a function of distance 1 week and 4 weeks post implantation. (F, H) Quantification of total GFAP fluorescent intensity 1 week and 4 weeks post implantation (n = 4). *P < 0.05 compared with uncoated probes.
The intensity distribution as a function of distance for coated probes showed a similar pattern to the uncoated probes: weak GFAP staining around the probe-brain interface (40 μm), followed by an intensive region (40–165 μm), and a drop of GFAP intensity after 165 μm (Fig. 2B and E). DEX treatment significantly reduced total GFAP intensity (Fig. 2F). Compared with uncoated probes, astrocytes subjected to DEX treatment still remained stellate in the GFAP intensive region, although they were hypertrophied compared to astrocytes in the non-injured area (Fig. 2B, inset). Astrocyte processes were less dense in this region compared to the same region for the uncoated probes (Fig. 2A and B).
Four weeks post implantation, there was still a region of less intensive GFAP staining around the probe-brain interface for uncoated probes (Fig. 2C). GFAP intensity peaked between 40 to 60 μm (compared with 60 to 110 μm at 1 week), and decreased rapidly after 120 μm (Fig. 2G). The GFAP intensive region (20–120 μm) became more compact and closer to the probe-brain interface compared with 1 week (40–200 μm). Most astrocytes in the GFAP intensive area were hypertrophied compared with normal astrocytes (Fig. 2C, inset); astrocyte processes were interwoven to form a dense meshwork. Compared with uncoated probes, astrocytes in the same region around DEX coated probes were less hypertrophied, and the processes were less densely distributed (Fig. 2D, inset). Qualitatively, GFAP peak intensity and reactive area were reduced for coated probes compared with uncoated probes (Fig. 2G). Total GFAP intensity was also significantly lower for DEX coated probes compared with uncoated probes (Fig. 2H).
2.3. Presence of chondroitin sulfate proteoglycans
CS56 staining was used to identify the presence of chondroitin sulfate proteoglycans around the probe. Similar to ED1 localization, intensive CS56 staining was concentrated around the probe-brain interface 1 week post implantation (Fig. 3A and B). For uncoated probes, the area of intensive staining extended about 44 μm from the electrode-brain interface, followed by a extensive but much less intensive region (44–135 μm), beyond which CS56 intensity started to approach background intensity (Fig. 3E). It is noteworthy that the CS56 intensive region was correlated with the weakly GFAP stained region, and the ED1 intensive region (Fig. 1A and E, and Fig. 2A and E). In the less intensive CS56 staining region, individual cells with a hypertrophied stellate morphology could be identified (Fig. 3A), and double staining with GFAP showed that these cells are hypertrophic reactive astrocytes (data not shown).
Fig. 3.
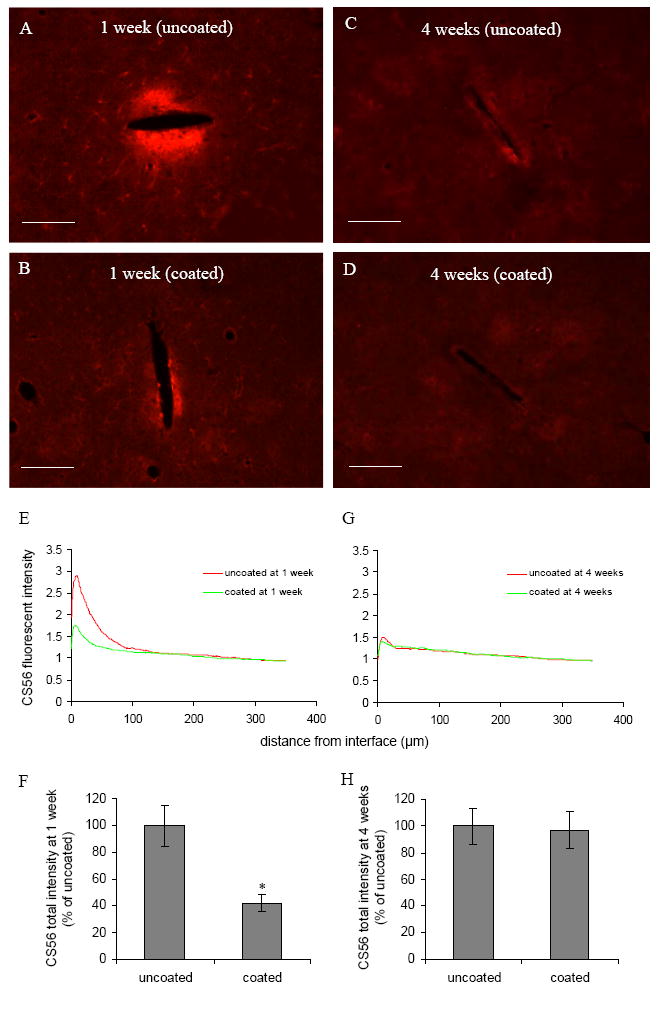
(A)–(D) Representative images of CS56 staining for CSPGs in the horizontal brain sections 1 week and 4 weeks post implantation for both uncoated and DEX coated probes. Scale bar = 100 μm. (E, G) CS56 fluorescent intensity profiles as a function of distance 1 week and 4 weeks post implantation. (F, H) Quantification of total CS56 fluorescent intensity 1 week and 4 weeks post implantation (n = 4). *P < 0.05 compared with uncoated probes.
In comparison with uncoated probes, CS56 staining intensity was significantly reduced for DEX coated probes. The total intensity for coated probes was significantly lower than uncoated probes (Fig. 3F). Despite the reduction of CS56 staining for the coated probes, the pattern of CS56 distribution was similar to that of the uncoated probes: intensive CS56 staining in the vicinity of the electrode-brain interface (27 μm), followed by a less intensive region with cells positive for both CS56 and GFAP staining (Fig. 3E).
Four weeks post implantation, CS56 staining was significantly reduced for uncoated probes compared with one week (Fig. 3 A and C). The intensity around the probe-brain interface was only slightly higher than background intensity (Fig. 4G). There was no significant difference in the total CS56 intensity between the uncoated and coated probes at this time point (Fig. 3H).
