

Abstract
Objective
To test the hypothesis that some antiphospholipid antibodies (aPL) in patients with the Antiphospholipid Syndrome (APS) recognize a conformational epitope shared by β2 glycoprotein I (β2GPI, the major autoantigen for the antiphospholipid antibodies) and the homologous catalytic domains of several serine proteases (such as thrombin, activated protein C and plasmin) in hemostasis.
Methods
We generated four new IgG monoclonal aPL (including two screened against β2GPI, one against thrombin and one against protein C) from two APS patients. The monoclonal antibodies (mAb) were analyzed for binding to β2GPI, thrombin, activated protein C (APC) and plasmin, and for anti-cardiolipin antibody (aCL) activity. To demonstrate a shared epitope between β2GPI and a serine protease, one mAb was studied by cross-inhibition.
Results
Both IgG anti-β2GPI mAb bound to thrombin, APC and plasmin. On the other hand, one anti-thrombin mAb and one anti-protein C mAb also bound to β2GPI. Moreover, the binding of one crossreactive mAb to β2GPI was inhibited by α-thrombin (that contains only the catalytic domain of thrombin). All four mAb displayed aCL activity.
Conclusion
Taken together with the findings that some aCL bind to several serine proteases that participate in hemostasis and share homologous catalytic domains, these data demonstrate that some aCL in APS patients recognize one or more conformational epitopes shared by β2GPI and the catalytic domains of disease-relevant serine proteases.
INTRODUCTION
Antiphospholipid antibodies (aPL) are associated with thrombosis and fetal loss in some patients, and their combined presence is recognized as the antiphospholipid syndrome (APS) (1-7). APL include anticardiolipin antibodies (aCL, as detected by enzyme-linked immunosorbent assay) and lupus anticoagulants (LAC, as detected by their abilities to prolong certain in vitro phospholipid-restricted blood clotting tests). Immunologic studies of aPL show that aPL represent a heterogeneous group of immunologically distinct antibodies (Ab) that recognize various phospholipids (PL), PL-binding plasma proteins and/or PL-protein complexes (8-13). The involved plasma proteins include β2 glycoprotein-I (β2GPI), prothrombin (PT), thrombin, protein C (PC), activated PC (APC), protein S, annexin A5, plasminogen, plasmin and tissue-type plasminogen activator (tPA) (9-23). Of these plasma proteins, β2GPI has emerged to play a major role in aCL activity, serving either as the major autoantigen or as a necessary co-factor. Ab against β2GPI and its complexes with cardiolipin (CL) probably account for most of the positive findings on tests for aCL in APS (24), while anti-PT Ab (aPT) and anti-β2GPI Ab are responsible for the majority of the LAC activity (11, 25).
On the other hand, thrombin, APC, plasmin and tPA belong to the trypsin-like serine protease superfamily; and the catalytic domains of these four enzymes are homologous (26-29). At the amino acid levels, human thrombin and human APC share a 50.5% similarity, while human thrombin and human plasmin share a 48% similarity (19, 20). Recently, we showed that 5/7 patient-derived IgG monoclonal aCL reacted with human thrombin, APC, plasmin and tPA; and that one patient-derived IgG monoclonal aPT also bound to CL, thrombin, APC, plasmin and tPA (Table 1) (17, 19, 20, 23). Moreover, the binding of the CL15 monoclonal antibody (mAb) to tPA could be inhibited by α-thrombin (which contains only the catalytic domain), indicating that the shared homologous catalytic domains of the reactive proteases are the structural basis of the observed crossreactivity (23). Of note, in addition to the catalytic domain, tPA contains two Kringle domains plus two epidermal growth factor (EGF) domains. Furthermore, of the protease-reactive mAb, CL24 could interfere with inactivation of thrombin by antithrombin, while CL15 could inhibit the functional activities of APC, plasmin, and tPA (17, 19, 20, 23). Combined, these data indicate that some aCL bind to the homologous catalytic domains of several serine proteases that are involved in coagulation.
Table 1.
Summary of 12 monoclonal IgG aPL from four APS patientsa
| Antigens:
mAb: |
screening | CL/BS | β2GPI | thrombin | APC | plasmin |
|---|---|---|---|---|---|---|
| IS1 | CL/BS | +++ | - | - | - | - |
| IS2 | CL/BS | ++ | - | - | - | - |
| IS3 | CL/BS | +++ | ++ | +++ | +++ | + |
| IS4 | CL/BS | ++ | + | ++ | +++ | +++ |
| IS6 | prothrombin | ++ | ++ | ++ | ++ | ++ |
| CL1 | CL/BS | ++ | ++ | +++ | +++ | + |
| CL15 | CL/BS | ++ | - | ++ | +++ | ++ |
| CL24 | CL/BS | ++ | + | ++ | + | ++ |
| B1 | β2GPI | + | +++ | + | + | + |
| B2 | β2GPI | ++ | + | ++ | ++ | +++ |
| T1 | thrombin | + | + | + | + | ++ |
| P1 | protein C | ++ | +++ | ++ | ++ | +++ |
aBinding to all test antigens are expressed in relative term for each antigen by all mAbs. Binding to CL in the presence of bovine serum (BS) and to human β2GPI are from references (30, 32, 33) and the present study. Binding to human thrombin, APC, and plasmin are from references (17, 19, 20) and the present study. Although initial study of IS6 binding to β2GPI in Tris-buffered saline was negative (33), subsequent study of IS6 binding to β2GPI in PBS revealed positive interaction (data not shown).
In this context, it was tempting to speculate that some aCL in APS patients recognize a conformational epitope shared by β2GPI and the homologous catalytic domains of several serine proteases in hemostasis, as there is no meaningful amino acid sequence homology between β2GPI and the catalytic domain of any reactive serine proteases. Intriguingly, this speculation was supported by the fact that 5/8 aforementioned patient-derived IgG monoclonal aCL/aPT also react with β2GPI (Table 1). Thus, to test the aforementioned hypothesis, we generated and analyzed four new patient-derived IgG monoclonal aPL, including two mAb that were initially screened against β2GPI, one against thrombin and one against PC.
MATERIALS AND METHODS
Hybridoma donor patients
Patient #1 was a Hispanic female APS patient with no history of pregnancy. In 1994, she was diagnosed with APS at age 21 when she presented with recurrent episodes of deep venous thromboses, elevated aCL and a positive LAC, as determined by the Kaolin Clotting Time (KCT) test. Her course was complicated by the development of systemic lupus erythematosus (SLE) at age 26 manifested by arthritis, hemolytic anemia, leukopenia, persistently positive LAC (as determined by the dilute Russell’s Viper Venom Time - DRVVT test), high titer IgG aCL > 100 GPL (one GPL unit is equivalent to 1 μg of affinity purified standard IgG aCL), a positive anti-nuclear Ab (ANA) at 1:160 dilution and elevated anti-DNA Ab. Furthermore, she suffered from recurrent pulmonary emboli and secondary pulmonary hypertension, pulmonary hemorrhage, recurrent subdural hematomas, sinus thrombosis and cerebrovascular accidents. Despite aggressive therapy with anticoagulation, intravenous cyclophosphamide, high dose corticosteroid, Rituximab and plasma exchange, she expired at age 32. At the time of the blood collection (February 2001), she was just on prednisolone at 4.5 mg a day and warfarin.
