

Abstract
Several mammalian carboxylesterases were shown to activate the prodrug irinotecan (CPT-11) to produce SN-38, a topoisomerase inhibitor used in cancer therapy. However, the potential use of bacterial carboxylesterases, which have the advantage of high stability, has not been explored. We present the crystal structure of the carboxyesterase Est55 from Geobacillus stearothermophilus and evaluation of its enzyme activity on CPT-11. Crystal structures were determined at pH 6.2 and 6.8 and resolution of 2.0 and 1.58 Å, respectively. Est55 folds into three domains, a catalytic domain, an α/β domain and a regulatory domain. The structure is in an inactive form; the side chain of His409, one of the catalytic triad residues, is directed away from the other catalytic residues Ser194 and Glu310. Moreover, the adjacent Cys408 is triply oxidized and lies in the oxyanion hole, which would block the binding of substrate, suggesting a regulatory role. However, Cys408 is not essential for enzyme activity. Mutation of Cys408 showed that hydrophobic side chains were favorable, while polar serine was unfavorable for enzyme activity. Est55 was shown to hydrolyze CPT-11 into the active form SN-38. The mutant C408V provided a more stable enzyme for activation of CPT-11. Therefore, engineered thermostable Est55 is a candidate for use with irinotecan in enzyme-prodrug cancer therapy.
Keywords: carboxylesterase, crystal structure, α/β hydrolase, cancer therapy, cysteine oxidation
Introduction
Carboxylesterases (carboxylic ester hydrolase, EC 3.1.1.1) are members of the α/β hydrolase family that hydrolyze short chain aliphatic and aromatic esters.1 They share a common α/β hydrolase fold,2; 3; 4 and the catalytic triad of Ser, His, and Asp/Glu. We are characterizing two carboxylesterases Est30 and Est55 from the Gram-positive bacterium Geobacillus stearothermophilus. These enzymes are stable and catalytically active at high temperatures.5 The crystal structure of the 248 residue Est30 contained a tetrahedral reaction intermediate, which provided information about the reaction mechanism of serine hydrolases. 6 Est30 and Est55 are only distantly related with 14.3% sequence identity. The 498 residue Est55 showed optimal activity for short chain acyl derivatives at a temperature of 60°C.5 Interestingly, Est55 shares 32% sequence identity with human carboxylesterase CE1 (hCE1) and 31% with rabbit liver carboxylesterase (rCE).
Human carboxylesterases are important for the hydrolysis and detoxification of peptides and many drugs containing ester groups, such as cocaine, heroin, aspirin, and steroids. 7 Carboxylesterases have been evaluated for activation of the prodrug CPT-11 (Irinotecan) in cancer gene therapy. CPT-11 is activated by a carboxylesterase to generate SN-38 (7-ethyl-10-hydroxycamptothecin), a topoisomerase I poison used in cancer therapy. CPT-11 has shown significant anti-tumor activity in human tumor xenograft models, especially for colon cancer.8 Although SN-38 can be detected in the plasma of patients within minutes after administration of CPT-11,9 only a small percentage of CPT-11 is converted into SN-38.10 Therefore, cDNAs encoding carboxylesterases from rabbit liver (rCE), human small intestine (hiCE) and human liver (hCE2) were isolated and their enzymatic roles for CPT-11 activation were explored.11; 12; 13 To date, rCE is the most efficient enzyme, activating CPT-11 within 22 hours under the conditions of applying 2000 units of the enzyme.14 The possibility of using a bacterial enzyme to activate CPT-11 has not been explored previously. Mammalian enzymes have the advantage of low immunogenicity, which supports longer periods of prodrug activation.15 However, they lack the advantage of the large therapeutic index and high thermostability that can be achieved with enzymes from thermostable bacterial origins. While immunoreactivity may be a general concern, several bacterial enzymes have been used in cancer gene therapy, for example, E. coli cytosine deaminase for activation of 5-fluorocytosine, E. coli purine nucleoside phosphorylase for 6 methyl purine -2′-deoxynucleoside and E. coli nitroreductase for 5-aziridn-1-yl-2–4-dinitrobenzamide. 15
The crystal structures of the mammalian carboxylesterases hCE1 and rCE and Bacillus subtilis carboxylesterase (pnb CE) have been determined. 16; 17; 18; 19; 20; 21 These enzymes share similar sequences, three structural domains: a catalytic domain, an α/β domain and a regulatory domain, and can activate CPT-11. The hCE1 structures were determined in complexes with various substrates, substrate analogues and inhibitors. 18; 19; 20; 21 However, no human carboxylesterase structure has been determined with the substrate CPT-11 or products SN38 and 4PP. The rCE crystal structure has been determined with the product 4-piperdino-piperdine (4PP).17 However, 4PP is bound on the rCE surface, rather than in the active site, and the active site is partially disordered. The crystal structure of Torpedo californica acetylcholinesterase (TcAChE) was determined with CPT-11, which is an inhibitor of this enzyme. 22 Several hydrophobic residues in the active site interact with CPT-11, but the carbon of the carbamate linkage in CPT-11 was 9.3 Å away from catalytic residue Ser200. Therefore, the molecular basis for activation of CPT-11 is not understood.
In this study, we have assessed the possibility of using the thermostable Est55 as the initial model for engineering a more efficient and stable enzyme to activate the CPT-11 pro-drug to the SN-38 drug in cancer treatment. We have determined the crystal structure of Est55 and demonstrated that the enzyme can activate CPT-11. Unexpectedly, the Est55 structure showed that the catalytic triad residue His409 was rotated away from the active conformation. Moreover, entry to the active site was blocked by oxidized Cys408, which is not conserved in the mammalian carboxylesterases. Therefore, mutational analysis was used to explore the role of Cys408 in the Est55 enzymatic activity on p-nitrophenyl substrates and CPT-11.
Results and Discussion
Crystal structures of Est55
Est55 was crystallized under two different conditions in the presence of the iodine ion. No diffraction quality crystals were obtained in the presence of substrates, products, inhibitors, reducing agents, or for tested Est55 mutants. Crystals with similar morphology were obtained from conditions at two different pHs. Est55 was crystallized in 20% PEG3350, 0.2 M ammonium iodide (pH 6.2), or in 20% PEG3350, 0.2 M potassium iodide (pH 6.8). The higher the pH, the longer the crystals took to grow. The crystallographic statistics for data collection and refinement are shown in Table 1. Both of the Est55 crystal structures were observed in the space group P212121 with one molecule per asymmetric unit. The structures were solved by molecular replacement using Pnb esterase (1QE3) as a search model.16 The pH 6.2 structure (Est55-pH6.2) was refined to an R-factor of 20.0 % at 2.02 Å resolution. The pH 6.8 structure (Est55-pH6.8) was refined to R-factor of 17.2% at 1.58 Å resolution. The higher resolution structure of Est55-pH6.8 had more disorder than the lower resolution structure of Est55-pH6.2. 479 and 463 residues of 498 amino acids of the Est55 structure were positioned in the pH 6.2 and pH 6.8 structures, respectively. Two residues at both termini were not visible in the electron density maps. Residues were not visible in one or two surface loops, and before His409 of the catalytic triad. Diffraction data were analyzed for several crystals but none showed fully ordered density at the active site. Residues 66–76 in a surface loop and 401–404 near the catalytic His were not visible in Est55-pH6.2. In Est55-pH6.8 disorder occurred in the surface loops of residues 66–79, 349–358 and 401–407.
Table 1.