Fig. 4.
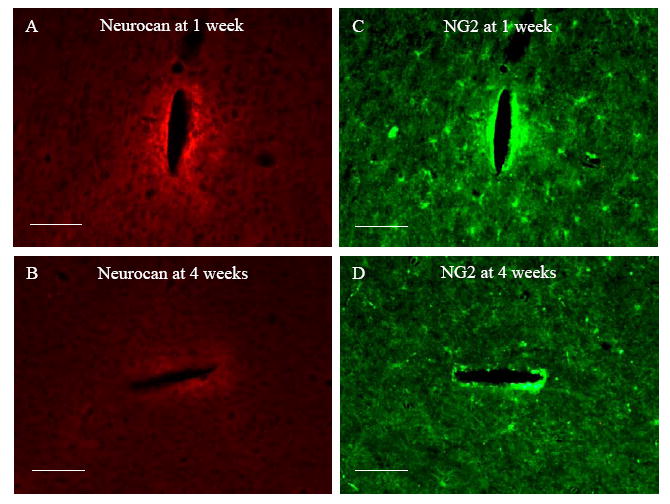
Representative fluorescent images of horizontal brain sections immunostained for Neurocan and NG2 1 week and 4 weeks post implantation. Scale bar = 100 μm.
2.4. Examining the presence of specific CSPGs Neurocan and NG2
Immunostainings for neurocan and NG2 were carried out to investigate the specific nature of proteoglycan expression level with time. Brain sections were double stained with GFAP or Iba-1 for astrocytes and microglia to identify the cellular sources of the proteoglycans.
At the end of 1 week, elevated neurocan staining was observed around the probebrain interface (Fig. 4A). Although there was positive GFAP staining in the same region, the GFAP peak area was outside this region (Fig. 2A). Double staining with Iba-1 showed aggregation of microglia in the neurocan intensive region (Fig. 5A). However, the intensity distribution of neurocan staining did not strictly follow Iba-1 staining.
Fig. 5.
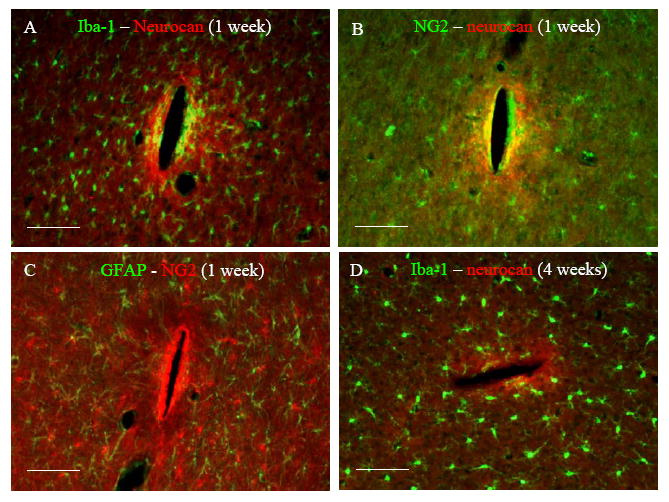
Representative fluorescent images of horizontal brain sections double stained with (A) Iba-1 – neurocan at 1 week, (B) NG2 – neurocan at 1 week, (C) GFAP – NG2 at 1 week, and (D) Iba-1 – neurocan 4 weeks post implantation. Scale bar = 100 μm.
Double staining with NG2 antibody (marker for OPCs) showed upregulation of NG2 staining intensity around the probe-brain interface as well (Fig. 4C). Neurocan and NG2 staining distribution partly overlapped. While positive neurocan staining was only observed around the probe-brain interface (Fig. 4A), NG2 staining was distributed throughout the whole brain sections in the form of NG2 positive cells except around the probe-brain interface where no individual cells could be identified (Fig. 4C). Double staining with GFAP antibody revealed no correlation between GFAP staining and NG2 staining (Fig. 5C).
Four weeks post implantation, both neurocan and NG2 staining were considerably reduced compared with 1 week (Fig. 4). Neurocan staining was still concentrated around the probe-brain interface with weak GFAP staining (Fig. 4B). Iba-1 staining showed that the distribution and morphology of microglia around the probe-brain interface almost reduced to normal levels compared to that observed at 1 week (Fig. 5 A and D), and there was no correlation between Iba-1 staining and neurocan staining (Fig. 5D).
2.5. Characterizing neural presence around the electrodes
Neurofilament (NF) staining is a marker for neuron cell bodies and neural processes. Reduced NF staining intensity around the probe-brain interface was observed 1 week post implantation (Fig. 6A). The average width of the NF loss region was about 37 μm for uncoated probes and 17 μm for DEX coated probes (Fig. 6A, B, and E). The NF loss around uncoated probes was significantly higher than coated probes (Fig. 6F). The majority of NF loss was observed in the ED1 and CS56 intensive area (Fig. 1 A and E, and Fig. 3 A and E).
Fig. 6.