Patient #2 is a 26-year-old female APS patient with no history of pregnancy. In 1999, at age 20, she developed severe headache, pseudotumor cerebri, optic neuritis and multiple ischemic white matter lesions on her magnetic resonance imaging (MRI) of her brain. Her cerebrospinal fluid (CSF) was unremarkable with negative cultures, with normal protein levels and cell counts, negative oligoclonal bands, and negative myelin basic protein. Her IgM aCL was elevated at 69 MPL (one MPL unit is equivalent to 1 μg of affinity purified standard IgM aCL; normal < 10), and her IgM anti-β2GPI was at 43, with normal < 10. Her DRVVT was negative. She was treated with a course of high dose steroids tapered over 3 months and anti-coagulation. She has remained on warfarin without recurrent thrombotic events and continues to have persistently elevated IgM aCL and anti-β2GPI Ab. She donated her blood in January 2001.
Generation and preparation of mAb
Four new mAb were generated as described previously (30). Briefly, peripheral blood mononuclear cells from the patients were transformed with Epstein-Barr virus, and cultured in 96-well plates. Supernatants were screened for desired Ab by appropriate enzyme-linked immunosorbent assays (ELISA) (17, 19, 30). For anti-β2GPI Ab (30), high-binding ELISA plates (Costar, Cambridge, MA) were coated with 10 μg/ml of human β2GPI (Haematologic Technologies, Essex Junction, VT) in phosphate-buffered saline (PBS, pH 7.2). After incubating overnight at 4 °C, plates were blocked with PBS containing 0.25% gelatin. Supernatants were distributed to wells in duplicate and incubated for 2 hours at room temperature. After washing with PBS, bound human IgG was detected with affinity purified horseradish peroxidase labeled goat anti-human IgG (γ-chain specific; BioSource International, Camarillo, CA). After an additional incubation for one hour at room temperature, 50 μl of the substrate tetramethylbenzidine/H2O2 (Kirkegard & Perry Labs, Gaithersburg, MD) was added, and the reaction terminated with 50 μl of 1 M H3PO4. Results were read at a wavelength of 450 nm against a background of 650 nm in an Emax plate reader (Molecular Devices, Sunnyvale, CA).
The ELISA for anti-thrombin Ab and anti-PC Ab were similar except for the following modifications. For anti-thrombin Ab, wells were coated with human thrombin (Haematologic Technologies) at 5 μg/ml in Tris-buffered saline (TBS, 50 mM Tris/150 mM NaCl, pH 7.5), and were blocked with 0.3% gelatin in TBS (17). For anti-PC Ab, wells were coated with 5 μg/ml of human PC (Haematologic Technologies) in PBS, and were blocked with 0.25% gelatin in PBS.
Positive cells were subcloned to one cell per well, and then fused with the Oubain resistant K6H6/B5 human-mouse heterohybridoma cell line (31). Positive hybridomas were subcloned twice at 1 cell/well. To ensure the monoclonality of each mAb, their light chain isotypes and IgG subclasses were determined by ELISA using isotype and subclass-specific reagents.
To purify mAb, each hybridoma was first switched to a serum-free culture medium. Culture supernatants were passed through a HiTrap Protein G column (Pharmacia, Piscataway, NJ), and the bound IgG was eluted with 0.1 M glycine HCl (pH 2.8), and dialyzed against PBS (32).
In addition to the four new mAb generated here, seven IgG monoclonal aCL and one IgG monoclonal aPT were analyzed in the present study. The aCL included CL1, CL15, CL24, IS1, IS2, IS3 and IS4 (30, 32), and the single aPT was IS6 (33). Their generation and characterization had been reported previously (30, 32, 33).
ELISA for Ab against APC and plasmin, and competitive inhibition assays
The ELISA for anti-APC Ab was similar to that for anti-PC Ab except that plates were coated with human APC (5 μg/ml; Haematologic Technologies) in PBS, pH 7.4. The ELISA for anti-plasmin Ab was done similarly except that plates were coated with human plasmin (5 μg/ml; Haematologic Technologies) in PBS (20).
Competitive inhibition assays were used to study the binding properties of mAb to four concerned antigens: β2GPI, α-thrombin, APC, and plasmin (17, 19, 20). Briefly, each mAb at the chosen concentration (in the linear range of its titration curve) was preincubated for 1.5 hours at RT with various concentrations of the indicated inhibitor in PBS (for β2GPI, APC, and plasmin) or TBS (for α-thrombin) containing 0.1% gelatin. Of note, we have previously found that the enzymes prepared in the buffer containing 0.1% gelatin remain stable during incubation at RT.
The amount of inhibition for a mAb at a given concentration of soluble inhibitor was calculated as follows: % inhibition of mAb binding to a test antigen = [(OD from a test mAb alone) − (OD from the same mAb plus the same antigen at the given concentration)] /(OD from the same mAb alone) × 100. The inhibition data of each mAb for each test antigen were used to calculate its relative Kd toward this test antigen (34).
For inhibition with APC, the recombinant human APC (rhAPC, Xigris, Eli Lilly, Indianapolis, IN) was used in the competitive inhibition, as it was immunologically indistinguishable from purified human APC (data not shown) but was much cheaper than purified human APC. For inhibition with plasmin, a few studies reported plasmin digestion of some human IgG (35, 36), suggesting that some of the observed plasmin inhibition of mAb binding to plasmin on plates might actually be due to plasmin digestion of mAb. To address this issue, plasmin was irreversibly inactivated with Nα-Tosyl-L-lysine chloromethyl ketone-HCl (TLCK, Sigma-Aldrich), which forms a chemical bond with His622 (one of the three conserved catalytic residues of all serine proteases). Briefly, 12 μM plasmin was first incubated with 6 mM TLCK in PBS (pH 7.4) for 100 min at RT, which eliminated > 99% of the plasmin amidolytic activity using S-2403. After incubation, the excess TLCK was removed by dialyzing the plasmin-TLCK mixture against PBS at 4 °C overnight. Thereafter, the TLCK-inactivated plasmin was used as a soluble inhibitor. In addition, an active-site blocked thrombin, α-thrombin-diisopropylfluorophosphate (DFP; Haematologic Technologies) was used to study the role of active site in the binding of mAb to α-thrombin. DFP reacts with Ser195 of thrombin, which is one of the conserved catalytic triad.