Crystallographic Data Collection and Refinement Statistics
| Est55-pH6.2 | Est55-pH6.8 | |
|---|---|---|
| Data collection | ||
| Space group | P212121 | P212121 |
| Unit cell Å | a=69.53, b=73.49, c=99.03 | a=69.36, b=74.430, c=98.60 |
| Unique reflections | 67530 | 71770 |
| Rmerge (last shell) | 0.07 (0.5) | 0.08 (0.4) |
| Redundancy | 5.3 | 5.1 |
| Completeness (last shell) % | 99.1 (95.6) | 97.8 (91.5) |
| <I/σ> | 17.6 | 17.5 |
| Refinement | ||
| Resolution Å | 10–2.02 | 10–1.58 |
| Rfactor /Rfree % | 20.0/23.0 | 17.2/23.7 |
| No. residues | 479 | 463 |
| No. water molecules | 151 | 159 |
| No. Iodine atoms | 3 | 1 |
| rms deviation from ideality | ||
| Bond length (Å) | 0.006 | 0.018 |
| Bond angles | 1.3a | 0.028b |
| Ramachandran plot | ||
| Most favored regions (%) | 89.3 | 89.4 |
| Additional allowed regions (%) | 10.2 | 10.1 |
| Averaged B-values (Å2) | ||
| Main chain atoms | 35.4 | 27.9 |
| Side chain atoms | 39.4 | 33.5 |
| Solvent atoms | 36.1 | 39.4 |
degree
angle distances
Est55 is folded into three domains, a catalytic domain, an α/β domain and a regulatory domain (Figure 1). The catalytic domain shows the typical α/β hydrolase fold, with a central antiparallel β sheet surrounded by α helices. The catalytic triad comprises Ser194, Glu310 and His409. The two structures of Est55-pH6.2 and Est55-pH6.8 superimposed with a RMSD value of 0.2 Å for Cα atoms, which indicated that there were no significant conformational changes in the overall structure. The Est55-pH6.2 structure was compared with the crystal structure of hCE1 in complex with the Alzheimer’s drug Tacrine, which was determined at 2.4 Å resolution.19 hCE1 and Est55 share 35 % sequence identity and 2.3 Å RMSD over 458 Cα atoms, although hCE1 has extended surface loops and the regulatory domain differs in conformation and length (Figure 2). A detailed comparison of the active sites is given in a later section.
Figure 1.
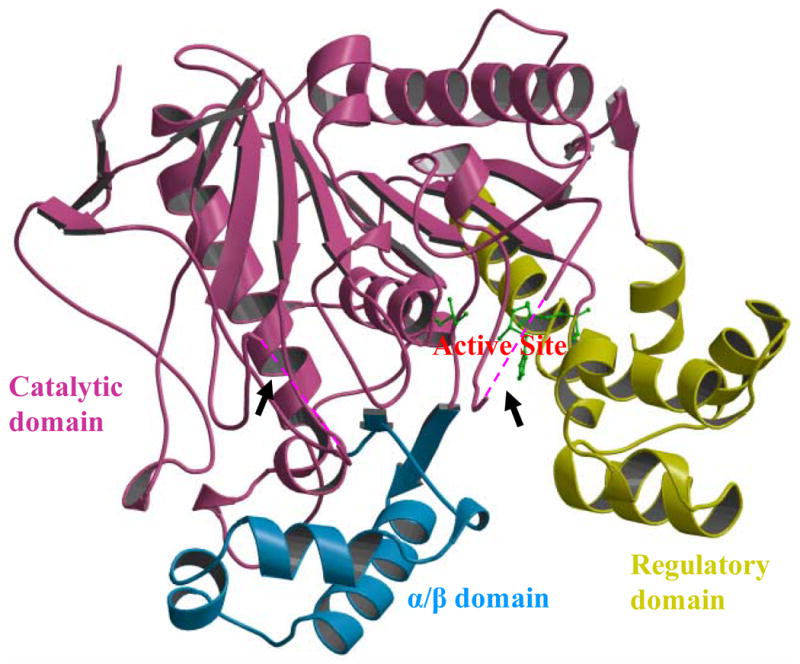
Overall structure of carboxylesterase Est55 comprising three domains. The catalytic domain is in magenta, the α/β domain is in blue, and the regulatory domain is in yellow. The catalytic residues Ser194, Glu310, and His409 are shown in green ball and stick representation. Black arrows point to two disordered loops in the Est55-pH6.2 structure, which are indicated by the dashed lines.
Figure 2.
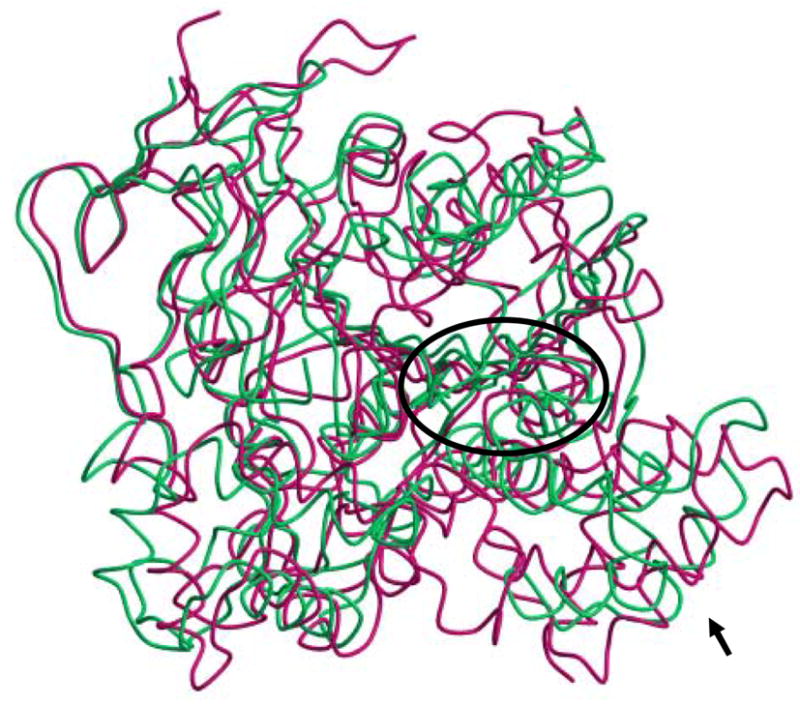
Superposition of Est55 and human carboxylesterase hCE1 structures. Est55 is green and hCE1 is red. The active sites are within the ellipse. The black arrow indicates the regulatory domain, which shows significant differences between the two structures.
Iodine in the Est55 structure
Iodine was necessary to produce large well-diffracting crystals. Three iodine atoms were identified in the Est55-pH6.2 structure, and single iodine in the Est55-pH6.8 structure. One iodine binding site was conserved in both structures, with 0.65 iodine occupancy in the Est55-pH6.2 structure and 0.34 occupancy in the Est55-pH6.8 structure, respectively. The iodine atom was identified by the very high level of electron density, even in the 2Fo-Fc map contoured at 10σ level, as shown in Fig 3A. The iodine had a hydrogen bond interaction with the amide of Asp309 and van der Waals interactions with the hydrophobic side chains of Phe488, Val372, and Thr367 and Arg393. Two more iodines were observed in the Est55-pH6.2 structure. The second iodine atom with 0.65 occupancy was located near the active site and had hydrogen bond interactions with amides of Ala407 and Cys408, the carbonyl oxygen of Cys408 and two water molecules. It formed van der Waals interactions with the side chains of Phe112, Lys406, Cys408 and the main chain of Gly108 (Fig 3B). The interactions of this iodine with Lys406 and Cys408 appear to stabilize the active site loop of Est55-pH6.2 that is disordered in Est55-pH6.8. However, iodine does not interact with the side chain of His409 in the catalytic triad. It is not obvious why the Est55-pH6.8 structure did not show iodine at this site. The third iodine was refined with partial occupancy and was located on the surface of the protein with an ionic interaction with the side chain of Arg377, a hydrogen bond interaction with amide of Ala400, and van der Waals interactions with main chain atoms of Pro399 and side chain of Glu290. This iodine was located next to the disordered loop of residues 401–404 in the active site of Est55-pH6.2 and interacted with Ala400 in a symmetry-related molecule. It was positioned partly in the density for one alternate conformation of the side chain of Arg293, indicating that the Arg293 side chain is in the other conformation in the presence of the iodine ion. The hydrogen bond interactions observed between the three iodine atoms and the amides of the protein have N….I distances in the range of 3.3–3.9 Å. Similar hydrogen bond distances have been observed for iodine in small molecule and protein crystal structures. 22; 23; 24; 25
Figure 3.