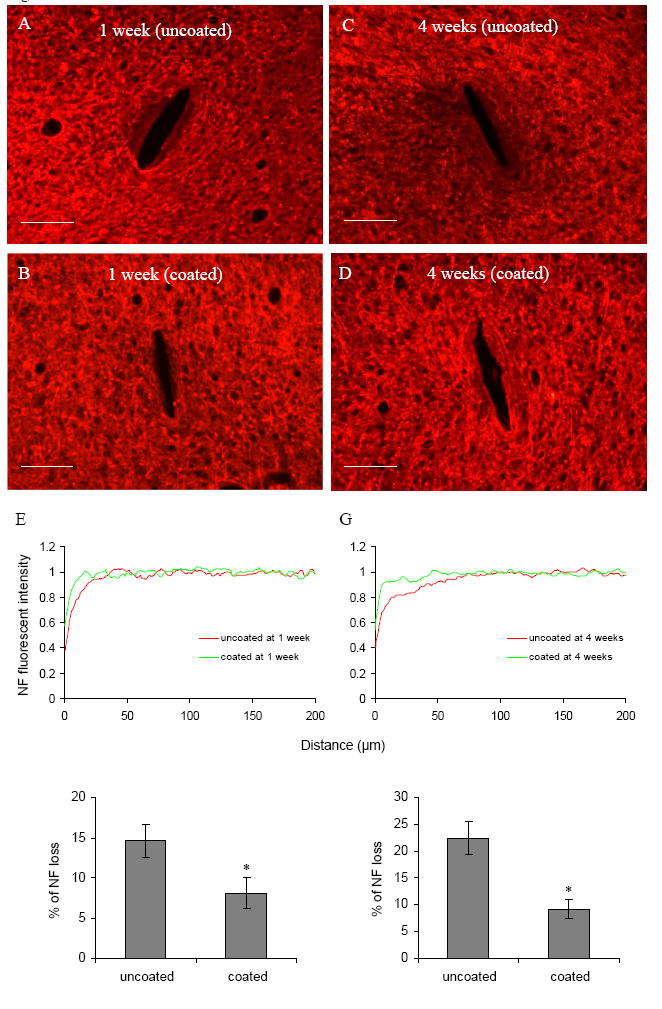
(A)–(D) Representative images of NF160 staining for neurons in the horizontal brain sections 1 week and 4 weeks post implantation for both uncoated and DEX coated probes. Scale bar = 100 μm. (E, G) NF fluorescent intensity profiles as a function of distance 1 week and 4 weeks post implantation. (F, H) Quantification of % of NF loss 1 week and 4 weeks post implantation (n = 4). *P < 0.05 compared with uncoated probes.
The area of NF loss increased at the end of 4 weeks compared with 1 week for both uncoated and coated probes (Fig. 6 C and D). The average radius of this NF loss region was 100 μm for uncoated probes and 45 μm for DEX coated probes (Fig. 6G), compared to NF loss radius of 37 and 17 μm respectively at 1 week (both measured as distance from the probe interface). The NF loss around uncoated probes was significantly higher than coated probes at 4 weeks (Fig. 6H). The NF loss region overlapped the ED1 and CS56 positive area with weak GFAP staining, and extended to the GFAP intensive area (Fig. 1 C and G, Fig.2 C and G, and Fig. 3 C and G).
3. Discussion
To maintain long term functional recording from silicon neural probes, the neural elements of recording interest need to be preserved in the vicinity of the implanted electrodes. We have previously reported the fabrication of a nitrocellulose-DEX coating for Si neural probes (Zhong et al., 2005). In vitro dexamethasone release was observed over 16 days. The coating thickness was 1.72 ± 0.16 μm. Coating adhesion test showed that the coatings still remained attached on the probes after insertion implantation (Zhong, 2006). We have reported that this micron-scale coating mildly reduced the electrode impedance, possibly due to the increase of capacitance as evidenced by impedance spectroscopy (Zhong et al., 2005). Low electrode impedance potentially improves the signal transport across the neural interface and helps to increase the detection sensitivity to neural activity, therefore the electrical performance of the electrode was not adversely affected by the polymer coating. This current study demonstrates that local release of DEX from neural probe coatings can attenuate the cellular inflammatory responses to implanted neural probes, suppress the expression of inhibitory molecules (CSPGs), and significantly reduce neuronal loss around the neural probes. Tissue reaction around probes coated with nitrocellulose alone did not exhibit any difference from uncoated probes as evidenced by GFAP and ED1 staining (data not shown).
In this study, the cellular and molecular responses to the implanted silicon neural probes were investigated to give us insight into the cellular and molecular environment that local DEX delivery helps create around the probe-brain interface. The glial cell types involved in glial scar formation include astrocytes, microglia/blood-borne macrophages, and OPCs. Microglia/macrophages and OPCs respond very quickly to brain injuries (Fawcett and Asher, 1999;Kato and Walz, 2000;Hampton et al., 2004). Following injury to the adult CNS, a large number of microglia/macrophages and OPCs are recruited to the injury site. The activated microglia become more macrophage-like with an amoeboid morphology, and they proliferate and migrate to injury sites (Fawcett and Asher, 1999). Activated microglia/blood-borne macrophages release neurotoxic molecules such as free radicals, nitric oxide (NO), as well as proinflammatory cytokines including interleukin-1 (IL-1), tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) (Bruccoleri et al., 1998;Hays, 1998;Kyrkanides et al., 2001;Takeuchi et al., 2001), which subsequently activate astrocytes (Merrill and Benveniste, 1996; John et al., 2005). Astrocyte activation is also mediated by blood-borne factors including growth factors and hormone, albumin, thrombin, angiotensin II and cAMP (Logan and Berry, 2002). The reactive astrocytes undergo hypertrophy, proliferation, and upregulate trophic factors, cytokines, as well as extracellular matrix (Fawcett and Asher, 1999;Polikov et al., 2005).