RESULTS
Generation of four monoclonal IgG aPL from two APS patients
To test the hypothesis that β2GPI shares a conformational epitope with several serine proteases that are recognized by aPL, we initiated the efforts to generate IgG anti-β2GPI mAb from APS patients with high titers of anti-β2GPI Ab. This was in part due to present lack of any patient-derived IgG anti-β2GPI mAb, considering that the major autoantigen for aPL is thought to be β2GPI and that the IgG aPL is more disease relevant than the IgM aPL. After extensive efforts, we obtained two IgG anti-β2GPI mAb from a patient with primary APS by screening the culture supernatants initially for binding to β2GPI and then subcloning each positive hybridoma to a monoclonal population that secret an IgG anti-β2GPI Ab. The mAb were designated B1 and B2. Figure 1A shows the binding reactivity of B1 and B2 to β2GPI.
Figure 1. Binding characteristics of four new patient-derived IgG monoclonal aPL.
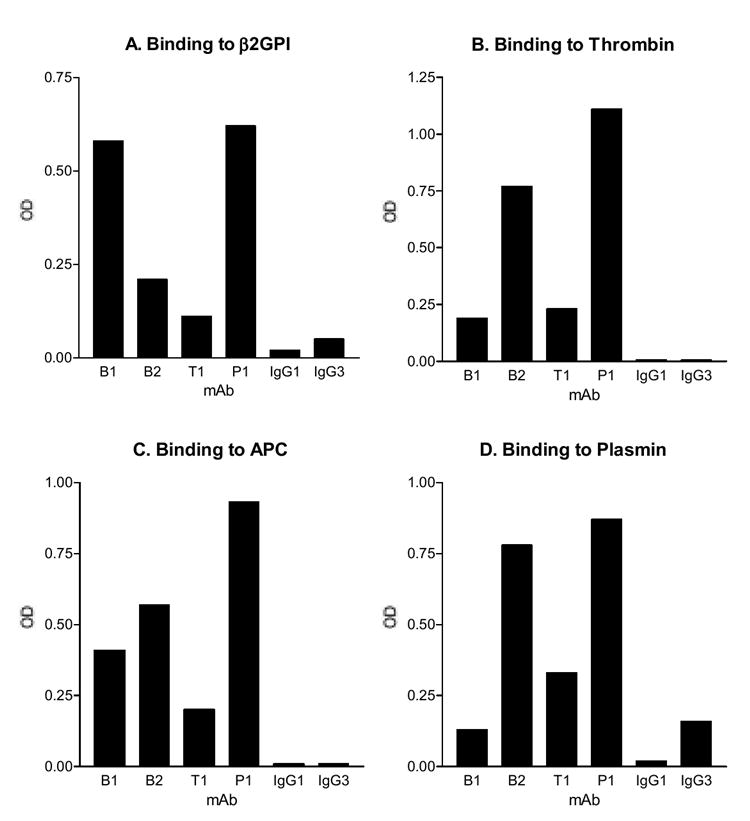
The mAb were analyzed for their binding to β2GPI (panel A), thrombin (B), APC (C) and plasmin (D). Microtiter wells were coated with the indicated autoantigens, and the test mAb, or the monoclonal isotype controls (IgG1 or IgG3) were analyzed at 1 μg/ml. Except for B2 (which is IgG3), all mAb are IgG1. Bound IgG were measured and expressed in OD. One of two experiments with similar results is shown.
On the other hand, as noted in Introduction, our recent studies of 7 patient-derived IgG monoclonal aCL revealed broad reactivity with thrombin, APC and plasmin, all share homologous catalytic domains. Although we have 5-crossreactive monoclonal IgG aCL, these mAb were generated by screening initially against CL in the presence of bovine serum and thus might not represent all IgG Ab against the same serine proteases in APS patients. Therefore, we initiated the efforts to generate new IgG mAb by screening initially against the concerned protease autoantigens. We obtained two mAb from an APS patient: one anti-thrombin Ab and one anti-PC Ab; the mAb were designated T1 and P1, respectively. Figure 1 B shows the binding of T1 to thrombin, and Figure 1 C shows the binding of P1 to APC. Of note, we elect to present the binding data to APC, which is a serine protease, while PC is a zymogen.
Since some autoimmune patients are known to have Ab against gelatin, all mAb were analyzed for binding to wells that were not coated with antigens but were blocked with buffers containing 0.3% gelatin only. The results showed that bindings of all mAb to gelatin were negligible (data not shown).
To ensure the monoclonality of each mAb, the heavy chain subclass and light chain isotype of each mAb were determined. The results showed that each mAb had only one light chain isotype and one IgG subclass. Specifically, B1, and B2 have κ light chains, while P1 and T1 have λ light chains. For heavy chains, B1, P1 and T1 are of the γ1 subclass, while B2 is of the γ3 subclass.
Immunological characterization of four patient-derived IgG monoclonal antibodies
Subsequently, we analyzed reactivity of both anti-β2GPI mAb toward thrombin, APC and plasmin, as well as the reactivity of the T1 and P1 toward β2GPI. The results showed that both anti-β2GPI mAb bound to thrombin, APC, and plasmin (Figure 1 B, C and D, and Table 1). Although the binding activities of B1 to thrombin and plasmin were weak, they were still significantly higher than the IgG1 isotype control (Figure 1 B and D, and Table 1). On the other hand, both T1 and P1 bound to β2GPI when compared against the IgG1 isotype control (Figure 1A, and Table 1). Moreover, P1 binding to β2GPI was even stronger than that of the B2 anti-β2GPI mAb (Figure 1A).
To determine the significance of the above crossreactivity of mAb, we used competitive inhibition to determine binding affinity of mAb to all reactive antigens. When mAb binding to β2GPI was inhibited by soluble β2GPI up to 12 μM, only B1 could be partially inhibited (about 16%), suggesting that both bound to β2GPI with low affinity (Figure 2A). Surprisingly, at the same concentration of 12 μM, P1 binding to β2GPI was inhibited by about 47%, indicating that P1 bound to β2GPI with an affinity that is higher than that of B1 (Figure 2A and Table 2).
Figure 2. Competitive inhibition of mAb binding to four APS-relevant autoantigens.