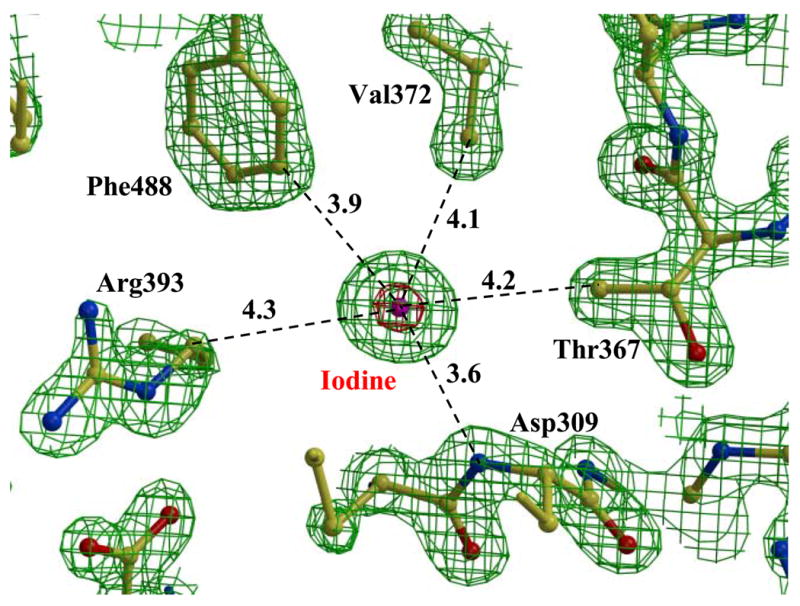
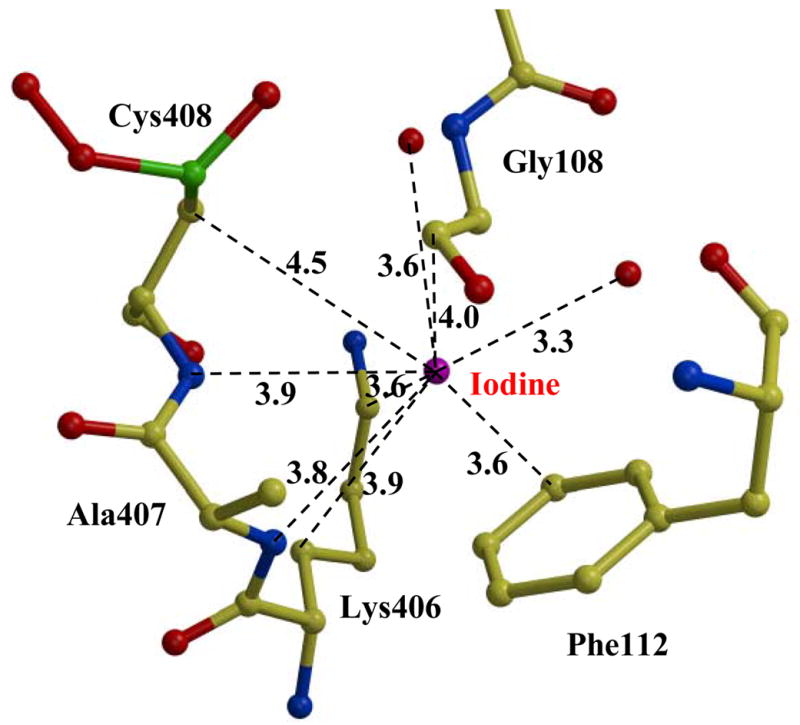
Iodine in the Est55 crystal structure. A) The conserved iodine site. 2Fo-Fc electron density map contoured at 2.0 σ (green) and 10.0 σ (red). Water is indicated by red spheres. Interatomic interactions are shown as dashed lines with distances in Å. B) The second iodine site in Est55-pH6.2.
Flexibility of the active site
The electron density in the active site region showed partial disorder in Est55-pH6.8, while more residues were visible in Est55-pH6.2. In the Est55-pH6.8 structure, the loop containing the catalytic His409 was partially disordered and residues 401–407 were not visible. Only one water molecule was observed, which formed hydrogen bond interactions with the side chains of Ser220 and two of catalytic triad residues Ser194 and Glu310. However, the structure of Est55-pH6.2 showed more ordered density near the active site. The loop around His409 showed clear electron density and residues 405–409 were fit unambiguously. The conformation of this loop was stabilized by several hydrogen bond and van der Waals interactions with the surrounding residues in the active site (Fig 4). No symmetry related molecules are close to the disordered loop in the active site. The active site residues are in similar positions in both Est55-pH6.8 and Est55-pH6.2 structures, with the exception of the disorder of residues 405–409 and a small rotation of the His409 side chain in Est55-pH6.8. The His409 side chain has hydrophobic interactions with Phe314 and Leu313 in both structures. In the pH6.2 structure the His409 side chain has weak hydrogen bonds with the main chain carbonyl oxygens of Leu313 and Lys406. In the pH6.8 structure the His side chain forms a good hydrogen bond with the carbonyl oxygen of Leu313, however, Lys406 is not visible due to the disorder. It is not clear whether the disorder and small structural changes are related to the protonation state of His409.
Figure 4.
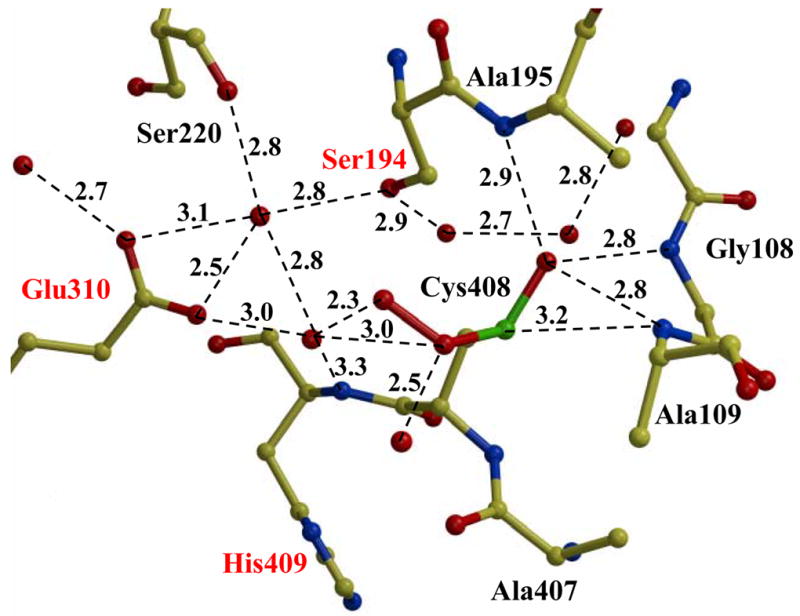
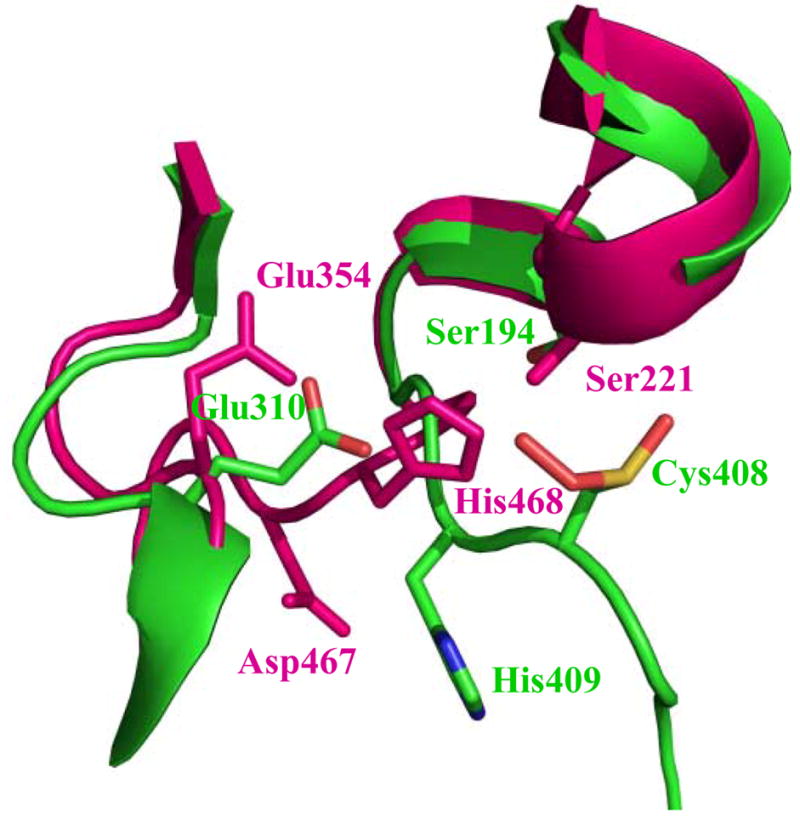
The active site region of Est55. Interatomic interactions are shown as dashed lines with distances in Å. The catalytic residues Ser194, Glu310 and His409 are labeled in red. B) Comparison of active sites of Est55 (green) and hCE1 (red). The catalytic triad residues of hCE1 are Ser221, Glu354, and His468. Cys408 in Est55 is equivalent to Asp467 in hCE1.