Chondroitin sulfate proteoglycans are important inhibitory molecules in the glial scar (Fawcett and Asher, 1999). Upregulated CSPGs have differential sulfation patterns (Properzi et al., 2003; Properzi and Fawcett, 2005) which differentially inhibit neurite outgrowth (Gilbert et al., 2005). Following chondroitinase treatment, glycosaminoglycan removal enhances neurite outgrowth, suggesting an inhibitory role for CSPGs (Bradbury et al., 2002). Neurocan and NG2 are identified as two important proteoglycans that are inhibitory for neurite extention (Asher et al., 2000; Jones et al., 2002;Tang et al., 2003;Alonso, 2005). In this study, immunostaining for neurocan and NG2 1 week and 4 weeks post implantation was carried out to investigate the individual proteoglycan expression level with time. One week post implantation, staining intensity for glial cells (astrocytes, microglia, and OPCs) as well as inhibitory molecules (CSPGs including neurocan and NG2) reached peak level. It is noteworthy that for all the inhibitory molecules, intensive staining was concentrated around the probe-brain interface and appeared to be inversely correlated with NF staining intensity. Interestingly, GFAP staining was weak in this region starting from day 1 (data not shown). A similar observation has been previously reported (Fitch and Silver, 1997), and the authors suggested that CSPGs in the area lacking GFAP-positive cells might be produced by astrocytes that subsequently died, migrated away from the injury site, or lost their GFAP immunoreactivity. The authors also suggested that microglia/macrophages might be another source of inhibitory CSPGs. Since neurocan staining was correlated with both Iba-1 staining and NG2 staining, it is possible that microglia and OPCs also contributed to neurocan upregulation. In the intensive NG2 staining area, it was difficult to identify individual NG2 positive cells. In the normal tissue of the CNS, NG2 is found almost exclusively on the surfaces of OPCs (Levine et al., 2001;Ughrin et al., 2003). Therefore, the intensive NG2 staining might be a marker for the proliferated and aggregated OPCs. However, since NG2 can be shed from the cell surface and secreted into extracellular matrix, there is the possibility that the intensive NG2 staining was not only a cell marker, but also the marker for NG2 proteoglycan in extracellular matrix secreted by OPCs as well as other types of cells. Double staining with ED1 antibody showed that the intensity distribution of NG2 staining was also correlated to ED1 staining (data not shown), therefore reactive microglia/macrophages might be another cell source for upregulation of NG2. Upregulation of Neurocan and NG2 staining was reduced for the DEX treated group at 1 week, and little upregulation of neurocan and NG2 staining was observed at 4 weeks for both uncoated and coated probes (data now shown).
It is interesting that neural loss increased with time while the level of microglia/macrophage activation and CSPG expression was much lower at 4 weeks compared with 1 week. One possible explanation is that neural loss was mediated by the inhibitory and neurotoxic molecules secreted by activated glial cells, and progressive neuronal loss might continue long after the activation of glial cells and the expression of inhibitory molecules. As the inflammatory tissue response was high at 1 week, the neuronal loss was expected to increase after 1 week, which is consistent with the result that neural loss at 4 weeks was higher than 1 week. This result further suggests that neuronal loss follows the initial inflammatory response. Four weeks post implantation, the intensity of ED1 staining was reduced compared with 1 week. CS56, neurocan and NG2 staining intensity declined to near normal levels after 4 weeks, and it appeared that the inflammatory responses started to stabilize at 4 weeks. This observation is consistent with a recent study showing reduced reactivity around implanted neural probes at 4 weeks compared with 1 week as evidenced by ED1, vimentin and collagen staining (McConnell et al., 2007).
Local release of DEX significantly attenuated the inflammatory responses at 1 week, and reduced neuronal loss both at 1 week and 4 weeks as evidenced by NF staining. A recent study shows that sustained release of DEX from nanoparticles of poly(lactic-coglycolic acid) (PLGA) embedded in alginate hydrogel (HG) coatings prevents the increase of electrode impedance in vivo, which is presumably due to the reduced amount of glial inflammation in the immediate vicinity of DEX-modified neural probe (Kim and Martin, 2006). Our study confirms this reduction of glial inflammation around DEXcoated neural probes. It has been previously reported that DEX effectively reduces the inflammatory tissue reaction around the neural implant when DEX concentration at the implant-tissue interface immediately after insertion is calculated to be 0.2 μM (Shain et al., 2003). In another study, 0.5 μg/cm2 DEX release is calculated to result in an average concentration of 1 μM within a 500 μm radius from the electrode (Wadhwa et al., 2006). Our in vitro study showed 0.18 μg/cm2 DEX release on day 16, corresponding to 0.36 μM in vivo. However, in all these studies DEX amounts in vivo are calculated values, as quantifying these low levels (due to very low surface areas of the implanted electrodes) is technically very difficult at this point and indeed the actual in vivo values may be different from these estimated values. The evaporation technique in this study only allows for coating one side of neural probes (the side with recording sites). However, we didn’t observe any significant difference of tissue reaction on different sides. One possible explanation might be that DEX is a small molecule (MW 392 Da), and the thickness of the probe is only 15 μm, therefore DEX can diffuse easily to the back of the neural probe.
DEX is known as a potent anti-inflammatory drug, and it has previously been shown to be capable of reducing inflammatory responses in the CNS (Holmins and Mathiesen, 1996;Hermens and Verhaagen, 1998;Spataro et al., 2005). Its anti-inflammatory effects have usually been attributed to its effects on microglia/macrophages, which knowingly express high levels of glucocorticoid receptors (Tanaka et al., 1997). DEX has been shown to inhibit iNOS synthesis and cytokine production by microglia, as well as microglia proliferation (Chao et al., 1992;Tanaka et al., 1997;Golde et al., 2003). A recent study shows that DEX strongly inhibits proliferation of NG2 positive cells, which preferentially differentiate into astrocytes in injured brain (Alonso, 2005). As these NG2 positive cells do not express the glucocorticoid receptors, the authors suggests that DEX may have indirect effects on these cells via modification of glutamate release and/or interaction with microglia. DEX also directly inhibits astrocyte proliferation (Crossin et al., 1997). It is possible that DEX exerts its anti-inflammatory effects on the glial cells through direct interactions with cells as well as the interactions among different types of cells.
This study demonstrates that DEX coatings on Si probes not only can reduce the cellular inflammatory response, but also can reduce the expression of chondroitin sulfate proteoglycans, the important inhibitory molecules in glial scar. There was no significant difference between the uncoated probes and DEX coated probes for ED1 and CS56 staining at 4 weeks. This could be due to either drug depletion, or the stabilization of the inflammatory response. However, DEX treatment significantly reduced neuronal loss both at 1 week and 4 weeks after implantation, which suggests that reducing inflammation immediately after implantation may have long term beneficial consequences. Reactive glial cells may contribute to neuronal loss through the secretion of neurotoxic molecules including glutamate, pro-inflammatory cytokines, prostaglandins, NO and free radical species, as well as inhibitory molecules such as CSPGs. Therefore, inhibition of glial cell activation may reduce the expression of these molecules, which results in less neuronal damage.