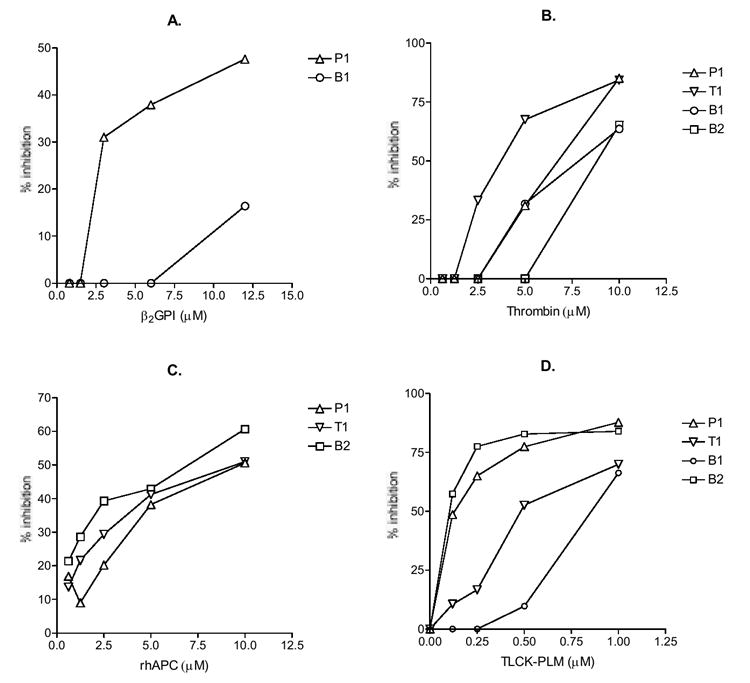
The test mAb were preincubated with β2GPI (panel A), thrombin (B), rhAPC (C) or the TLCK-inactivated plasmin (D) at the indicated concentrations, and then the mixtures were distributed to wells coated with the corresponding autoantigens. The results are expressed in % inhibition. One of two experiments with similar results is shown.
Table 2.
Relative Kd of IgG monoclonal aPL toward β2GPI and relevant serine proteases
| mAb:
Antigens: |
B1 | B2 | T1 | P1 |
|---|---|---|---|---|
| β2GPI | > 10-5 M | Not inhibitable | Not inhibitable | 10-5 M |
| Thrombin | 10-5 M | 2 × 10-5 M | 4 × 10-6 M | 8 × 10-6 M |
| rhAPC | Not inhibitable | 5 × 10-6 M | 7 × 10-6 M | 9 × 10-6 M |
| TLCK-Plasmin | 1 × 10-6 M | 9 × 10-8 M | 6 × 10-7 M | 1 × 10-7 M |
When mAb binding to thrombin was inhibited by soluble thrombin, all four mAb, including B1 and B2, were inhibited (Figure 2B). The relative Kd values were 4 × 10-6 M for T1, 8 × 10-6 M for P1, 1 × 10-5 M for B1, and 2 × 10-5 M for B2 (Table 2). Similar to inhibition by thrombin, all four mAb binding to APC, including B1 and B2, were inhibited by soluble APC (Figure 2B). The relative Kd values were 5 × 10-6 M for B2, 7 × 10-6 M for T1, and 9 × 10-6 M for P1 (Table 2).
The competitive inhibition of mAb binding to plasmin was done with the soluble plasmin that had been inactivated with TLCK, which forms a chemical bond with His622 of plasmin (one of the three conserved catalytic residues of all serine proteases). This was because a few studies had reported plasmin digestion of some human IgG (35, 36), which would lead to lower bound IgG mAb and inaccurate inhibition data. As can be seen in Figure 2D, all four mAb were inhibited by TLCK-inactivated plasmin. The relative Kd values are 9 × 10-8 M for B2, 1 × 10-7 M for P1, 6 × 10-7 M for T1, 1 × 10-6 M for B1 (Table 2).
Combined, these data show that anti-β2GPI mAb often bind to various relevant members of serine proteases, while the mAb generated against the concerned serine proteases frequently react with β2GPI. These findings suggest that β2GPI shares an epitope with the concerned serine proteases. Of the four studied autoantigens, plasmin displays the highest relative binding affinity to all four patient-derived mAb, with the B2 anti-β2GPI mAb having the highest binding affinity (relative Kd = 9 × 10-8 M). In contrast, β2GPI has the lowest relative binding affinity to all four patient-derived mAb.
β2GP1 shares conformational epitopes with the catalytic domain of thrombin
To test the hypothesis that β2GPI shares an epitope with the concerned serine proteases, we performed a cross inhibition assay with the P1 mAb, which binds strongly to β2GPI and all three test serine proteases. As noted in the Introduction, the three test serine proteases are homologous in their catalytic domains, while they differ greatly from each other in other regions. For example, in addition to the catalytic domain, APC contains one Gla (for γ-carboxyglutamate) domain and two EGF domains, while plasmin has 1-5 kringle domains depending on the type of plasminogen from which it originated (whether Glu-1, Lys-77 after a cleavage between K76-K77 by plasmin, or Val-442 after a cleavage by elastase). Therefore, the shared epitope is most likely to reside in the catalytic domains of the reactive serine proteases. Consequently, α-thrombin (that contains only the catalytic domain of thrombin) was used in the cross-inhibition assay.
The results showed that P1 binding to β2GPI was inhibited by β2GPI and α-thrombin (Figure 3A); and that P1 binding to α-thrombin was inhibited by α-thrombin and β2GPI (Figure 3B). Since α-thrombin is a protease, the reduced mAb binding to solid phase antigen in the competitive inhibition assay may, in part, be due to digestion of IgG by α-thrombin. To address this issue, we incubated P1 mAb (10-100 μg) with either α-thrombin (10 μM) or plasmin (10 μM) in PBS for 1 hour at RT, and analyzed the mixtures by SDS-PAGE and a silver staining. The results showed that mixture of P1 plus plasmin contained the expected bands of IgG and plasmin plus additional bands of 10 & 20 KD, while the mixture of P1 plus α-thrombin contained only the expected bands of IgG and α-thrombin (data not shown). The data indicated digestion of IgG by plasmin, but no digestion of IgG by α-thrombin, and thus demonstrated that inhibition by α-thrombin was mainly due to antigenic competition.
Figure 3. Cross inhibition of P1 binding to β2GPI (panel A) or α-thrombin (B).
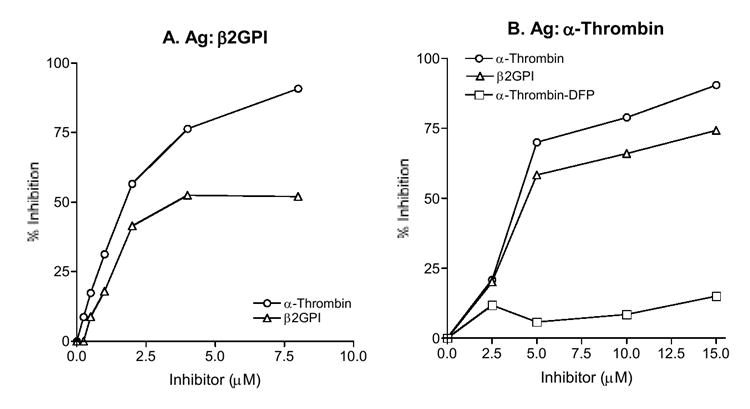
The P1 mAb was preincubated with β2GPI, α-thrombin or α-thrombin-DFP (an active site-blocked α-thrombin) at the indicated concentrations, and then the mixtures were distributed to wells coated with β2GPI (panel A) or α-thrombin (B). The results are expressed in % inhibition. A representative of two experiments is shown.