The catalytic triad of Est55 consists of Ser194, Glu310, and His409. The catalytic Ser194 was located at the bottom of a deep active site cleft and formed water-mediated hydrogen bond interactions with the carboxylate side chain of Glu310 (Fig 4). Although, the side chain of His409 was positioned unambiguously in the Est55-pH6.2 and Est55-pH6.8 structures, it was directed away from the other members of the catalytic triad, Ser194 and Glu310, and did not show the expected hydrogen bond interactions with their side chains. The rearrangement of the catalytic triad residues in Est55 is shown by comparison with the structure of hCE1 containing the catalytic triad of Ser221, Glu354, and His468 in an active conformation (Figure 4B). The catalytic triads of the two structures have distinct conformations. The serines are in similar positions. However, the side chains of the catalytic Glu and His of Est55 have moved away from the positions in hCE1. In particular, the His side chain is rotated by about 90 degrees from the His of hCE1, and the main chain continues in a different direction. The catalytic triad of Est55 appears to be in an inactive conformation in contrast to the active form in hCE1, suggesting that the partially disordered loop containing the catalytic His undergoes conformational changes when substrates or inhibitors bind. These observations suggested that the flexibility of the loop where His409 is located plays an important role in substrate binding and catalysis. It is likely that the active site is not fully formed until the substrate is bound within the catalytic gorge. The rCE structure also showed flexibility near the catalytic site.17 Disorder was observed for two loops in the rCE structure adjacent to the catalytic triad residues Glu353 and His467. Moreover, the catalytic Glu353 was rotated away from the active site relative to the conserved orientations observed in other esterases. Therefore, the existence of disordered active sites suggested that both Est55 and rCE enzymes bind the substrate by an induced fit mechanism.
Est55 contains an oxidized cysteine in the active site
Extra electron density was observed near the side chain of Cys408 in the Est55-6.2 structure (Figure 5A). This density indicated the presence of three oxygen atoms close to the cysteine sulfur. Refinement confirmed that the cysteine was oxidized to the sulfinylperoxy form of SO2O− (Figure 5C). This oxidation state is unusual, however, similar forms have been observed in oxidized small molecules at low temperatures. The small molecule sulfinylperoxy intermediate (−SO2O−) and cysteine sulfonyl peroxyl radicals (−SO2− and SO2OO−) have been reported.26; 27 The loop containing Cys408 and the catalytic His409 is not in the active conformation observed in other carboxylesterases such as hCE1 (Figure 4B). Instead, the His409 is rotated away and the Cys408 side chain lies closer to the Ser and His side chains in the catalytic triad of hCE1. The Cys408 side chain lies in the oxyanion hole of Est55 blocking the entrance of the active site cavity, which appears to mimic an inhibitor. The oxidized cysteine was stabilized by hydrogen bond interactions with amides of Gly108, Ala109, and Ala195, and with two water molecules in the oxyanion hole, as shown in Figure 4. A total of five water molecules occupied the active site and made hydrogen bond interactions with the side chains of Ser194, Ser220, Glu310, Cys408 and the amide of His409.
Figure 5.
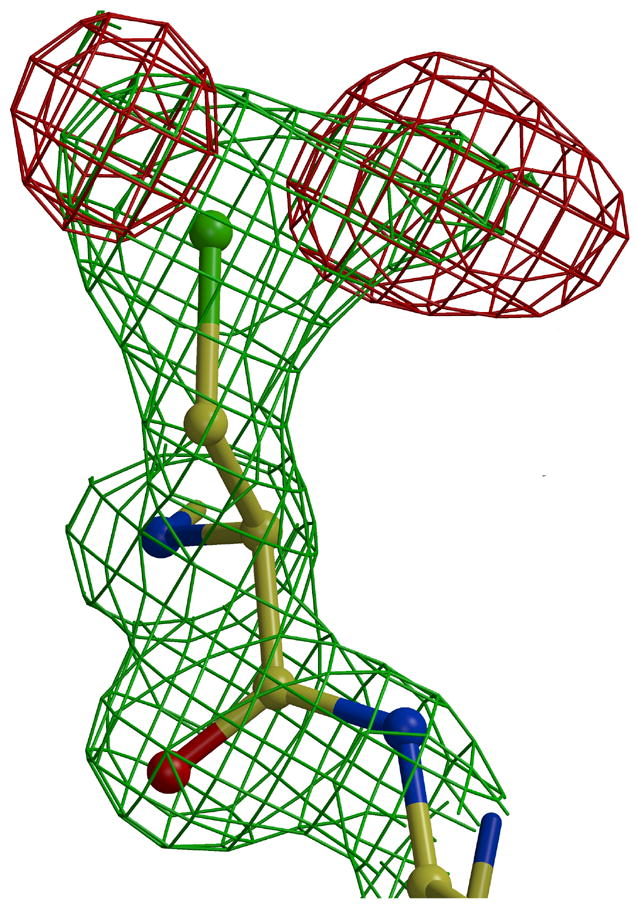
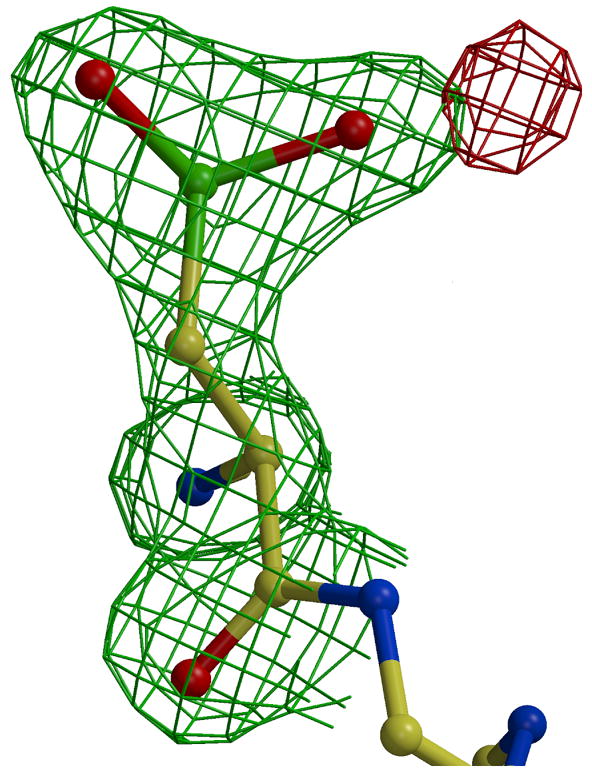
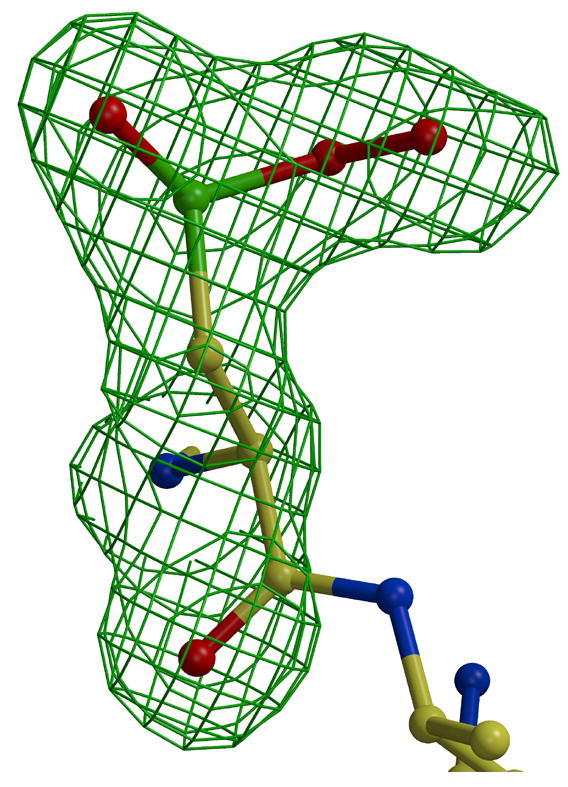
Oxidized Cys408. 2Fo-Fc electron density map contoured at 1.6 σ (green) and Fo-Fc electron density map contoured at 3.5 σ (red). A) without oxygen atoms. B) after addition and refinement of two oxygen atoms, and C) omit map after addition and refinement of three oxygen atoms.