Our previous in vitro release study revealed sustained release of DEX from nitrocellulose coatings for over 16 days (Zhong et al., 2005). It is not known how long drug release needs to last in order to obtain long-term stable neural recording. The most conservative approach would be to facilitate continuous, local drug release for the lifetime of the implant. However, this is technically not feasible, and it may not be necessary. The experience from drug eluting stents which also aim to manage the acute injury response, albeit in the blood vessels, suggests that the release of anti-inflammatory agents in the early stage of implantation inhibits the long-term tissue reaction (De Scheerder et al., 1996;Huang et al., 2002). Following brain injury, the first cells to respond and arrive are microglia and macrophages (Kreutzberg, 1996;Fawcett and Asher, 1999). The activated microglia/macrophages secrete proinflammatory molecules that subsequently activate adjacent microglia or other cell types, including astrocytes, via autocrine and paracrine passways, resulting in propagation and enhancement of the inflammatory response (Bruccoleri et al., 1998;Hays, 1998;Kyrkanides et al., 2001;Takeuchi et al., 2001). Consequently, astrocytes express additional inflammatory mediators that further contribute to the inflammatory response and eventually lead to glial scar formation (Benveniste, 1996;Kyrkanides et al., 2001;Hanisch, 2002; Merrill and John et al., 2005). Both activated microglia and astrocytes are capable of releasing neurotoxic molecules such as free radicals and NO (Lee et al., 1995;Kreutzberg, 1996). Therefore, administration of anti-inflammatory agents in the early stage of brain injury might be able to inhibit the expression of proinflammatory molecules that leads to progression of astroglosis and has long-term effect.
In conclusion, this study demonstrates that local delivery of DEX can reduce the cellular and molecular inflammatory responses to the implanted neural probes, as well as significantly reduce neural loss in the immediate vicinity of the probes. This might be a promising strategy to improve the long-term recording stability of silicon neural probes.
4. Experimental procedures
4.1. Fabrication of DEX loaded nitrocellulose coatings
Micromachined silicon neural recording probes (single shank, 16 recording sites, 5mm) were provided by the Center for Neural Communication Technology (CNCT) at the University of Michigan. The neural probes were mounted on polished Si wafers of 1 cm2 with a 10,000 Å oxide layer (University Wafer, MA). 33.3 mg nitrocellulose (Schleicher & Schuell BioScience) was dissolved in 12 ml methanol. 100 μg DEX (Sigma) powder was dissolved in 20 μl nitrocellulose (NC) solution and evaporated on the Si wafers. This DEX-nitrocellulose layer was subsequently coated with 3 additional layers of 20 μl pure nitrocellulose by evaporation. The DEX-nitrocellulose coated neural probes were removed from the Si wafers and used for the in vivo study.
4.2. In vivo implantation of DEX coated probes
All animal procedures were approved by the Institutional Animal Care and Use Committees (IACUC) at Georgia Institute of Technology. Eight adult male Sprague- Dawley rats weighing between 275–299 g were used in this study. The rats were anesthetized with isoflurane and immobilized in a stereotaxic frame. Following a midline incision, two 3 mm holes were created 0.2 mm anterior and 3 mm lateral to the bregma. After carefully removing the dura, two neural probes were inserted into the brain, with one DEX-coated probe on one side of the brain and one uncoated probe on the other side of the brain. The two holes were covered with agarose gel and dental acrylic, the skin was closed with suture, and then the animal was allowed to recover.
4.3. Tissue preparation and immunohistochemistry
At two distinct time points, one week and four weeks post surgery, the rats were perfused transcardially with 0.1 M PBS (pH7.4) followed by 4% paraformaldehyde in PBS. The brains were removed carefully so that the implanted neural probes remained intact. After post-fixation of the brains in 4% paraformaldehyde for 24 h, the implanted neural probes were carefully retrieved from the brain tissue. The brains were cryoprotected in 30% sucrose in PBS solution for approximately 48 h and then frozen in Tissue Tek OCT embedding compound. Cryostat sections 30 μm in thickness were cut in the horizontal plane.
The floating sections were blocked with 4% normal goat serum with 0.5% Triton X- 100 (Sigma) for 1h at room temperature and incubated overnight at 4°C with primary antibodies. Secondary antibody incubations were performed at room temperature for 1 h. All sections were counterstained with the nuclear dye 4’,6-diamidino-2-phenylindole (DAPI, Molecular Probes).
The following primary antibodies were used: rabbit polyclonal anti-glial fibrillary acidic protein (GFAP, Dako, 1:1000) to identify astrocytes, mouse monoclonal ED1 (Serotec, 1:1000) to identify reactive microglia and macrophages, rabbit polyclonal anti- Iba-1 to identify microglia/macrophages (Wako Chemicals, 1:500), mouse monoclonal anti-neurofilament 160 (NF160, Sigma, 1:500) to identify neuron cell body and processes, mouse monoclonal anti-chondroitin sulfate (CS56, Sigma, 1:100) to identify CSPGs, mouse monoclonal anti-neurocan (Chemicon, 1:1000) to identify neurocan, and rabbit polyclonal anti-NG2 (Chemicon, 1:500) to identify NG2. The secondary antibodies were diluted 1: 200 in blocking medium. In general, goat anti-rabbit IgG Alexa 488 (Molecular Probes) was used for polyclonal primary antibodies, and goat anti-mouse IgG1 Alexa 594 (Molecular Probes) was used for monoclonal primary antibodies. The secondary antibody for CS56 was goat anti-mouse IgM Alexa 594 (Molecular Probes). Sections treated without primary antibodies were used to distinguish specific staining from nonspecific antibody binding.