Subsequently, to define the epitope on α-thrombin, we performed comparative analyses of P1 reactivity with α-thrombin and the corresponding active site-blocked thrombin, α-thrombin-diisopropylfluorophosphate (DFP, which reacts with Ser195 of the conserved catalytic triad). In the direct binding assay, the binding of P1 to α-thrombin-DFP was significantly weaker than that to α-thrombin, indicating that the reactivity of α-thrombin with P1 was reduced significantly when Ser195 in its active site was modified (Fig. 4). The data suggested that the active site (or its vicinity) of α-thrombin was involved in its reactivity with P1. In the inhibition assay, P1 binding to α-thrombin was not inhibited by α-thrombin-DFP (Fig. 3B), suggesting again that the active site (or its vicinity) was involved in the reactivity of P1 to α-thrombin. Considering that there is no amino acid sequence homology between β2GPI and α-thrombin, these data indicate that β2GPI and the relevant serine proteases share conformational epitopes that reside at or around the active site in the catalytic domains of the reactive proteases.
Figure 4. Comparison of P1 binding to α-thrombin and α-thrombin-DFP.
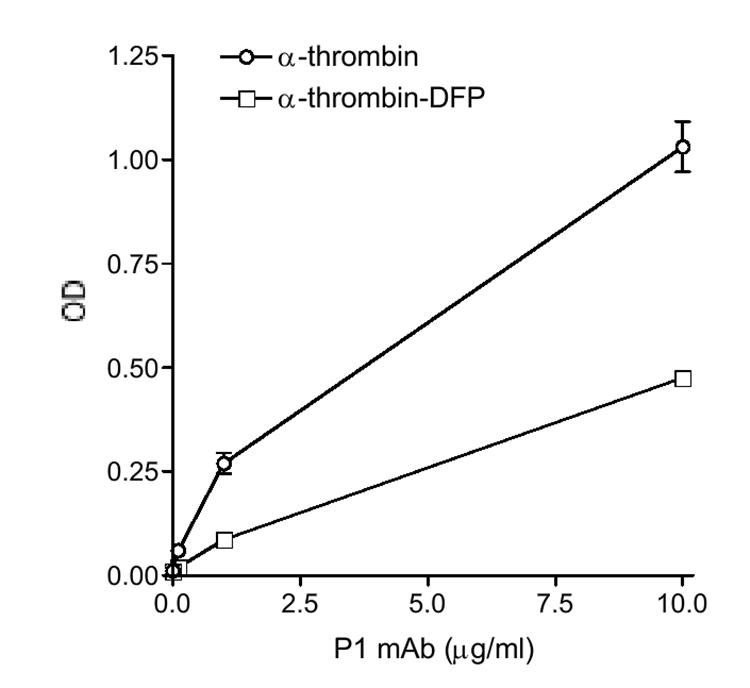
Wells were coated with each antigen at 5 μg/ml, and P1 mAb was analyzed at the indicated concentrations. Bound IgG were measured and expressed in OD. A representative of two experiments is shown.
All four patient-derived IgG mAb display standard aCL activity
Subsequently, to determine the clinical relevance of these mAb, we analyzed these mAb for binding to CL in the presence of bovine serum, the standard aCL assay. The results showed that all four mAb bound to CL when compared against the IgG1 and IgG3 isotype controls (Figure 5), although the aCL activities of B2 and P1 were stronger than those of B1 and T1. Since B2 and P1 bound to β2GPI and strongly to plasmin (Figures 1D and 2D), the data suggested that IgG aCL activity might reflect more the reactivity of the strong plasmin-reactive anti-β2GPI Ab than that of the weak plasmin-reactive anti-β2GPI Ab (like B1), and that IgG aCL include mainly Ab that recognize one or more epitopes shared by β2GPI and plasmin.
Figure 5. All four new patient-derived IgG monoclonal aPL bind to CL in the presence of bovine serum.
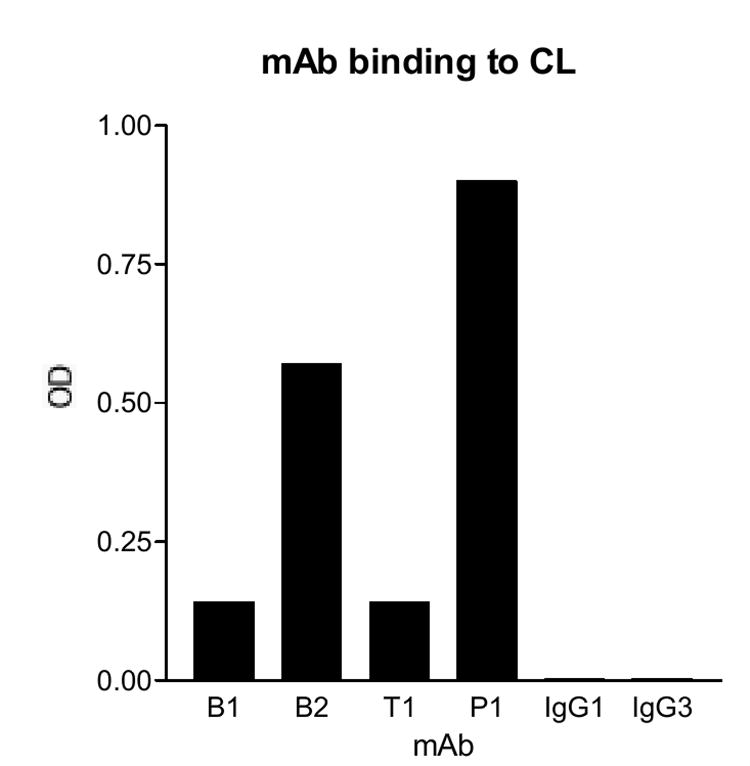
The mAb were analyzed for their aCL activity. Microtiter wells were coated with the CL, and the test mAb, or the monoclonal isotype controls (IgG1 or IgG3) were analyzed at 1 μg/ml. Except for B2 (which are IgG3), all mAb are IgG1. Bound IgG were measured and expressed in OD. A representative of two experiments is shown.