The presence of oxidized cysteine has been described in several protein structures, mainly in the form of cysteine sulfenic acids (Cys-SOHs), sulfinic acids (Cys-SO2 H) and sulfonic acids (Cys-SO3H). Some of these oxidized cysteines have been reported to play roles in enzyme catalysis28; 29; 30. In one example, Cdc25B phosphatase has been crystallized in three oxidation states of the active site cysteine: apo protein, sulfenic intermediate, and disulfide bond state. The P-loop in the active site has a distinct conformation in each state. The active conformation is formed in the apoenzyme, the P-loop partially blocks the active site cysteine in the sulfenic state, and the P-loop closes the active site in the disulfide bond conformation. 31 Therefore, the P-loop in the active site and oxidization state of cysteine regulate the conformation and the activity of this enzyme. Similarly, the active site of Est55 is blocked by the oxidized Cys408, which may have a role in regulating enzyme activity. However, it has proved difficult to assess the effect of oxidation on Est55 since reducing agents like DTT alter the substrate used in the activity assay.
Role of Cys408 in the enzymatic activity of Est55
The role of Cys408 on enzyme activity was assessed by mutagenesis. Cys408 is unlikely to be essential for the enzyme reaction since the corresponding residue is Asp in rCE and hCE1 (Figure 4B), although several other bacterial carboxylesterases have cysteine next to the catalytic His. Therefore, a random mutant library of C408 was constructed using the Quick-change site-directed mutagenesis kit with degenerate primers. Mutations to the hydrophobic amino acids C408A, C408P, C408V, the polar amino acid C408S, and the flexible amino acid C408G were selected to test the enzymatic roles of cysteine. The different mutants were identified by DNA sequencing. All mutant enzymes were purified to homogeneity and their enzymatic activities were compared with that of the wild type enzyme. The enzymatic activity for p-nitrophenyl substrate (p-nitrophenyl butyrate, p-NP butyrate) was measured for both the wild type and the mutants to evaluate the effect of the mutants on catalysis. The kinetic parameters are shown in Table 2. Little variation was observed in the Km for wild type Est55 and mutants, except for the C408S. Mutations to hydrophobic amino acids, C408A, C408P, and C408V, showed similar Km values as the wild type. However, mutation to the polar serine in C408S resulted in a 4-fold increased Km value, suggesting that a polar environment is unfavorable for substrate binding. Substitutions of Cys408 showed conserved or improved kcat values, except for C408G that showed a 4 fold decrease in kcat relative to wild type Est55. Overall, the C408S and C408G mutants demonstrated decreased catalytic efficiency for p-NP butyrate hydrolysis. The lower activity of the serine mutant is consistent with the hydrophobic nature of the Est55 substrate binding pocket, which is formed by the hydrophobic side chains of Phe112, Trp119, Leu225, Leu226, Met274, Leu313, Phe314, Leu316, Val370, Phe371, and Leu411. Moreover, the lower activity of C408G suggested that the very flexible amino acid glycine was unfavorable at this position. Overall, the analysis of these mutations suggested that Cys408 is not essential at 408, and the substitution of hydrophobic side chains of alanine or valine in fact produces higher enzyme activity. However, these results do not rule out a possible role of Cys408 in the regulation of Est55 activity in the bacillus.
Table 2.
Effect of mutation at Cys408 on the hydrolysis of p-NP butyrate
| Km (μM) | kcat (s−1) | kcat/Km (s−1 μM−1) | |
|---|---|---|---|
| Est55 | 24.2 ± 2.5 | (7.9 ± 0.3) × 105 | (3.3 ± 0.4) × 104 |
| C408A | 32.8 ± 2.9 | (13.7 ± 0.4) × 105 | (4.2 ± 0.4) × 104 |
| C408G | 23.1 ± 2.7 | (2.2 ± 0.09) × 105 | (0.95 ± 0.17) × 104 |
| C408P | 26.0 ± 3.2 | (6.5 ± 0.3) × 105 | (2.51 ± 0.3) × 104 |
| C408S | 97.5 ± 4.3 | (12.7 ± 0.6) × 105 | (1.1 ± 0.05) × 104 |
| C408V | 28.3 ± 3.8 | (10.7 ± 0.3) × 105 | (4.5 ± 0.6) × 104 |
Est55 Converts CPT-11 to SN-38
Freshly purified Est55 was used to measure the ability of Est55 to convert CPT-11 to SN-38. The enzymatic conversion of CPT-11 to SN-38 was quantified by HPLC separation of the substrate and product. The peak positions for CPT-11 and SN-38 were confirmed by using the pure compounds. Also, the presence of SN-38 was confirmed by mass spectrometry of the isolated fraction giving the molecular mass of 393.1. Est55 activated CPT-11 with a kcat/Km value of 0.84 ± 0.08 min−1 mM−1 (Table 3).
Table 3.
Hydrolysis of CPT-11 by Est55 and mutants of Cys408
| Km (μM) | kcat (min−1) | kcat/Km (min−1 mM−1) | |
|---|---|---|---|
| rCE# | 6.2 ± 0.63 | 180 | |
| hCE# | 82.8 ± 9.6 | 0.2 | |
| Est30 | 202 ± 2 | (17.2 ± 1.2)× 10−3 | 0.09 ± 0.01 |
| Est55 | 18.6 ± 1.8 | (15.6 ± 0.5) × 10−3 | 0.8 ± 0.08 |
| C408A | 45.3 ± 6.9 | (15.5 ± 0.35)× 10−3 | 0.34 ± 0.05 |
| C408G | 22.4 ± 1.4 | (5.0 ± 0.12) × 10−3 | 0.23 ± 0.02 |
| C408P | 20.6 ± 2.1 | (2.9 ± 0.1) × 10−3 | 0.14 ± 0.01 |
| C408S | N. D.* | N. D.* | N. D.* |
| C408V | 22.3 ± 2.5 | (13.5 ± 0.6) × 10−3 | 0.6 ± 0.07 |
Kinetic parameters were taken from 46
No detectable hydrolysis
The mutant enzymes were also evaluated for hydrolysis of CPT-11 to SN-38 (Table 3). Various effects on prodrug activation were observed for the different substitutions of Cys408. The most dramatic effect was observed for the C408S mutant. No product peak for SN-38 was detected even after doubling the incubation time. Therefore, mutation to the polar serine has eliminated the ability to hydrolyze CPT-11. Mutation to glycine resulted in ~four-fold decrease in kcat/Km. Substitution of hydrophobic amino acids had variable effects on kinetic parameters. C408A mutant showed more than 2-fold increase in Km, while C408P and C408V had ~3- to 5-fold decreased kcat relative to wild type Est55. The C408V mutant was more similar to the wild type enzyme in catalytic efficiency (70%). The other two mutants C408A and C408P had decreased catalytic efficiency of 40 and 17% of wild type kcat/Km.