4.4. Quantitative image analysis
Fluorescent images were captured from a Zeiss fluorescence upright light microscope (Wetzlar, Germany) equipped with an Olympus digital camera. The staining intensity was quantified using a custom-built Matlab-based image analysis program based on a method previously reported (Kim et al., 2004). This program generated 30 line intensity profiles starting from the probe-brain interface. The 30 line profiles were averaged while keeping track of distance from the probe to obtain the fluorescent intensity plots as a function of distance from the implanted probe surface. To account for variations in staining intensity due to immunohistochemical methods, the fluorescent intensity in noninjured regions was defined as background intensity for each brain section, and was normalized to 1. The fluorescent intensity along the line profiles was then quantified and plotted relative to the background intensity.
For quantification of GFAP, ED1 and CS56 staining, the total area under the intensity-distance curve subtracted by the background area (the shaded area in Fig. 7A) was used to calculate the total fluorescent intensity. Neurofilament (NF 160kDa) staining was used to assess neuronal loss. NF loss was defined as the percentage of the NF loss area (the shaded area in Fig. 7B) relative to the sum of NF loss area and the remaining NF area within a 50 μm radius from the surface of the inserted probe. The 50 μm radius was chosen because neurons more than 50 μm away from the recording sites are difficult to discriminate for single unit isolation (Henze et al., 2000). For each marker analyzed, at least 12 sections from 4 rats were used for quantitative data analysis, with a minimum of 3 sections taken from the cortical region for each brain (n = 4).
Fig. 7.
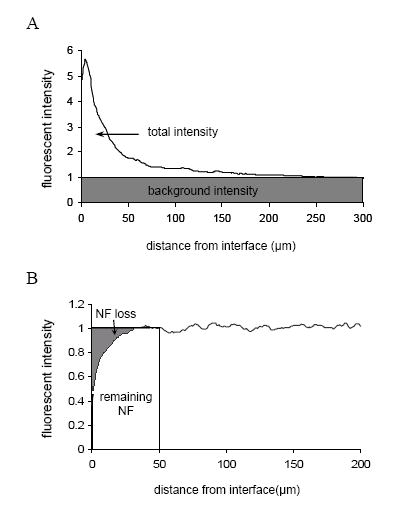
Diagram for quantification of (A) GFAP, ED1, and CS56 staining, the total area under the intensity-distance curve subtracted by the background (the shaded area) is defined as the total intensity, and (B) neurofilament staining, the percentage of the NF loss area relative to the sum of the NF loss area and the remaining NF area is defined as % of NF loss.
4.5. Statistical analysis
Data are represented as the average value ± the standard error of the mean (S.E.M). A general linear ANOVA model was used to compare mean values of the different conditions. Pairwise comparisons were conducted using Tukey 95% simultaneous confidence intervals, and P < 0.05 was used to indicate statistical significance.
Acknowledgments
This research was funded by the National Institutes of Health (R01 DC06849-01 and R01 NS45072 to RVB) and by GTEC, an NSF Engineering Research Center based at Georgia Tech/Emory. The authors thank George McConnell, Matthew Ward, and Matt Davis for their technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alonso G. NG2 proteoglycan-expressing cells of the adult rat brain: possible involvement in the formation of glial scar astrocytes following stab wound. Glia. 2005;49:318–338. doi: 10.1002/glia.20121. [DOI] [PubMed] [Google Scholar]
- Asher RA, Morgenstern DA, Fidler PS, Adcock KH, Oohira A, Braistead JE, Levine JM, Margolis RU, Rogers JH, Fawcett JW. Neurocan is upregulated in injured brain and in cytokine-treated astrocytes. J Neurosci. 2000;20:2427–2438. doi: 10.1523/JNEUROSCI.20-07-02427.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biran R, Martin DC, Tresco PA. Neuronal cell loss accompanies the brain tissue response to chronically implanted silicon microelectrode arrays. Exp Neurol. 2005;195:115–126. doi: 10.1016/j.expneurol.2005.04.020. [DOI] [PubMed] [Google Scholar]
- Bradbury EJ, Moon LD, Popat RJ, King VR, Bennett GS, Patel PN, Fawcett JW, McMahon SB. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature. 2002;416:636–640. doi: 10.1038/416636a. [DOI] [PubMed] [Google Scholar]
- Bruccoleri A, Brown H, Harry GJ. Cellular localization and temporal elevation of tumor necrosis factor-alpha, interleukin-1 alpha, and transforming growth factor-beta 1 mRNA in hippocampal injury response induced by trimethyltin. J Neurochem. 1998;71:1577–1587. doi: 10.1046/j.1471-4159.1998.71041577.x. [DOI] [PubMed] [Google Scholar]
- Chao CC, Hu S, Close K, Choi CS, Molitor TW, Novick WJ, Peterson PK. Cytokine release from microglia: differential inhibition by pentoxifylline and dexamethasone. J Infect Dis. 1992;166:847–53. doi: 10.1093/infdis/166.4.847. [DOI] [PubMed] [Google Scholar]
- Crossin KL, Tai MH, Krushel LA, Mauro VP, Edelman GM. Glucocorticoid receptor pathways are involved in the inhibition of astrocyte proliferation. Proc Natl Acad Sci. 1997;94(6):2687–2692. doi: 10.1073/pnas.94.6.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Scheerder I, Wang K, Wilczek K, van Dorpe J, Verbeken E, Desmet W, Schacht E, Piessens J. Local methylprednisolone inhibition of foreign body response to coated intracoronary stents. Coron Artery Dis. 1996;7(2):161–166. doi: 10.1097/00019501-199602000-00011. [DOI] [PubMed] [Google Scholar]
- Donoghue JP. Connecting cortex to machines: recent advances in brain interfaces. Nat Neurosci. 2002;5(Suppl):1085–1088. doi: 10.1038/nn947. [DOI] [PubMed] [Google Scholar]