DISCUSSION
To test the hypothesis that some aPL in APS patients recognize a conformational epitope shared by β2GPI and the homologous catalytic domains of several serine proteases in hemostasis, we generated and analyzed four new IgG monoclonal aPL from two APS patients. Of the four IgG mAb, two were generated by screening against β2GPI, one against thrombin and one against PC. Importantly, immunological analyses of these four monoclonal aPL showed that both anti-β2GPI mAb bound to thrombin, APC and plasmin. On the other hand, one anti-thrombin mAb and one anti-PC mAb reacted with β2GPI (Figure 1 and Table 1). Moreover, the B2 anti-β2GPI mAb bound to APC and plasmin with affinities that were even higher than those of T1 (anti-thrombin mAb) and P1 (anti-PC mAb), while the P1 mAb displayed the highest affinity toward β2GPI (Figure 2 and Table 2). Most importantly, P1 binding to β2GPI was inhibited by α-thrombin (that contains only the catalytic domain of thrombin, Figure 3A). Taken together with the fact that there is no amino acid sequence homology between β2GPI and the involved serine proteases, these data demonstrate that some aPL in APS patients recognize conformational epitopes shared by β2GPI and the homologous catalytic domains of several serine proteases in hemostasis.
When the four new IgG monoclonal aPL were analyzed for aCL activity, all bound to CL in the presence of bovine serum (Figure 5). Previously, we generated seven IgG mAb generated from two APS patients by screening against CL in the presence of bovine serum. Interestingly, 5/7 bound to thrombin, APC, plasmin and tPA (Table 1) (23). Of the 5 protease-reactive mAb, CL1, CL24, IS3, and IS4 also bound to β2GPI (Table 1) (32). In addition, analyses of the IS6 patient-derived IgG monoclonal aPT showed that it displayed aCL activity, bound to all four above serine proteases, and reacted with β2GPI (Table 1). Combined, these data showed that 10 of 12 IgG monoclonal aPL from four APS patients bound to thrombin, APC, plasmin and tPA (Table 1); and, of the 10 protease-reactive mAb, 9 (90%) also reacted with β2GPI (Table 1). On the other hand, of the 9 patient-derived β2GPI-reactive IgG aCL, all interacted with the concerned serine proteases (Table 1). Thus, these data indicate that IgG anti-β2GPI Ab overlap extensively with Ab against certain serine proteases in hemostasis, and that β2GPI shares epitopes with these serine proteases. Moreover, considering that 9/12 (75%) patient-derived IgG aCL react with β2GPI and the concerned serine proteases, these findings also show that the majority of IgG aCL recognize one or more epitopes shared by β2GPI and the concerned serine proteases.
While the above data are from patient-derived IgG monoclonal aPLs, a similar finding was reported recently among polyclonal aPL in APS patients (37). Specifically, when aPL were studied in 120 APS patients, the presence of IgG anti-β2GPI Ab was closely related to the presence of IgG anti-thrombin Ab. On the other hand, the contention of a shared epitope between β2GPI and the relevant serine proteases is supported by our previous analyses of five monoclonal IgG aPL Fab fragments from a phage displayed-Ab library, including B14, B22 and B27 anti-β2GPI from panning with β2GPI; and P11 and P13 anti-PT from panning with PT (38). The results revealed that the P11 aPT Fab also reacted with β2GPI, and had comparable binding affinity to both antigens, with relative Kd values of 3 × 10-6 M for PT versus 2 × 10-6 M for β2GPI (38). Moreover, similar findings were also reported very recently by other investigators (39). In particular, when 39 single chain FV (scFv) clones from an APS patient were isolated and analyzed, 10 clones were found to react with PT and to crossreact with β2GPI. Taken together with the present data in Figures 1-3, these data clearly demonstrate that β2GPI shares epitopes with the homologous catalytic domains of certain serine proteases in hemostasis.
It is interesting to note that, of the four currently studied autoantigens, the mAb bind to plasmin with the highest affinity, with the relative Kd values ranging from 1 × 10-6 M to 9 × 10-8 M (Table 2). Similar high affinity interaction was also observed between plasmin and six other patient-derived, protease-reactive monoclonal IgG aPL (20). Specifically, CL1 and CL15 bind to plasmin with relative Kd values of 6 × 10-8 M and 1 × 10-7 M, respectively. These affinities are significantly higher than the reported affinity of IgG anti-β2GPI Ab (in 5 APS patients) toward β2GPI, with the relative Kd values ranging from 3 to 7 × 10-6 M (40). Viewed as a whole, these data suggest that plasmin may be an important autoantigen that drives certain IgG aPL in some APS patients, and that plasmin may even be the driving autoantigen for some IgG anti-β2GPI Ab (like the P1 mAb).
Since our seven monoclonal IgG aCL from two APS patients were generated and reported in 1999 (32), they have been studied extensively by several investigators, and the results have contributed significantly toward our understanding of immunopathogenesis of autoantibody-mediated thrombosis and fetal loss in APS. In addition to our analyses of these mAb, Pierangeli and her colleagues examined the prothrombotic potentials and functional activities of these mAb (41). Using a pinch-induced in vivo murine thrombosis model, which allowed for continuous and quantitative monitoring of a focally induced non-occlusive mural thrombosis in an exposed femoral vein (42), five aCL (including CL15 and CL24) were found to be prothrombotic (41). In addition, CL15 was shown to induce human umbilical vein endothelial cells to express highest level of E-selectin and vascular cell adhesion molecule-1 (41). Furthermore, Pierangeli and her colleagues employed an in vivo microcirculation model to examine aCL-induced leukocyte adhesion to endothelium in venules (41). The results showed that IS2, CL15 and IS4 increased significantly the number of endothelium-adhering leukocytes (41). On the other hand, Rand and his associates studied the effects of these mAb on annexin A5. Using atomic force microscopy, a method previously used to study the crystallization of annexin A5, IS3 together with β2GPI were shown to disrupt the annexin A5 crystallization pattern over the bilayers and to increase generation of thrombin (43). Along this line, IS4 together with β2GPI were found to reduce annexin A5 binding to PL and to inhibit the annexin A5 anticoagulant activity (16). Therefore, we expect the availability of four new patient-derived IgG monoclonal aPL will similarly contribute toward our progress in delineating immunopathogenesis of autoantibody-mediated thrombosis and fetal loss in APS.
Acknowledgments
We thank Dr. John FitzGerald for critical review of the manuscript and helpful discussion, and Brian Chung and Tony Hsu for their technical assistance.
This work was supported by a research award from the Alliance for Lupus Research, a grant from the Southern California Chapter of the Arthritis Foundation and a grant AR42506 from the National Institutes of Health. W. Lin was supported by a Research Training grant from the Taiwan government.