The effects of the Cys408 substitutions on Est55 activity differed for the two substrates, p-NP butyrate and CPT-11, as shown in Figure 6. Hydrolysis of p-NP butyrate showed relatively little change (76–140% of wild type Est55 activity) for the mutations to hydrophobic amino acids, alanine, valine and proline, whereas substitution of glycine or serine was unfavorable and resulted in only about 30% of wild type catalytic efficiency. In contrast, hydrolysis of CPT-11 was substantially reduced for all the tested mutants of Cys408, with the exception of C408V that showed 70% of wild type Est55 activity. Moreover, the serine mutant had no detectable activity. These results suggested that the hydrophobicity of residue 408 was more critical for binding and hydrolysis of CPT-11 compared with p-NP butyrate.
Figure 6.
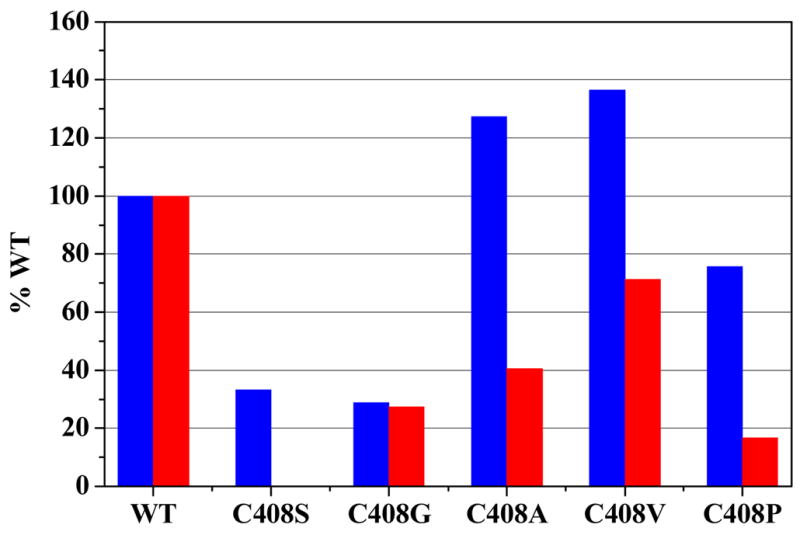
Relative activity of Est55 Mutants on p-NP butyrate (blue) and CPT-11 (red).
In comparison with the published kinetic data for CPT-11 activation by other carboxylesterases, Est55 showed an increased catalytic efficiency compared to hCE, although rabbit carboxylesterase was about 200-fold more efficient (Table 3). The Est55 mutants containing hydrophobic substitutions for Cys408 showed hydrolysis of CPT-11 Also, CPT-11 activation was assessed for Est30 from Geobacillus stearothermophilus, which is only distantly related to Est55 5; 6. Est30 showed relatively little activity with about 10-fold lower catalytic efficiency than Est55, mainly due to an increased Km. CPT-11 is a bulky molecule comprising the 5-ring system of the product SN-38 and the ester link to the 4PP group. Therefore, it is likely that the low activity of most tested enzymes arises at least partly from the difficulty of fitting such a large molecule in the active site. Unfortunately, there are no structural data to support analysis of CPT-11 binding in the active sites of carboxylesterases. First, no structures are available for the CPT-11 substrate or the SN-38 product bound at the active site of a carboxylesterase. The crystal structure of rCE showed the product 4PP bound to the enzyme surface outside of the active site.17 Similarly, the inhibitory CPT-11 was bound 9 Å away from the catalytic serine of TcAChE. 22 In fact, no structures are available for ligands of similar size to CPT-11 bound in the active site of a related carboxylesterase. All structures of hCE show smaller ligands at the active site.18; 19; 20; 21 Second, the active sites are partially disordered in the structures of both Est55 and the most efficient enzyme rCE. The partial disorder of the active site and rotation of the loop containing the catalytic His409 away from the active conformation make it difficult to construct a model for CPT-11 bound in the active site of Est55.
Conclusions
We have confirmed the feasibility of using Est55 as a model in the development of a more efficient enzyme for therapeutic activation of the prodrug CTP-11 to the drug SN-38. Est55 showed similar activity to hCE for hydrolysis of CPT-11, although rCE is still the most efficient enzyme. However, the bacterial enzyme Est55 has the advantages of high thermostability and catalytic efficiency at mesophilic temperatures.5 The other advantages of proteins of bacterial origin compared to those of mammalian cells are relatively easy overexpression, purification and engineering. These advantages suggest that Est55 can provide an attractive starting point to engineer an enzyme with higher stability for more efficient conversion of prodrug CPT-11 into the active drug during cancer therapy. Future engineering of mutants with higher affinity for CPT-11 would benefit from a structural model for CPT-11 bound to Est55. However, the current crystal structure does not contain CPT-11, the catalytic traid residues are not in the active conformation, and oxidized Cys408 blocks entry to the active site. Mutant C408V may provide a more stable construct by removing the only cysteine residue, which can oxidize as shown in the crystal structure and potentially alter Est55 activity.
METHODS
Site-directed mutagenesis, expression and purification of recombinant Est55
Random mutations of C408 were generated by the QuickChange mutagenesis kit (Stratagene) using the following degenerate primers: CYS408_f (5′-GCTGAAAGCANNNCACGCGCTCGAGCTGCCGTTTGTG-3′) and CYS408_r (5′-CGAGCGCGTGNNNTGCTTTCAGCTGGCCGCCGAACAC-3′). The mutation sites were confirmed by DNA sequencing. Est55 and the Cys408 mutants were expressed and purified to homogeneity as described previously.5 Briefly, the enzyme was expressed using E. coli Top 10 cells carrying the recombinant clone pHE55. The wild type Est55 and the mutants were purified in two steps, ion exchange using high-load Q-Sepharose HR 26/10 (Pharmacia) and gel filtration using Sephacryl S200 (HR26/60) columns. The protein concentration was measured by the Bradford method with bovine serum albumin as the standard. 32
Carboxylesterase assay
The esterase activity was measured as described with modification.5 The substrate was p-nitrophenyl butyrate. The reaction mixture (2 ml) contained 10 mM Tris HCl (pH 8.5) and 0.5 mM substrate and 2 ng protein. The amount of p-nitrophenol liberated during the reaction was monitored continuously at 405 nm in a Carey 3E spectrophotometer (Varian) with a temperature control module. The molar extinction coefficient of p-nitrophenol is 18,380 M−1 cm−1 at 405 nm. Kinetic parameters were determined by fitting the data obtained at less than 20% substrate hydrolysis with the Michaelis-Menten equation using the SigmaPlot 8.0.2 program (SPSS, Inc.).
HPLC assay for enzymatic conversion of CPT-11 to SN-38
The reaction was carried out as described33 with minor modifications. In brief, freshly purified Est55 was incubated with different concentrations of CPT-11 between 1.67μM to 100 μM in a final volume of 300 μl of 50 mM HEPES (pH 7.4) at 37°C for 8hrs. The reaction was terminated with the addition of one volume of 75mM ammonium acetate (pH 4.0), vortexed, and centrifuged at 14,000 g for 2 min. The kinetic parameters for conversion of CPT-11 were obtained from HPLC separation of substrate and product as described14. An aliquot of the reaction was injected onto an Atlantis™ dC 18 chromatography column (4.6 × 50 mm) using an automatic injector (Amersham A-900) on an AKTA Purifier. A step gradient was run with 75mM ammonium acetate (pH 4.0), and 0 and 25% acetonitrile to separate the substrate and product. The reaction mixture was eluted at 0.75 ml/min. Under these conditions, the SN-38 and CPT-11 were eluted at 3.7 ml and 5.5 ml, respectively. The two compounds were detected by UV absorbance at wavelengths of 266 nm and 375 nm. The absorbance was converted to SN-38 concentration via the standard calibration curve of UV peak area generated by known concentrations of SN-38. Kinetic parameters were determined as described above for p-NP butyrate hydrolysis.