- Edell DJ, Toi VV, McNeil VM, Clark LD. Factors influencing the biocompatibility of insertable silicon microshafts in cerebral cortex. IEEE Trans Biomed Eng. 1992;39:635–643. doi: 10.1109/10.141202. [DOI] [PubMed] [Google Scholar]
- Fawcett JW, Asher RA. The glial scar and central nervous system repair. Brain Res Bull. 1999;49:377–391. doi: 10.1016/s0361-9230(99)00072-6. [DOI] [PubMed] [Google Scholar]
- Fitch MT, Silver J. Activated macrophages and the blood-brain barrier: inflammation after CNS injury leads to increase in putative inhibitory molecules. Exp Neurol. 1997;148:587–603. doi: 10.1006/exnr.1997.6701. [DOI] [PubMed] [Google Scholar]
- Gilbert RJ, McKeon RJ, Darr A, Calabro A, Hascall VC, Bellamkonda RV. CS-4,6 is differentially upregulated in glial scar and is a potent inhibitor of neurite extension. Mol Cell Neurosci. 2005;29:545–558. doi: 10.1016/j.mcn.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Golde S, Coles A, Lindquist JA, Compston A. Decreased iNOS synthesis mediates DEX-induced protection of neurons from inflammatory injury in vitro. Eur J Neurosci. 2003;18:2527–2537. doi: 10.1046/j.1460-9568.2003.02917.x. [DOI] [PubMed] [Google Scholar]
- Hampton DW, Rhodes KE, Zhao C, Franklin RJM, Fawcett JW. The responses of oligodendrocyte precursor cells, astrocytes and microglia to a cortical stab injury, in the brain. Neurosci. 2004;127:813–820. doi: 10.1016/j.neuroscience.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40(2):140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- Hays SJ. Therapeutic approaches to the treatment of neuroinflammatory diseases. Curr Pharm Des. 1998;4:335–348. [PubMed] [Google Scholar]
- Henze DA, Borhegyi Z, Csicsvari J, Mamiya A, Harris KD, Buzsaki G. Intracellular features predicted by extracellular recordings in the hippocampus in vivo. J Neurophysiol. 2000;84:390–400. doi: 10.1152/jn.2000.84.1.390. [DOI] [PubMed] [Google Scholar]
- Hermens WT, Verhaagen J. Suppression of inflammation by DEX prolongs adenoviral vector-mediated transgene expression in the facial nucleus of the rat. Brain Res Bull. 1998;47:133–140. doi: 10.1016/s0361-9230(98)00042-2. [DOI] [PubMed] [Google Scholar]
- Holmin S, Mathiesen T. DEX and colchicine reduce inflammation and delayed oedema following experimental brain contusion. Acta Neurochir. 1996;138:418–424. doi: 10.1007/BF01420304. [DOI] [PubMed] [Google Scholar]
- Huang Y, Wang L, Verweire I, Qiang B, Liu X, Verbeken E, Schacht E, De Scheerder I. Optimization of local methylprednisolone delivery to inhibit inflammatory reaction and neointimal hyperplasia of coated coronary stents. J Invasive Cardiol. 2002;14(9):505–513. [PubMed] [Google Scholar]
- John GR, Lee SC, Song X, Rivieccio M, Brosnan CF. IL-1-regulated responses in astrocytes: relevance to injury and recovery. Glia. 49:161–176. doi: 10.1002/glia.20109. [DOI] [PubMed] [Google Scholar]
- Jones LL, Yamaguchi Y, Stallcup WB, Tuszynski MH. NG2 is a major chondroitin sulfate proteoglycan produced after spinal cord injury and is expressed by macrophages and oligodendrocyte progenitors. J Neurosci . 22:2792–2803. doi: 10.1523/JNEUROSCI.22-07-02792.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaal EC, Vecht CJ. The management of brain edema in brain tumors. Curr Opin Oncol. 2004;16:593–600. doi: 10.1097/01.cco.0000142076.52721.b3. [DOI] [PubMed] [Google Scholar]
- Kato H, Walz W. The initiation of the microglial responses. Brain Pathol. 2000;10:137–143. doi: 10.1111/j.1750-3639.2000.tb00250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Martin DC. Sustained release of dexamethasone from hydrophilic matrices using PLGA nanoparticles for neural drug delivery. Biomaterials. 27:3031–3037. doi: 10.1016/j.biomaterials.2005.12.021. [DOI] [PubMed] [Google Scholar]
- Koehler PJ. Use of corticosteroids in neuro-oncology. Anticancer Drugs. 1995;6:19–33. doi: 10.1097/00001813-199502000-00002. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Kyrkanides S, O′Banion MK, Whiteley PE, Daeschner JC, Olschowka JA. Enhanced glial activation and expression of specific CNS inflammation-related molecules in aged versus young rats following cortical stab injury. J Neuroimmunol. 2001;119:269–277. doi: 10.1016/s0165-5728(01)00404-0. [DOI] [PubMed] [Google Scholar]
- Lee SC, Dickson DW, Brosnan CF. Interleukin-1, nitric oxide and reactive astrocytes. Brain Behav Immun. 1995;9(4):345–54. doi: 10.1006/brbi.1995.1032. [DOI] [PubMed] [Google Scholar]
- Levine JM, Reynolds R, Fawcett JW. The oligodendrocyte precursor cell in health and disease. Trends Neurosci. 2001;24:39–47. doi: 10.1016/s0166-2236(00)01691-x. [DOI] [PubMed] [Google Scholar]
- Liu X, McCreery DB, Carter RR, Bullara LA, Yuen TG, Agnew WF. Stability of the interface between neural tissue and chronically implanted intracortical microelectrodes. IEEE Trans Rehabil Eng. 1999;7:315–326. doi: 10.1109/86.788468. [DOI] [PubMed] [Google Scholar]
- Logan A, Berry M. Cellular and molecular determinants of glial scar formation. In: Alzheimer C, editor. Molecular and cellular biology of neuroprotection in the CNS. Kluwer Academic; 2002. pp. 115–137. [DOI] [PubMed] [Google Scholar]