References
- 1.Wilson WA, Gharavi AE, Koike T, Lockshin MD, Branch DW, Piette JC, Brey R, Derksen R, Harris EN, Hughes GRV, Triplett DA, Khamashta MA. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome. Arthritis Rheum. 1999;42:1309–1311. doi: 10.1002/1529-0131(199907)42:7<1309::AID-ANR1>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 2.Lockshin MD, Sammaritano LR, Schwartzman S. Validation of the Sapporo criteria for antiphospholipid syndrome. Arthritis Rheum. 2000;43:440–443. doi: 10.1002/1529-0131(200002)43:2<440::AID-ANR26>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 3.Levine J, Branch D, Rauch J. Medical progress: the antiphospholipid syndrome. N Engl J Med. 2002;346:752–763. doi: 10.1056/NEJMra002974. [DOI] [PubMed] [Google Scholar]
- 4.Passam F, Krilis S. Laboratory tests for the antiphospholipid syndrome: current concepts. Pathology. 2004;36:129–138. doi: 10.1080/00313020410001671966. [DOI] [PubMed] [Google Scholar]
- 5.De Groot PG, Derksen RH. Antiphospholipid antibodies: update on detection, pathophysiology, and treatment. Curr Opin Hematol. 2004;11:165–169. doi: 10.1097/01.moh.0000130313.95291.4a. [DOI] [PubMed] [Google Scholar]
- 6.Mackworth-Young CG. Antiphospholipid syndrome: multiple mechanisms. Clin Exp Immunol. 2004;136:393–401. doi: 10.1111/j.1365-2249.2004.02497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, Derksen RH, DEGroot PG, Koike T, Meroni PL, Reber G, Shoenfeld Y, Tincani A, Vlachoyiannopoulos PG, Krilis SA. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS) J Thromb Haemost. 2006;4:295–306. doi: 10.1111/j.1538-7836.2006.01753.x. [DOI] [PubMed] [Google Scholar]
- 8.Harris EN, Pierangeli SS. Antiphospholipid antibodies and the antiphospholipid syndrome. Springer Semin Immunopathol. 1994;16:223–245. doi: 10.1007/BF00197519. [DOI] [PubMed] [Google Scholar]
- 9.Matsuura E, Igarashi Y, Fujimoto M, Ichikawa K. Anticardiolipin cofactor(s) and differential diagnosis of autoimmune disease. Lancet. 1990;336:177–178. doi: 10.1016/0140-6736(90)91697-9. [DOI] [PubMed] [Google Scholar]
- 10.McNeil HP, Simpson RJ, Chesterman CN, Krilis SA. Anti-phospholipid antibodies are directed against a complex antigen that includes a lipid binding inhibitor of coagulation: beta 2-glycoprotein I (apolipoprotein H) Proc Natl Acad Sci USA. 1990;87:4120–4124. doi: 10.1073/pnas.87.11.4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fleck RA, Rapaport SI, Rao LV. Anti-prothrombin antibodies and the lupus anticoagulant. Blood. 1988;72:512–519. [PubMed] [Google Scholar]
- 12.Bevers EM, Galli M, Barui T, Comfurius P, Zwaal RFA. Lupus anticoagulant IgG’s (LA) are not directed to phospholipids only, but to a complex of lipid-bound human prothrombin. Thromb Haemost. 1991;66:629–632. [PubMed] [Google Scholar]
- 13.Oosting JD, Derksen RHWM, Bobbink IWG, Hackeng TM, Bouma BN, de Groot PG. Antiphospholipid antibodies directed against a combination of phospholipids with prothrombin, protein C, or protein S: An explanation for their pathogenic mechanism? Blood. 1993;81:2618–2625. [PubMed] [Google Scholar]
- 14.Erkan D, Zhang H, Shriky R, Merrill J. Dual antibody reactivity to beta(2)-glycoprotein i and protein s: increased association with thrombotic events in the antiphospholipid syndrome. Lupus. 2002;11:215–220. doi: 10.1191/0961203302lu178oa. [DOI] [PubMed] [Google Scholar]
- 15.Rand JH, Wu XX, Andree HAM, Lockwood CJ, Guller S, Scher J, Harpel PC. Pregnancy loss in the antiphospholipid-antibody syndrome - A possible thrombogenic mechanism. N Engl J Med. 1997;337:154–160. doi: 10.1056/NEJM199707173370303. [DOI] [PubMed] [Google Scholar]
- 16.Rand JH, Wu XX, Lapinski R, van Heerde WL, Reutelingsperger CP, Chen PP, Ortel TL. Detection of antibody-mediated reduction of annexin A5 anticoagulant activity in plasmas of patients with the antiphospholipid syndrome. Blood. 2004;104:2783–2790. doi: 10.1182/blood-2004-01-0203. [DOI] [PubMed] [Google Scholar]
- 17.Hwang KK, Grossman JM, Visvanathan S, Chukwuocha RU, Woods VL, Le DT, Hahn BH, Chen PP. Identification of anti-thrombin antibodies in the antiphospholipid syndrome that interfere with the inactivation of thrombin by antithrombin. J Immunol. 2001;167:7192–7198. doi: 10.4049/jimmunol.167.12.7192. [DOI] [PubMed] [Google Scholar]
- 18.Lollar P. Pathogenic antibodies to coagulation factors. Part II. Fibrinogen, prothrombin, thrombin, factor V, factor XI, factor XII, factor XIII, the protein C system and von Willebrand factor. J Thromb Haemost. 2005;3:1385–1391. doi: 10.1111/j.1538-7836.2005.01228.x. [DOI] [PubMed] [Google Scholar]
- 19.Hwang KK, Yang CD, Yan W, Grossman JM, Hahn BH, Chen PP. A thrombin-cross-reactive anticardiolipin antibody binds to and inhibits the anticoagulant function of activated protein C. Arthritis Rheum. 2003;48:1622–1630. doi: 10.1002/art.10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang CD, Hwang KK, Yan W, Gallagher K, FitzGerald J, Grossman JM, Hahn BH, Chen PP. Identification of anti-plasmin antibodies in the antiphospholipid syndrome that inhibit degradation of fibrin. J Immunol. 2004;172:5765–5773. doi: 10.4049/jimmunol.172.9.5765. [DOI] [PubMed] [Google Scholar]
- 21.Cugno M, Dominguez M, Cabibbe M, Bisiani G, Galli M, Angles-Cano E, Agostoni A. Antibodies to tissue-type plasminogen activator in plasma from patients with primary antiphospholipid syndrome. Br J Haematol. 2000;108:871–875. doi: 10.1046/j.1365-2141.2000.01948.x. [DOI] [PubMed] [Google Scholar]
- 22.Cugno M, Cabibbe M, Galli M, Meroni PL, Caccia S, Russo R, Bottasso B, Mannucci PM. Antibodies to tissue-type plasminogen activator (tPA) in patients with antiphospholipid syndrome: evidence of interaction between the antibodies and the catalytic domain of tPA in 2 patients. Blood. 2004;103:2121–2126. doi: 10.1182/blood-2003-07-2422. [DOI] [PubMed] [Google Scholar]