Crystallization and X-ray data collection
Purified Est55 was dialyzed into Tris buffer at pH 8.0 and concentrated to 7–10 mg/ml. Est55 was crystallized by the hanging drop vapor diffusion method at 24°C using 1 μl of protein and 1 μl of mother liquor. Crystallization trials used the Hampton PEG/ION screen. Crystals grew after 7–15 days with 20% w/v polyethylene glycol 3350, 0.2 M ammonium iodide pH 6.2, or 0.2 M potassium iodide pH 6.8. Crystals were mounted on a nylon loop and cryo-protected in 20 % (v/v) PEG400 before flash cooling in liquid nitrogen. X-ray diffraction data were collected on a MAR CCD 225 detector at the SER-CAT beam line of the Advanced Photon Source (APS) at the Argonne National Laboratory. The crystal-to-detector distance was 142 mm. The oscillation angle was 0.25° and the rotation range was 180° for the complete dataset. X-ray diffraction data for the crystals grown at pH 6.8 and pH 6.2 were measured to 1.58 Å and 2.02 Å resolution, respectively.
Structure determination and refinement
The X-ray data were integrated using the HKL2000 program.34 The structure of Est55 at pH 6.8 (Est-pH6.8) was determined by molecular replacement with the CNS package. 35 The structure of Pnb esterase (1QE3) from Bacillus subtilis was used as a search model (40 % sequence identity).16 Data between 15 and 4 Å were used in the rotation research, resulting in one peak. The rotation solution was then applied in a translation search, yielding a distinct solution with a correction coefficient of 0.20. After rigid body refinement (CNS) and density modification using the phase probability distribution calculated from the model and the experimental amplitudes and solvent flipping, the figures-of-merit improved to 0.82. The Est55-pH6.2 structure was determined by molecular replacement with the Est55-pH6.8 structure using the program MolRep36 in CCP4i. 37
Crystallographic refinement of the Est55-pH6.8 structure was initially carried out using the CNS package. 35 The manual building of missing residues of structure was performed using the graphics program O. 38 After iteratively subjecting the model to several rounds of stimulated annealing and B-group refinement with CNS and manual rebuilding, the Rfactor and Rfree dropped to 26.4 % and 28.5 %, respectively. After manual building of missing residues, SHELX 9739 was used to refine the Est55-pH6.8 structure with solvent and anisotropic temperature factors and reduced the R values to the final Rfactor of 17.2% and Rfree of 23.7% Crystallographic refinement of the Est55-pH6.2 structure was carried out using CNS. The final Rfactor and Rfree were 20.0 % and 23.0 %, respectively.
Structural Analysis
The Est55 crystal structures at different pHs were superimposed on all Cα atoms using an implementation of the algorithm described in 40 to find the least square error coordinate transformation. The structures of Est55 and the human carboxylesterase were superimposed on all Cα atoms using the program DaliLite 41. Figures were generated with MolScript,42 BobScript,43 Raster3D,44 PyMOL.45
Protein Data Bank accession numbers
The structural data have been deposited in the Protein Data Bank with accession codes XXXX for Est55-pH6.8 and XXXX for Est55-pH6.2.
Acknowledgments
Ping Liu was supported by a Molecular Basis of Disease Fellowship. Irene Weber is a Distinguished Cancer Scholar. We thank the staff at the SER-CAT beamline at the Advanced Photon Source, Argonne National Laboratory, for assistance during X-ray data collection. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Basic Energy Sciences, Office of Science, under Contract No. W-31-109-Eng-38. The research was supported in part by the Georgia Research Alliance, the Georgia Cancer Coalition, and National Institutes of Health award GM 34776.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Potter PM, Wadkins RM. Carboxylesterases--detoxifying enzymes and targets for drug therapy. Curr Med Chem. 2006;13:1045–1054. doi: 10.2174/092986706776360969. [DOI] [PubMed] [Google Scholar]
- 2.Ollis DL, Cheah E, Cygler M, Dijkstra B, Frolow F, Franken SM, Harel M, Remington SJ, Silman I, Schrag J, Sussman JL. The alpha/beta hydrolase fold. Protein Eng. 1992;5:197–211. doi: 10.1093/protein/5.3.197. [DOI] [PubMed] [Google Scholar]
- 3.Heikinheimo P, Goldman A, Jeffries C, Ollis DL. Of barn owls and bankers: a lush variety of alpha/beta hydrolases. Struct Fold Des. 1999;7:141–146. doi: 10.1016/s0969-2126(99)80079-3. [DOI] [PubMed] [Google Scholar]
- 4.Nardini M, Dijkstra BW. a/ß hydrolase fold enzymes: the family keeps growing. Curr Opin Struct Biol. 1999;9:732–737. doi: 10.1016/s0959-440x(99)00037-8. [DOI] [PubMed] [Google Scholar]
- 5.Ewis HE, Abdelal AT, Lu CD. Molecular cloning and characterization of two thermostable carboxyl esterases from Geobacillus stearothermophilus. Gene. 2004;329:187–195. doi: 10.1016/j.gene.2003.12.029. [DOI] [PubMed] [Google Scholar]
- 6.Liu P, Wang YF, Ewis HE, Abdelal AT, Lu CD, Harrison RW, Weber IT. Covalent reaction intermediate revealed in crystal structure of the Geobacillus stearothermophilus carboxylesterase Est30. J Mol Biol. 2004;342:551–561. doi: 10.1016/j.jmb.2004.06.069. [DOI] [PubMed] [Google Scholar]
- 7.Tang BK, Kalow W. Variable activation of lovastatin by hydrolytic enzymes in human plasma and liver. Eur J Clin Pharmacol. 1995:449–451. doi: 10.1007/BF00196860. [DOI] [PubMed] [Google Scholar]
- 8.Fuchs C, Mitchell EP, Hoff PM. Irinotecan in the treatment of colorectal cancer. Cancer Treat Rev. 2006;32:491–503. doi: 10.1016/j.ctrv.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 9.Gupta E, Mick R, Ramirez J, Wang X, Lestingi TM, Vokes EE, Ratain MJ. Pharmacokinetic and pharmacodynamic evaluation of the topoisomerase inhibitor irinotecan in cancer patients. J Clin Oncol. 1997;15:1502–1510. doi: 10.1200/JCO.1997.15.4.1502. [DOI] [PubMed] [Google Scholar]
- 10.Rivory LP, Haaz MC, Canal P, Lokiec F, Armand JP, Robert J. Pharmacokinetic interrelationships of irinotecan (CPT-11) and its three major plasma metabolites in patients enrolled in phase I/II trials. Clin Cancer Res. 1997;3:1261–1266. [PubMed] [Google Scholar]
- 11.Potter PM, Pawlik CA, Morton CL, Naeve CW, Danks MK. Isolation and partial characterization of a cDNA encoding a rabbit liver carboxylesterase that activates the prodrug irinotecan (CPT-11) Cancer Res. 1998;58:2646–2651. [PubMed] [Google Scholar]
- 12.Khanna R, Morton CL, Danks MK, Potter PM. Proficient metabolism of irinotecan by a human intestinal carboxylesterase. Cancer Res. 2000;60:4725–4728. [PubMed] [Google Scholar]
- 13.Humerickhouse R, Lohrbach K, Li L, Bosron WF, Dolan ME. Characterization of CPT-11 hydrolysis by human liver carboxylesterase isoforms hCE-1 and hCE-2. Cancer Res. 2000;60:1189–1192. [PubMed] [Google Scholar]
- 14.Danks MK, Morton CL, Krull EJ, Cheshire PJ, Richmond LB, Naeve CW, Pawlik CA, Houghton PJ, Potter PM. Comparison of activation of CPT-11 by rabbit and human carboxylesterases for use in enzyme/prodrug therapy. Clin Cancer Res. 1999;5:917–924. [PubMed] [Google Scholar]