- Merrill JE, Benveniste EN. Cytokines in inflammatory brain lesions: helpful and harmful. Glia. 1996;19:331–338. doi: 10.1016/0166-2236(96)10047-3. [DOI] [PubMed] [Google Scholar]
- McConnell GC, Schneider TM, Owens DJ, Bellamkonda RV. Extraction force and cortical tissue reaction of silicon microelectrode arrays implanted in the rat brain. IEEE Biomed Eng. 2007 doi: 10.1109/TBME.2007.895373. accepted. [DOI] [PubMed] [Google Scholar]
- Norton WT. Cell reactions following acute brain injury: a review. Neurochem Res. 1999;24:213–218. doi: 10.1023/a:1022505903312. [DOI] [PubMed] [Google Scholar]
- Otto KJ, Vetter RJ, Marzullo TC, Kipke DR. Brain-machine interfaces in rat motor cortex. 2003 Proceedings of the 1st international IEEE EMBS Conference on Neural Engineering Capri Island, Italy. [Google Scholar]
- Polikov VS, Tresco PA, Reichert WM. Response of brain tissue to chronically implanted neural electrodes. J Neurosci Methods. 2005;148:1–18. doi: 10.1016/j.jneumeth.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Properzi F, Asher RA, Fawcett JW. Chondroitin sulphate proteoglycans in the central nervous system: changes and synthesis after injury. Biochem Soc Trans. 2003;31:335–336. doi: 10.1042/bst0310335. [DOI] [PubMed] [Google Scholar]
- Properzi F, Fawcett JW. Proteoglycans and brain repair. News Physiol Sci. 2004;19:33–38. doi: 10.1152/nips.01449.2003. [DOI] [PubMed] [Google Scholar]
- Sanchez JC, Carmena JM, Lebedev MA, Nicolelis MAL, Harris JG, Principle JC. Ascertaining the importance of neurons to develop better brain-machine interfaces. IEEE Trans Biomed Eng. 2004;51:943–953. doi: 10.1109/TBME.2004.827061. [DOI] [PubMed] [Google Scholar]
- Schwartz AB. Cortical neural prosthetics. Annu Rev Neurosci. 2004;27:487–507. doi: 10.1146/annurev.neuro.27.070203.144233. [DOI] [PubMed] [Google Scholar]
- Shain W, Spataro L, Dilgen J, Haverstick K, Retterer S, Isaacson M, Saltzman M, Turner JN. Controlling cellular reactive responses around neural prosthetic devices using peripheral and local intervention strategies. IEEE Trans Neural Syst Rehabil Eng. 2003;11:186–188. doi: 10.1109/TNSRE.2003.814800. [DOI] [PubMed] [Google Scholar]
- Spataro L, Dilgen J, Retterer S, Spence AJ, Isaacson M, Turner JN, Shain W. DEX treatment reduces astroglia responses to inserted neuroprosthetic devices in rat neocortex. Exp Neurol. 2005;194:289–300. doi: 10.1016/j.expneurol.2004.08.037. [DOI] [PubMed] [Google Scholar]
- Szarowski DH, Andersen MD, Retterer S, Spence AJ, Isaacson M, Craighead HG, Turner JN, Shain W. Brain responses to micro-machined silicon devices. Brain Res. 2003;983:23–35. doi: 10.1016/s0006-8993(03)03023-3. [DOI] [PubMed] [Google Scholar]
- Takeuchi A, Miyaishi O, Kiuchi K, Isobe K. Macrophage colony-stimulating factor is expressed in neuron and microglia after focal brain injury. J Neurosci Res. 2001;65:38–44. doi: 10.1002/jnr.1125. [DOI] [PubMed] [Google Scholar]
- Tanaka J, Fujita H, Matsuda S, Toku K, Sakanaka M, Maeda N. Glucocorticoid- and mineralocorticoid receptors in microglial cells: the two receptors mediate differential effects of corticosteroids. Glia. 1997;20:23–37. [PubMed] [Google Scholar]
- Tang X, Davies JE, Davies SJA. Changes in distribution, cell associations, and protein expression levels of NG2, neurocan, phosphacan, brevican, versican V2, and tenascin-C during acute to chronic maturation of spinal cord scar tissue. J Neurosci Res. 2003;71:427–444. doi: 10.1002/jnr.10523. [DOI] [PubMed] [Google Scholar]
- Tatsumi K, Haga S, Matsuyoshi H, Inoue M, Manabe T, Makinodan M, Wanaka A. Characterization of cells with proliferative activity after a brain injury. Neurochem Int. 2005;46:381–389. doi: 10.1016/j.neuint.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Turner JN, Shain W, Szarowski DH, Andersen M, Martins S, Isaacson M, Craighead H. Cerebral astrocyte response to micromachined silicon implants. Exp Neurol. 1999;156:33–49. doi: 10.1006/exnr.1998.6983. [DOI] [PubMed] [Google Scholar]
- Twycross R. The risks and benefits of corticosteroids in advanced cancer. Drug Saf. 1994;11:163–178. doi: 10.2165/00002018-199411030-00003. [DOI] [PubMed] [Google Scholar]
- Ughrin YM, Chen ZJ, Levine JM. Multiple regions of the NG2 proteoglycan inhibit neurite growth and induce growth cone collapse. J Neurosci. 2003;23:175–186. doi: 10.1523/JNEUROSCI.23-01-00175.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadhwa R, Lagenaur CF, Cui XT. Electrochemically controlled release of dexamethasone from conducting polymer polypyrrole coated electrode. J Control Release. 2006;110:531–541. doi: 10.1016/j.jconrel.2005.10.027. [DOI] [PubMed] [Google Scholar]
- Zhong Y, McConnell GC, Ross JD, DeWeerth SP, Bellamkonda RV. A novel DEX-releasing, anti-inflammatory coating for neural implants. 2005:522–525. Proceedings of the 2nd international IEEE EMBS Conference on Neural Engineering Arlington, USA. [Google Scholar]
- Zhong Y. Development and characterization of anti-inflammatory coatings for implanted neural probes. 2006 Ph.D. dissertation, Georgia Institute of Technology, Atlanta, GA. [Google Scholar]