- 23.Lu CS, Horizon AA, Hwang KK, Fitzgerald J, Lin WS, Hahn BH, Wallace DJ, Metzger AL, Weisman MH, Chen PP. Identification of polyclonal and monoclonal antibodies against tissue plasminogen activator in the antiphospholipid syndrome. Arthritis Rheum. 2005;52:4018–4027. doi: 10.1002/art.21485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roubey RAS, Hoffman M. From antiphospholipid syndrome to antibody-mediated thrombosis. Lancet. 1997;350:1491–1493. doi: 10.1016/S0140-6736(05)63936-0. [DOI] [PubMed] [Google Scholar]
- 25.Permpikul P, Rao LVM, Rapaport SI. Functional and binding studies of the roles of prothrombin and b2-glycoprotein I in the expression of lupus anticoagulant activity. Blood. 1994;83:2878–2892. [PubMed] [Google Scholar]
- 26.St Charles R, Matthews J, Zhang E, Tulinsky A. Bound structures of novel p3-p1 ’beta-strand mimetic inhibitors of thrombin. J Med Chem. 1999;42:1376–1383. doi: 10.1021/jm980052n. [DOI] [PubMed] [Google Scholar]
- 27.Mather T, Oganessyan V, Hof P, Huber R, Foundling S, Esmon C, Bode W. The 2.8 Angstrom crystal structure of gla-domainless activated protein C. EMBO J. 1996;15:6822–6831. [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X, Lin X, Loy JA, Tang J, Zhang XC. Crystal structure of the catalytic domain of human plasmin complexed with streptokinase. Science. 1998;281:1662–1665. doi: 10.1126/science.281.5383.1662. [DOI] [PubMed] [Google Scholar]
- 29.Pennica D, Holmes WE, Kohr WJ, Harkins RN, Vehar GA, Ward CA, Bennett WF, Yelverton E, Seeburg PH, Heyneker HL, Goeddel DV, Collen D. Cloning and expression of human tissue-type plasminogen activator cDNA in E. coli. Nature. 1983;301:214–221. doi: 10.1038/301214a0. [DOI] [PubMed] [Google Scholar]
- 30.Olee TW, Pierangeli SS, Handley HH, Le DT, Wei X, Lai CJ, En J, Novotny W, Harris EN, Woods VL, Jr, Chen PP. A monoclonal IgG anticardiolipin antibody from a patient with the antiphospholipid syndrome is thrombogenic in mice. Proc Natl Acad Sci USA. 1996;93:8606–8611. doi: 10.1073/pnas.93.16.8606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carroll WL, Thielemans K, Dilley J, Levy R. Mouse x human heterohybridomas as fusion partners with human B cell tumors. J Immunol Methods. 1986;89:61–72. doi: 10.1016/0022-1759(86)90032-3. [DOI] [PubMed] [Google Scholar]
- 32.Zhu M, Olee T, Le DT, Roubey RAS, Hahn BH, Woods VL, Jr, Chen PP. Characterization of IgG monoclonal anti-cardiolipin/antib2GP1 antibodies from two patients with the anti-phospholipid syndrome reveals three species of antibodies. Br J Haematol. 1999;105:102–109. [PubMed] [Google Scholar]
- 33.Zhao Y, Rumold R, Ahmed AE, Le DT, Hahn BH, Woods VL, Jr, Chen PP. An IgG anti-prothrombin antibody enhances prothrombin binding to damaged endothelial cells and shortens plasma coagulation times. Arthritis Rheum. 1999;42:2132–2138. doi: 10.1002/1529-0131(199910)42:10<2132::AID-ANR13>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 34.Friguet B, Chaffotte AF, Djavadi-Ohaniance L, Goldberg ME. Measurements of the true affinity constant in solution of antigen-antibody complexes by enzyme-linked immunosorbent assay. J Immunol Methods. 1985;77:305–319. doi: 10.1016/0022-1759(85)90044-4. [DOI] [PubMed] [Google Scholar]
- 35.Harpel PC, Sullivan R, Chang TS. Binding and activation of plasminogen on immobilized immunoglobulin G. Identification of the plasmin-derived Fab as the plasminogen-binding fragment. J Biol Chem. 1989;264:616–624. [PubMed] [Google Scholar]
- 36.Chuba JV. Susceptibility of monoclonal IgG paraproteins to plasmic cleavage using glycerin-stabilized human plasmin. Biochem Biophys Res Commun. 1994;202:367–373. doi: 10.1006/bbrc.1994.1937. [DOI] [PubMed] [Google Scholar]
- 37.Miesbach W, Matthias T, Scharrer I. Identification of thrombin antibodies in patients with antiphospholipid syndrome. Ann N Y Acad Sci. 2005;1050:250–256. doi: 10.1196/annals.1313.026. [DOI] [PubMed] [Google Scholar]
- 38.Chukwuocha RU, Hsiao ET, Shaw P, Witztum JL, Chen PP. Isolation, characterization and sequence analysis of five IgG monoclonal anti-b2-glycoprotein-1 and anti-prothrombin antigen-binding fragments generated by phage display. J Immunol. 1999;163:4604–4611. [PubMed] [Google Scholar]
- 39.Languren M, Becerril B, Cabral AR, Fernandez-Altuna LE, Pascual V, Hernandez-Ramirez DF, Cabiedes J. Characterization of monoclonal anti-beta2-glycoprotein-I and anti-prothrombin antibody fragments generated by phage display from a patient with primary antiphospholipid syndrome. J Autoimmun. 2006;26:57–65. doi: 10.1016/j.jaut.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 40.Tincani A, Spatola L, Prati E, Allegri F, Ferremi P, Cattaneo R, Meroni P, Balestrieri G. The anti-b2-clycoprotein I activity in human anti-phospholipid syndrome sera is due to monoreactive low-affinity autoantibodies directed to epitopes located on native b2-glycoprotein I and preserved during species’ evolution. J Immunol. 1996;157:5732–5738. [PubMed] [Google Scholar]
- 41.Pierangeli SS, Liu XW, Espinola R, Olee T, Zhu M, Harris NE, Chen PP. Functional analyses of patient-derived IgG monoclonal anticardiolipin antibodies using in vivo thrombosis and in vivo microcirculation models. Thromb Haemost. 2000;84:388–395. [PubMed] [Google Scholar]
- 42.Pierangeli SS, Barker JH, Stikovac D, Ackerman D, Anderson G, Barquinero J, Acland R, Harris EN. Effect of human IgG antiphospholipid antibodies on an in vivo thrombosis model in mice. Thromb Haemost. 1994;71:670–674. [PubMed] [Google Scholar]
- 43.Rand JH, Wu XX, Quinn AS, Chen PP, McCrae KR, Bovill EG, Taatjes DJ. Human monoclonal antiphospholipid antibodies disrupt the annexin A5 anticoagulant crystal shield on phospholipid bilayers: evidence from atomic force microscopy and functional assay. Am J Pathol. 2003;163:1193–1200. doi: 10.1016/S0002-9440(10)63479-7. [DOI] [PMC free article] [PubMed] [Google Scholar]