- 15.Aghi M, Hochberg F, Breakefield XO. Prodrug activation enzymes in cancer gene therapy. J Gene Med. 2000;2:148–164. doi: 10.1002/(SICI)1521-2254(200005/06)2:3<148::AID-JGM105>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 16.Spiller B, Gershenson A, Arnold FH, Stevens RC. A structural view of evolutionary divergence. Proc Natl Acad Sci U S A. 1999;96:12305–12310. doi: 10.1073/pnas.96.22.12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bencharit S, Morton CL, Howard-Williams EL, Danks MK, Potter PM, Redinbo MR. Structural insights into CPT-11 activation by mammalian carboxylesterases. Nat Struct Biol. 2002;9:337–342. doi: 10.1038/nsb790. [DOI] [PubMed] [Google Scholar]
- 18.Bencharit S, Morton CL, Xue Y, Potter PM, Redinbo MR. Structural basis of heroin and cocaine metabolism by a promiscuous human drug-processing enzyme. Nat Struct Biol. 2003;10:345–356. doi: 10.1038/nsb919. [DOI] [PubMed] [Google Scholar]
- 19.Bencharit S, Edwards CC, Morton CL, Howard-Williams EL, Kuhn P, Potter PM, Redinbo MR. Crystal structure of human carboxylesterase 1 complexed with the Alzheimer’s drug tacrine: from binding promiscuity to selective inhibition. Chem Biol. 2003;10:341–349. doi: 10.1016/s1074-5521(03)00071-1. [DOI] [PubMed] [Google Scholar]
- 20.Fleming CD, Bencharit S, Edwards CC, Hyatt JL, Tsurkan L, Bai F, Fraga C, Morton CL, Howard-Williams EL, Potter PM, Redinbo MR. Structural insights into drug processing by human carboxylesterase 1: tamoxifen, mevastatin, and inhibition by benzil. J Mol Biol. 2005;352:165–177. doi: 10.1016/j.jmb.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 21.Bencharit S, Edwards CC, Morton CL, Howard-Williams EL, Kuhn P, Potter PM, Redinbo MR. Multisite promiscuity in the processing of endogenous substrates by human carboxylesterase 1. J Mol Biol. 2006;363:201–214. doi: 10.1016/j.jmb.2006.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harel M, Hyatt JL, Brumshtein B, Morton CL, Yoon KJ, Wadkins RM, Silman I, Sussman JL, Potter PM. The crystal structure of the complex of the anticancer prodrug 7-ethyl-10-[4-(1-piperidino)-1-piperidino]-carbonyloxycamptothecin (CPT-11) with Torpedo californica acetylcholinesterase provides a molecular explanation for its cholinergic action. Mol Pharmacol. 2005;67:1874–1881. doi: 10.1124/mol.104.009944. [DOI] [PubMed] [Google Scholar]
- 23.Sheldrick GM, Sheldrick WS. Crystal structure of trimethylammonium iodine; refinement of absorption for a laminar crystal. Acta Crystallogr B. 1970;26:1334–1338. [Google Scholar]
- 24.Dale SH, Elsegood MR, Holmes KE, Kelly PF. The effect of hydrogen-bonding anions on the structure of metal-sulfimide complexes. Acta Crystallogr C. 2005;61:m34–m39. doi: 10.1107/S0108270104029051. [DOI] [PubMed] [Google Scholar]
- 25.Ghosh D, Erman M, Sawicki M, Lala P, Weeks DR, Li N, Pangborn W, Thiel DJ, Jornvall H, Gutierrez R, Eyzaguirre J. Determination of a protein structure by iodination: the structure of iodinated acetylxylan esterase. Acta Crystallogr D Biol Crystallogr. 1999;55:779–784. doi: 10.1107/s0907444999000244. [DOI] [PubMed] [Google Scholar]
- 26.Sevilla MD, Becker D, Yan M. The formation and structure of the sulfoxyl radicals RSO-, RSOO-, RSO2-, and RSO2OO- from the reaction of cysteine, glutathione and penicillaminethiyl radicals with molecular oxygen. Int J Raciat Biol. 1990;57:65–81. doi: 10.1080/09553009014550351. [DOI] [PubMed] [Google Scholar]
- 27.Kim YH, Yoon DC. Efficient oxidation of sulfides to the sulfoxides using a new sulfinylperoxy intermediate generated from 2-nitrobenzenesulfinyl chloride and superoxide. Tetrahedron Lett. 1988;29:6453–6456. [Google Scholar]
- 28.Stehle T, Claiborne A, Schulz GE. NADH binding site and catalysis of NADH peroxidase. Eur J Biochem. 1993;211:221–226. doi: 10.1111/j.1432-1033.1993.tb19889.x. [DOI] [PubMed] [Google Scholar]
- 29.Mande SS, Parsonage D, Claiborne A, Hol WG. Crystallographic analyses of NADH peroxidase Cys42Ala and Cys42Ser mutants: active site structures, mechanistic implications, and an unusual environment of Arg 303. Biochemistry. 1995;34:6985–6992. doi: 10.1021/bi00021a010. [DOI] [PubMed] [Google Scholar]
- 30.Mizohata E, Sakai H, Fusatomi E, Terada T, Murayama K, Shirouzu M, Yokoyama S. Crystal structure of an archaeal peroxiredoxin from the aerobic hyperthermophilic crenarchaeon Aeropyrum pernix K1. J Mol Biol. 2005;354:317–329. doi: 10.1016/j.jmb.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 31.Buhrman G, Parker B, Sohn J, Rudolph J, Mattos C. Structural mechanism of oxidative regulation of the phosphatase Cdc25B via an intramolecular disulfide bond. Biochemistry. 2005;44:5307–5516. doi: 10.1021/bi047449f. [DOI] [PubMed] [Google Scholar]
- 32.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;7:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 33.Guichard SM, Morton CL, Krull EJ, Stewart CF, Danks MK, Potter PM. Conversion of the CPT-11 metabolite APC to SN-38 by rabbit liver carboxylesterase. Clin Cancer Res. 1998;4:3089–3094. [PubMed] [Google Scholar]
- 34.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter JCW, Sweet RM, editors. Methods in Enzymology. Vol. 276. Academic Press; New York: 1997. [DOI] [PubMed] [Google Scholar]
- 35.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 36.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Cryst. 1997;30:1022–1025. [Google Scholar]
- 37.Potterton E, Briggs P, Turkenburg M, Dodson E. A graphical user interface to the CCP4 program suite. Acta Crystallogr D Biol Crystallogr. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 38.Jones TA, Zou JY, Cowan SW, Kjeldgaard Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 39.Sheldrick GM. Program for Crystal Structure Analysis. University of Gottingen; Germany: 1997. SHELX97. [Google Scholar]
- 40.Ferro DR, Hermans J. A different best rigid-body molecular fit routine. Acta Cryst A. 1977;33:345–347. [Google Scholar]
- 41.Holm L, Park J. DaliLite workbench for protein structure comparison. Bioinformatics. 2000;16:566–567. doi: 10.1093/bioinformatics/16.6.566. [DOI] [PubMed] [Google Scholar]
- 42.Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Cryst. 1991;24:946–950. [Google Scholar]
- 43.Esnouf RM. Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crystallogr D Biol Crystallogr. 1999;55:938–940. doi: 10.1107/s0907444998017363. [DOI] [PubMed] [Google Scholar]
- 44.Merritt EA, Bacon DJ. Raster3D photorealistic Molecular Graphics. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 45.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]
- 46.Wadkins RM, Morton CL, Weeks JK, Oliver L, Wierdl M, Danks MK, Potter PM. Structural constraints affect the metabolism of 7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin (CPT-11) by carboxylesterases. Mol Pharmacol. 2001;60:355–362. doi: 10.1124/mol.60.2.355. [DOI] [PubMed] [Google Scholar]